Selective Modification of Streptozotocin at the C3 Position to Improve Its Bioactivity as Antibiotic and Reduce Its Cytotoxicity towards Insulin-Producing β Cells

 ,
,  ,
,  and
and
Abstract
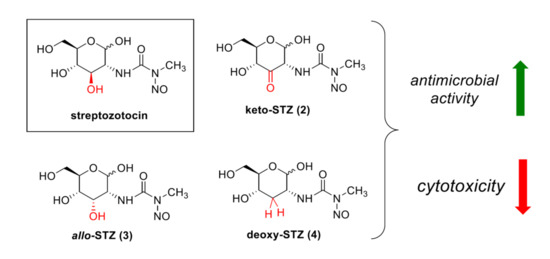
:
1. Introduction
2. Results
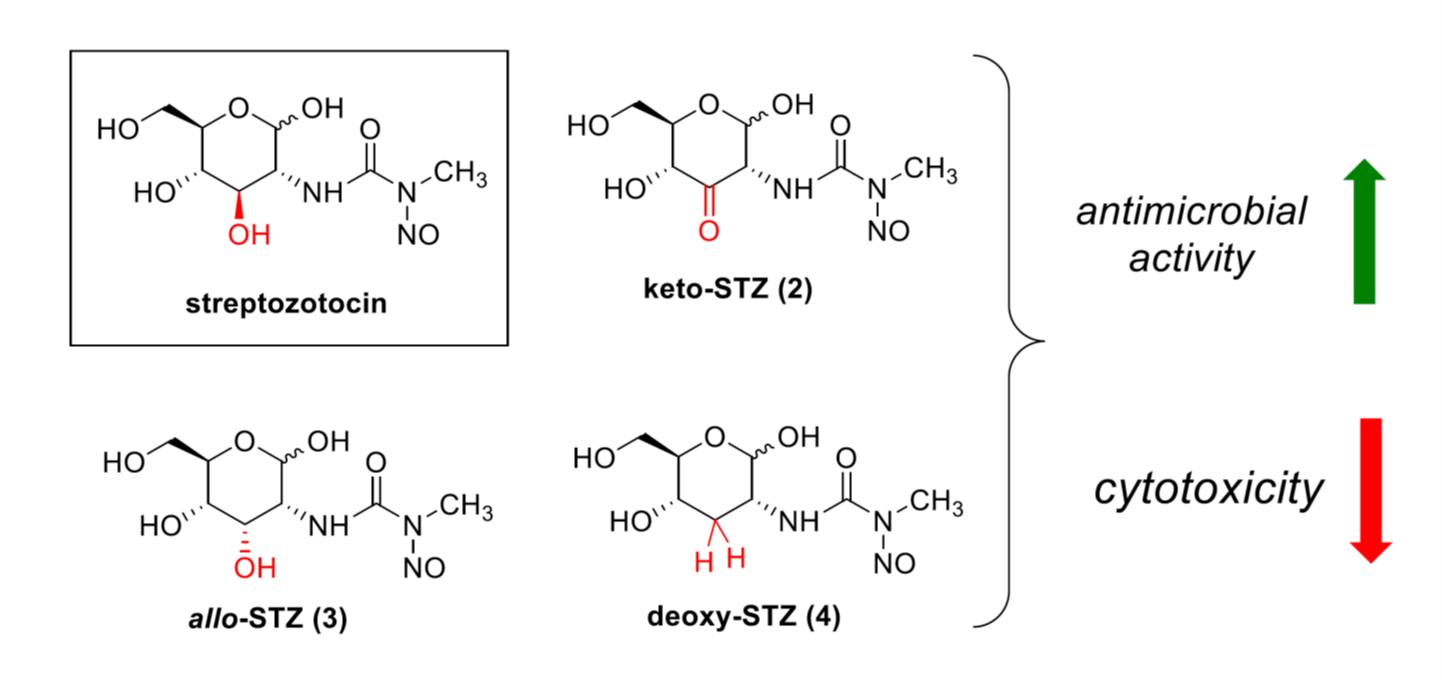
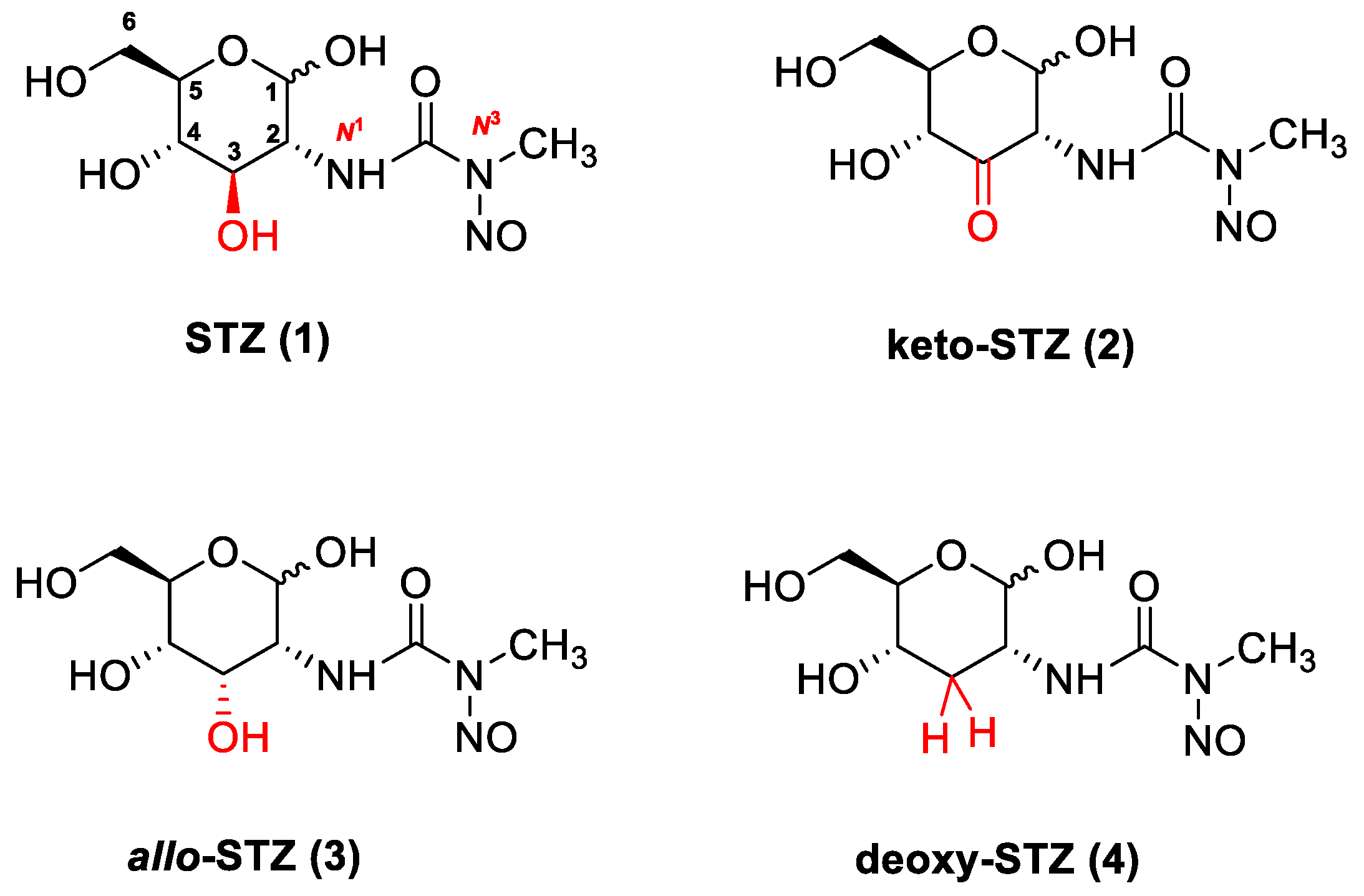
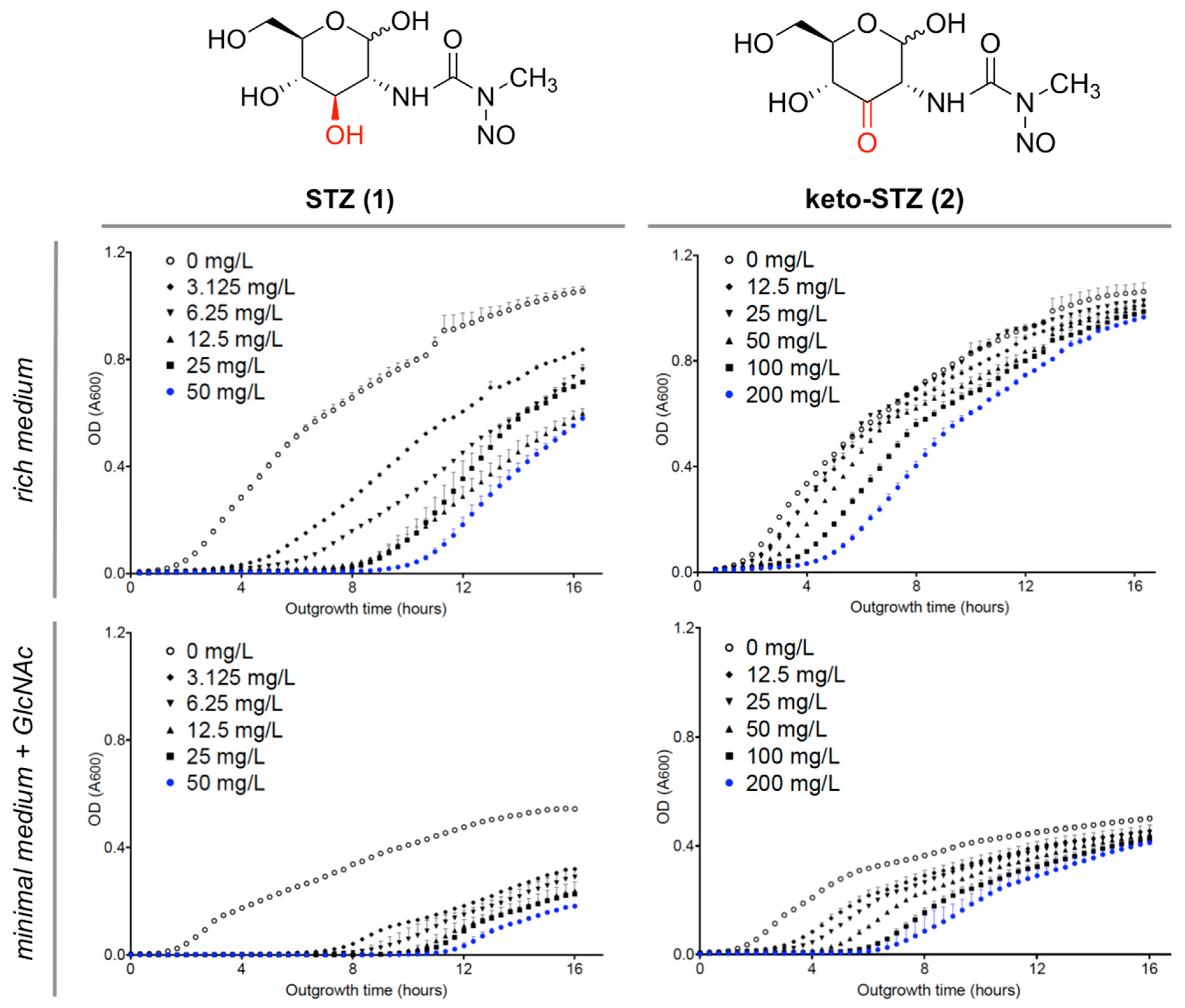
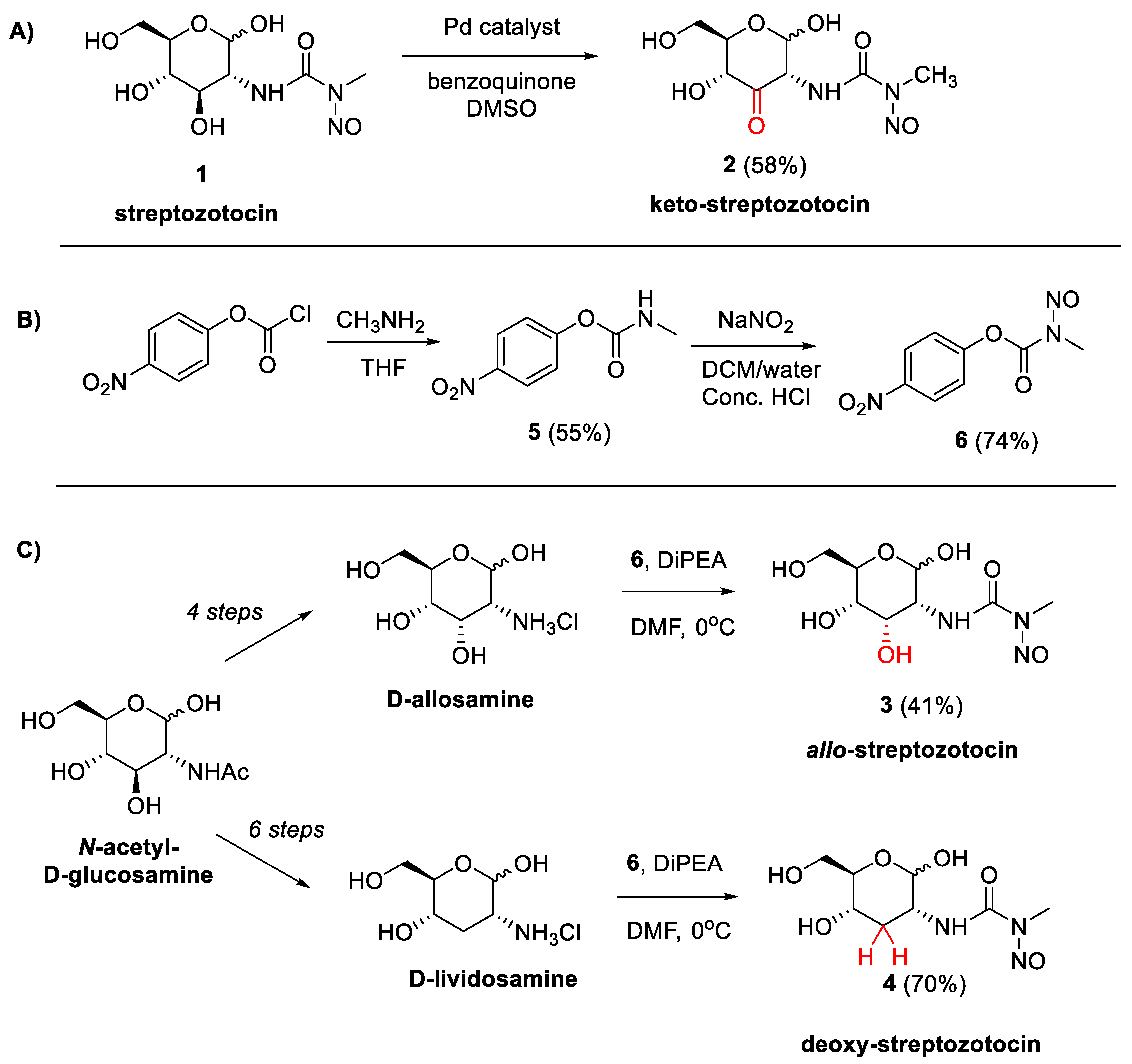
2.1. Chemical Synthesis of STZ Derivatives 2–4
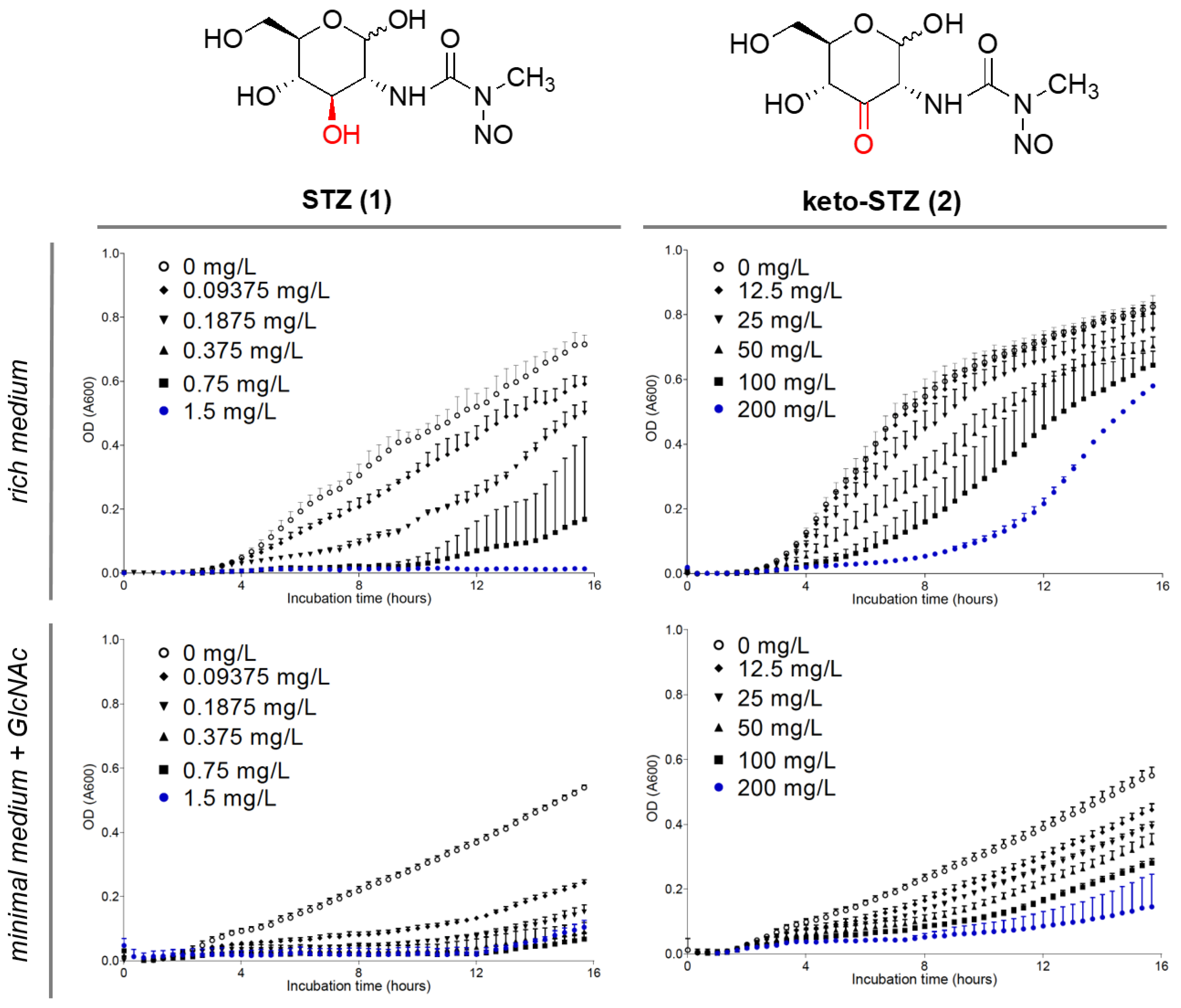
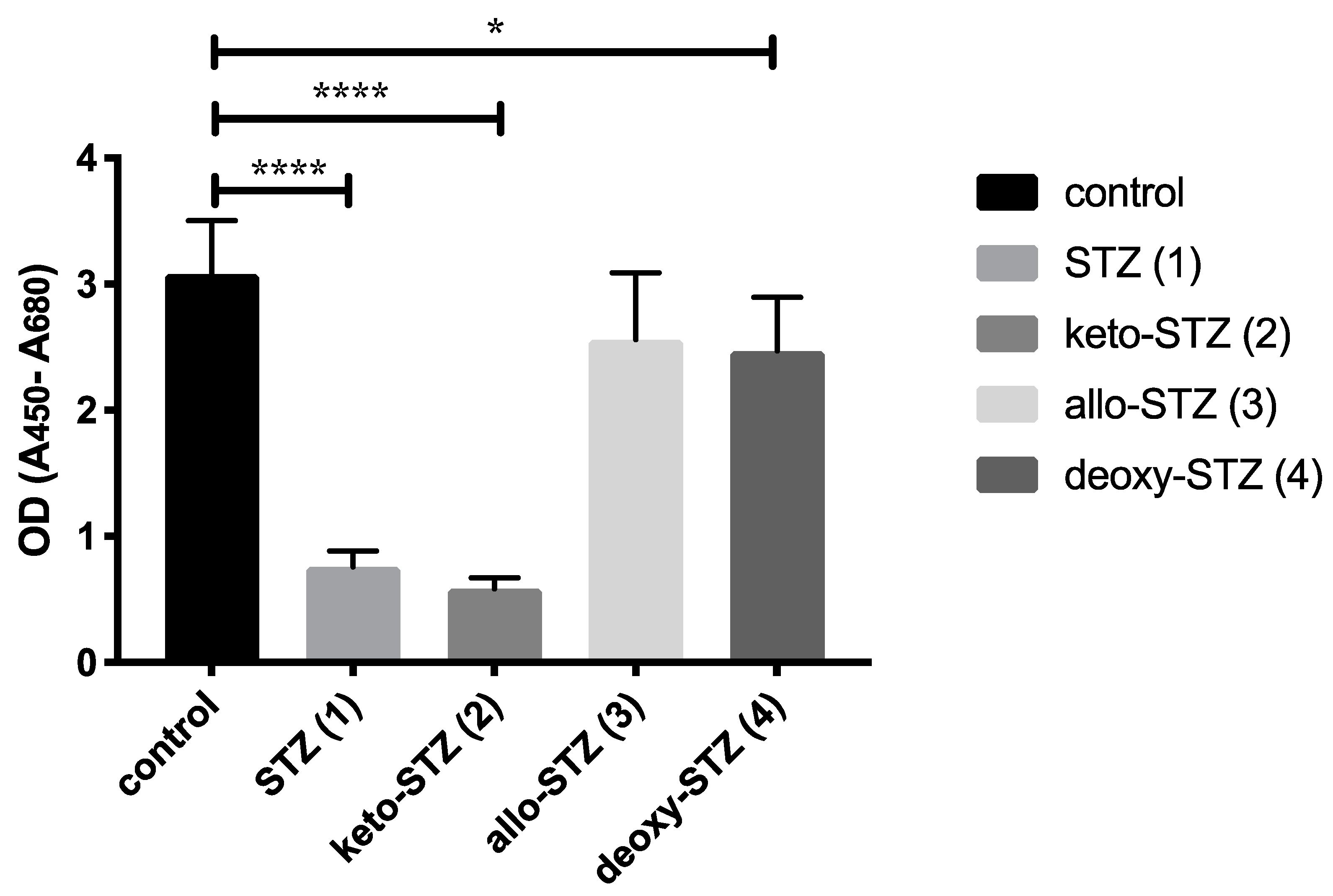
2.2. Biological Evaluation of STZ Compounds 1–4
3. Discussion
4. Materials and Methods
4.1. Determination of MIC Values
4.2. Growth-Based Viability Assays
4.3. Cytotoxicity Assays
Supplementary Materials
Author Contributions
Funding
Acknowledgments
Conflicts of Interest
References
- Mühlberg, E.; Umstätter, F.; Kleist, C.; Domhan, C.; Mier, W.; Uhl, P. Renaissance of vancomycin: Approaches for breaking antibiotic resistance in multidrug-resistant bacteria. Can. J. Microbiol. 2020, 66, 11–16. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Khan, F.; Pham, D.T.N.; Kim, Y.-M. Alternative strategies for the application of aminoglycoside antibiotics against the biofilm-forming human pathogenic bacteria. Appl. Microbiol. Biotechnol. 2020, 104, 1955–1976. [Google Scholar] [CrossRef] [PubMed]
- Chandrika, N.T.; Garneau-Tsodikova, S. Comprehensive review of chemical strategies for the preparation of new aminoglycosides and their biological activities. Chem. Soc. Rev. 2018, 47, 1189–1249. [Google Scholar] [CrossRef] [PubMed]
- Kotra, L.P.; Haddad, J.; Mobashery, S. Aminoglycosides: Perspectives on Mechanisms of Action and Resistance and Strategies to Counter Resistance. Antimicrob. Agents Chemother. 2000, 44, 3249–3256. [Google Scholar] [CrossRef] [Green Version]
- Ramirez, M.S.; Tolmasky, M.E. Aminoglycoside modifying enzymes. Drug Resist. Updat. 2010, 13, 151–171. [Google Scholar] [CrossRef] [Green Version]
- Kato, T.; Yang, G.; Teo, Y.; Juskeviciene, R.; Perez-Fernandez, D.; Shinde, H.M.; Salian, S.; Bernet, B.; Vasella, A.; Böttger, E.C.; et al. Synthesis and Antiribosomal Activities of 4′-O-, 6′-O-, 4″-O-, 4′,6′-O- and 4″,6″-O-Derivatives in the Kanamycin Series Indicate Differing Target Selectivity Patterns between the 4,5- and 4,6-Series of Disubstituted 2-Deoxystreptamine Aminoglycoside Antibiotics. ACS Infect. Dis. 2015, 1, 479–486. [Google Scholar] [CrossRef]
- Chen, W.; Matsushita, T.; Shcherbakov, D.; Boukari, H.; Vasella, A.; Böttger, E.C.; Crich, D. Synthesis, antiribosomal and antibacterial activity of 4′-O-glycopyranosyl paromomycin aminoglycoside antibiotics. MedChemComm 2014, 5, 1179–1187. [Google Scholar] [CrossRef]
- Matsushita, T.; Chen, W.; Juskeviciene, R.; Teo, Y.; Shcherbakov, D.; Vasella, A.; Böttger, E.C.; Crich, D. Influence of 4′-O-Glycoside Constitution and Configuration on Ribosomal Selectivity of Paromomycin. J. Am. Chem. Soc. 2015, 137, 7706–7717. [Google Scholar] [CrossRef]
- Sati, G.C.; Sarpe, V.A.; Furukawa, T.; Mondal, S.; Mantovani, M.; Hobbie, S.N.; Vasella, A.; Böttger, E.C.; Crich, D.; Mantovani, M. Modification at the 2′-Position of the 4,5-Series of 2-Deoxystreptamine Aminoglycoside Antibiotics To Resist Aminoglycoside Modifying Enzymes and Increase Ribosomal Target Selectivity. ACS Infect. Dis. 2019, 5, 1718–1730. [Google Scholar] [CrossRef] [Green Version]
- Sonousi, A.; Sarpe, V.A.; Brilkova, M.; Schacht, J.; Vasella, A.; Böttger, E.C.; Crich, D. Effects of the 1-N-(4-Amino-2S-hydroxybutyryl) and 6′-N-(2-Hydroxyethyl) Substituents on Ribosomal Selectivity, Cochleotoxicity, and Antibacterial Activity in the Sisomicin Class of Aminoglycoside Antibiotics. ACS Infect. Dis. 2018, 4, 1114–1120. [Google Scholar] [CrossRef]
- Herzog, I.M.; Feldman, M.; Eldar-Boock, A.; Satchi-Fainaro, R.; Fridman, M. Design of membrane targeting tobramycin-based cationic amphiphiles with reduced hemolytic activity. MedChemComm 2013, 4, 120–124. [Google Scholar] [CrossRef]
- Fosso, M.Y.; Zhu, H.; Green, K.D.; Garneau-Tsodikova, S.; Fredrick, K. Tobramycin variants with enhanced ribosome-targeting activity. ChemBioChem 2015, 16, 1565–1570. [Google Scholar] [CrossRef]
- Ogbonnaya, E.C.; Eleazu, K.; Chukwuma, S.; Essien, U.N. Review of the mechanism of cell death resulting from streptozotocin challenge in experimental animals, its practical use and potential risk to humans. J. Diabetes Metab. Disord. 2013, 12, 60. [Google Scholar] [CrossRef]
- Agarwal, M. Streptozotocin: Mechanisms of action—Proceedings of a Workshop Held on 21 June 1980, Washington, Dc. FEBS Lett. 1980, 120, 1–3. [Google Scholar] [CrossRef] [Green Version]
- Reusser, F. Mode of Action of Streptozotocin. J. Bacteriol. 1971, 105, 580–588. [Google Scholar] [CrossRef] [Green Version]
- King, A.J.F. The use of animal models in diabetes research. Br. J. Pharmacol. 2012, 166, 877–894. [Google Scholar] [CrossRef] [Green Version]
- Wang, Z.; Gleichmann, H. GLUT2 in pancreatic islets: Crucial target molecule in diabetes induced with multiple low doses of streptozotocin in mice. Diabetes 1998, 47, 50–56. [Google Scholar] [CrossRef]
- Schnedl, W.J.; Ferber, S.; Johnson, J.H.; Newgard, C.B. STZ transport and cytotoxicity. Specific enhancement in GLUT2-expressing cells. Diabetes 1994, 43, 1326–1333. [Google Scholar] [CrossRef]
- Elsner, M.; Guldbakke, B.; Tiedge, M.; Munday, R.; Lenzen, S. Relative importance of transport and alkylation for pancreatic beta-cell toxicity of streptozotocin. Diabetol. 2000, 43, 1528–1533. [Google Scholar] [CrossRef] [Green Version]
- Moertel, C.G.; Hanley, J.A.; Johnson, L.A. Streptozocin Alone Compared with Streptozocin plus Fluorouracil in the Treatment of Advanced Islet-Cell Carcinoma. N. Engl. J. Med. 1980, 303, 1189–1194. [Google Scholar] [CrossRef]
- Postma, P.W.; Lengeler, J.W.; Jacobson, G.R. Phosphoenolpyruvate:carbohydrate phosphotransferase systems of bacteria. Microbiol. Rev. 1993, 57, 543–594. [Google Scholar] [CrossRef] [Green Version]
- Ammer, J.; Brennenstuhl, M.; Schindler, P.; Holtje, J.V.; Zahner, H. Phosphorylation of streptozotocin during uptake via the phosphoenolpyruvate: Sugar phosphotransferase system in Escherichia coli. Antimicrob. Agents Chemother. 1979, 16, 801–807. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Lengeler, J. Analysis of the physiological effects of the antibiotic streptozotocin on Escherichia coli K 12 and other sensitive bacteria. Arch. Microbiol. 1980, 128, 196–203. [Google Scholar] [CrossRef] [PubMed]
- Bannister, B. Synthesis and Biological-Activities of some Analogs of Streptozotocin. J. Antibiot. 1972, 25, 377–386. [Google Scholar] [CrossRef] [Green Version]
- Iwasaki, M.; Ueno, M.; Ninomiya, K.; Sekine, J.; Nagamatsu, Y.; Kimura, G. Alkyl streptozotocin analogues with improved biological activities. J. Med. Chem. 1976, 19, 918–923. [Google Scholar] [CrossRef]
- Kim, C.; Song, S.; Park, C. The D-allose operon of Escherichia coli K-12. J. Bacteriol. 1997, 179, 7631–7637. [Google Scholar] [CrossRef] [Green Version]
- Jäger, M.; Hartmann, M.; De Vries, J.G.; Minnaard, A.J. Catalytic Regioselective Oxidation of Glycosides. Angew. Chem. Int. Ed. 2013, 52, 7809–7812. [Google Scholar] [CrossRef] [Green Version]
- Jumde, V.R.; Eisink, N.N.H.M.; Witte, M.D.; Minnaard, A.J. C3 Epimerization of Glucose, via Regioselective Oxidation and Reduction. J. Org. Chem. 2016, 81, 11439–11443. [Google Scholar] [CrossRef]
- Martinez, J.; Oiry, J.; Imbach, J.L.; Winternitz, F. A Selective Synthesis of N-Alkyl N-Nitrosoureas. Eur. J. Med. Chem. 1980, 15, 211–213. [Google Scholar]
- Martinez, J.; Oiry, J.; Imbach, J.L.; Winternitz, F. Activated N-nitrosocarbamates for regioselective synthesis of N-nitrosoureas. J. Med. Chem. 1982, 25, 178–182. [Google Scholar] [CrossRef]
- Gassmann, N.; Stoos, F.; Meier, A.; Helali, S.E.; Hardegger, E. Varianten im Zuckerteil des Streptozotocins. Helv. Chim. Acta 1975, 58, 182–185. [Google Scholar] [CrossRef] [PubMed]
- Zhang, J.; Eisink, N.N.H.M.; Witte, M.D.; Minnaard, A.J. Regioselective Manipulation of GlcNAc Provides Allosamine, Lividosamine, and Related Compounds. J. Org. Chem. 2018, 84, 516–525. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Goud, B.J.; Dwarakanath, V.; Chikka, B.K. Streptozotocin—A diabetogenic agent in animal models. Int. J. Pharm. Pharm. Res. 2015, 3, 253–269. [Google Scholar]
- Qiu, T.A.; Nguyen, T.H.T.; Hudson-Smith, N.V.; Clement, P.L.; Forester, D.-C.; Frew, H.; Hang, M.N.; Murphy, C.J.; Hamers, R.J.; Feng, Z.V.; et al. Growth-Based Bacterial Viability Assay for Interference-Free and High-Throughput Toxicity Screening of Nanomaterials. Anal. Chem. 2017, 89, 2057–2064. [Google Scholar] [CrossRef]
- Jacobson, G.R.; Poy, F.; Lengeler, J.W. Inhibition of Streptococcus mutans by the antibiotic streptozotocin: Mechanisms of uptake and the selection of carbohydrate-negative mutants. Infect. Immun. 1990, 58, 543–549. [Google Scholar] [CrossRef] [Green Version]
- Liu, H.M.; Sato, Y.; Tsuda, Y. Chemistry of Oxo-Sugars.2. Regioselective and Stereoselective Synthesis of Methyl D-Hexopyranosiduloses and Identification of their Forms Existing in Solutions. Chem. Pharm. Bull. 1993, 41, 491–501. [Google Scholar] [CrossRef] [Green Version]
- CLSI. Methods for Dilution Antimicrobial Susceptibility Tests for Bacteria that Grow Aerobically, 9th ed.; Clinical and Laboratory Standards Institute: Wayne, PA, USA, 2012. [Google Scholar]






{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
| Compound | MIC Value (mg/L) | ||
|---|---|---|---|
| LB a | MM b + GlcNAc | MM b + Ribose | |
| STZ (1) | 1.5 | >1.5 | n.d. |
| keto-STZ (2) | >200 | >200 | n.d. |
| allo-STZ (3) | >200 | >200 | >200 |
| deoxy-STZ (4) | >200 | >200 | >200 |
| Compound | Viability | ||
|---|---|---|---|
| LB a | MM b + GlcNAc | MM b + Ribose | |
| STZ (1) (50 mg/L) | 0.0035% | 0.0019% | n.d. |
| keto-STZ (2) (200 mg/L) | 8% | 0.1% | n.d. |
| allo-STZ (3) (200 mg/L) | 29% | 29% | 21% |
| deoxy-STZ (4) (200 mg/L) | 127% | 170% | 202% |
© 2020 by the authors. Licensee MDPI, Basel, Switzerland. This article is an open access article distributed under the terms and conditions of the Creative Commons Attribution (CC BY) license (http://creativecommons.org/licenses/by/4.0/).
Share and Cite
Zhang, J.; Yakovlieva, L.; de Haan, B.J.; de Vos, P.; Minnaard, A.J.; Witte, M.D.; Walvoort, M.T.C. Selective Modification of Streptozotocin at the C3 Position to Improve Its Bioactivity as Antibiotic and Reduce Its Cytotoxicity towards Insulin-Producing β Cells. Antibiotics 2020, 9, 182. https://doi.org/10.3390/antibiotics9040182
Zhang J, Yakovlieva L, de Haan BJ, de Vos P, Minnaard AJ, Witte MD, Walvoort MTC. Selective Modification of Streptozotocin at the C3 Position to Improve Its Bioactivity as Antibiotic and Reduce Its Cytotoxicity towards Insulin-Producing β Cells. Antibiotics. 2020; 9(4):182. https://doi.org/10.3390/antibiotics9040182
Chicago/Turabian StyleZhang, Ji, Liubov Yakovlieva, Bart J. de Haan, Paul de Vos, Adriaan J. Minnaard, Martin D. Witte, and Marthe T. C. Walvoort. 2020. "Selective Modification of Streptozotocin at the C3 Position to Improve Its Bioactivity as Antibiotic and Reduce Its Cytotoxicity towards Insulin-Producing β Cells" Antibiotics 9, no. 4: 182. https://doi.org/10.3390/antibiotics9040182
APA StyleZhang, J., Yakovlieva, L., de Haan, B. J., de Vos, P., Minnaard, A. J., Witte, M. D., & Walvoort, M. T. C. (2020). Selective Modification of Streptozotocin at the C3 Position to Improve Its Bioactivity as Antibiotic and Reduce Its Cytotoxicity towards Insulin-Producing β Cells. Antibiotics, 9(4), 182. https://doi.org/10.3390/antibiotics9040182