Thioporidiols A and B: Two New Sulfur Compounds Discovered by Molybdenum-Catalyzed Oxidation Screening from Trichoderma polypori FKI-7382

 , ,
, ,
Abstract
:1. Introduction
2. Results and Discussion
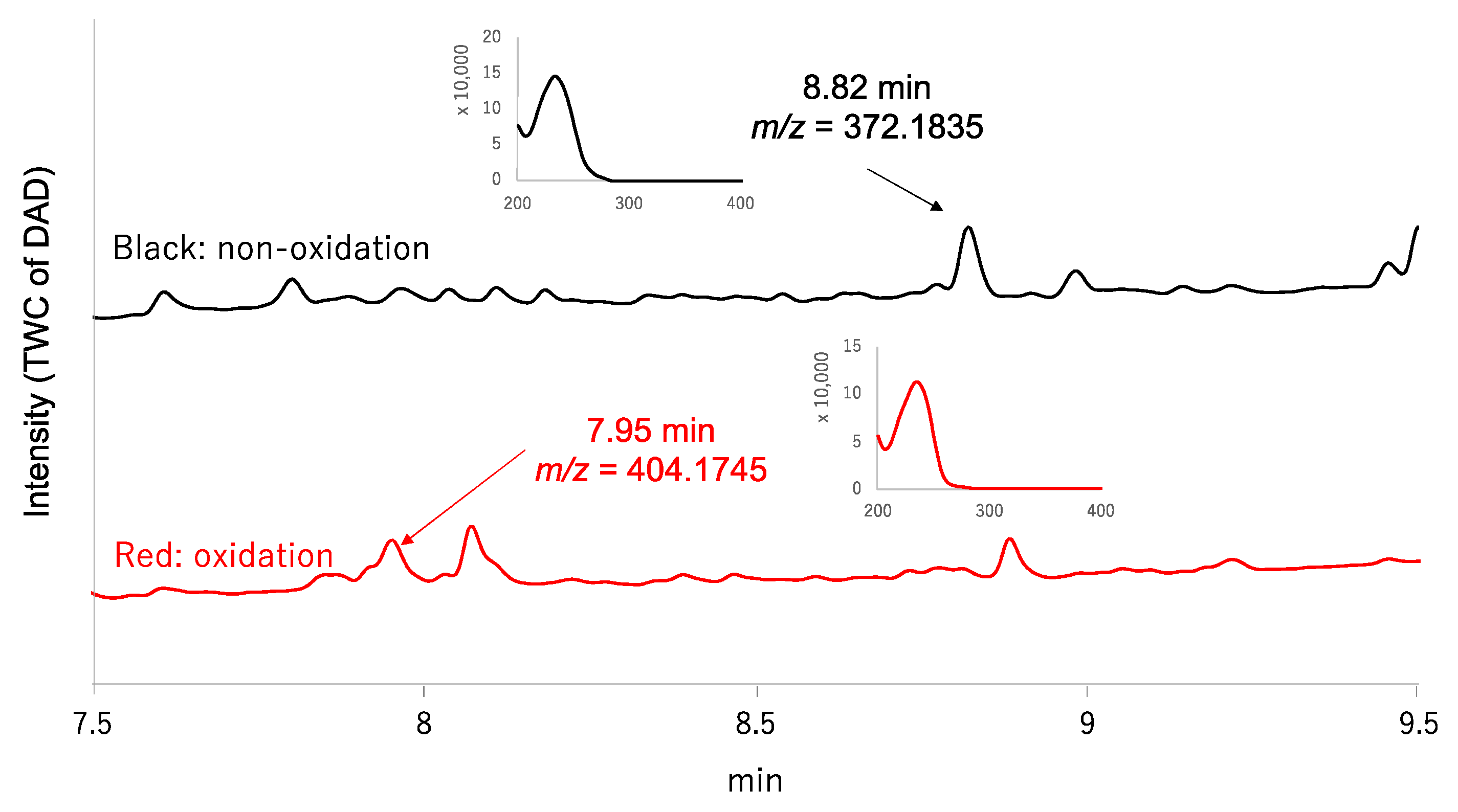
2.1. MoS-Screening of the Culture Broth of the Fungal Strain FKI-7382
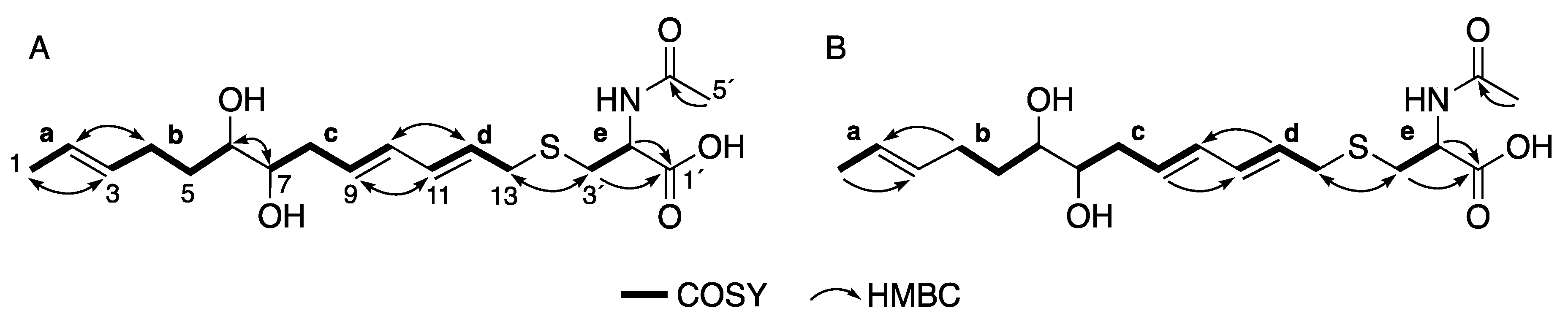
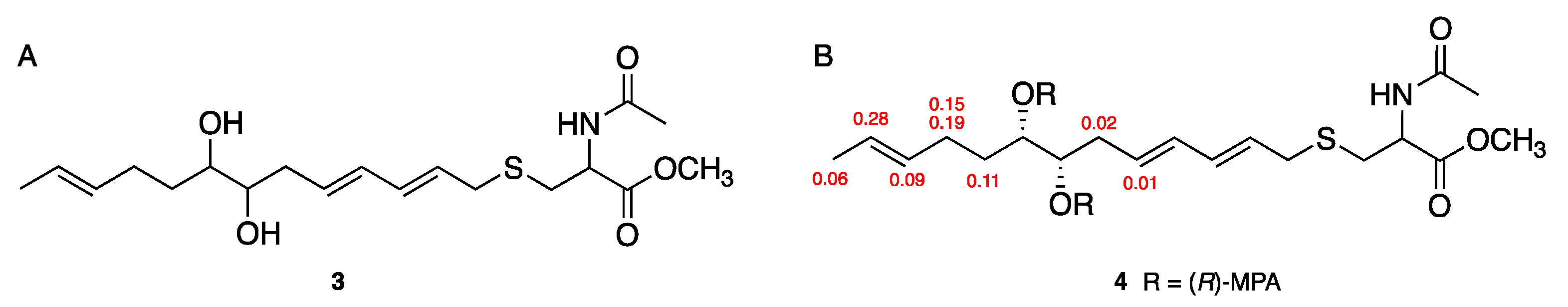
2.2. Structure Elucidation of 1 and 2
3. Materials and Methods
3.1. General Experimental Procedures
3.2. Fermentation of Strain FKI-7382 and Isolation of 1 and 2
3.3. Preparation of MPA Diester of 1
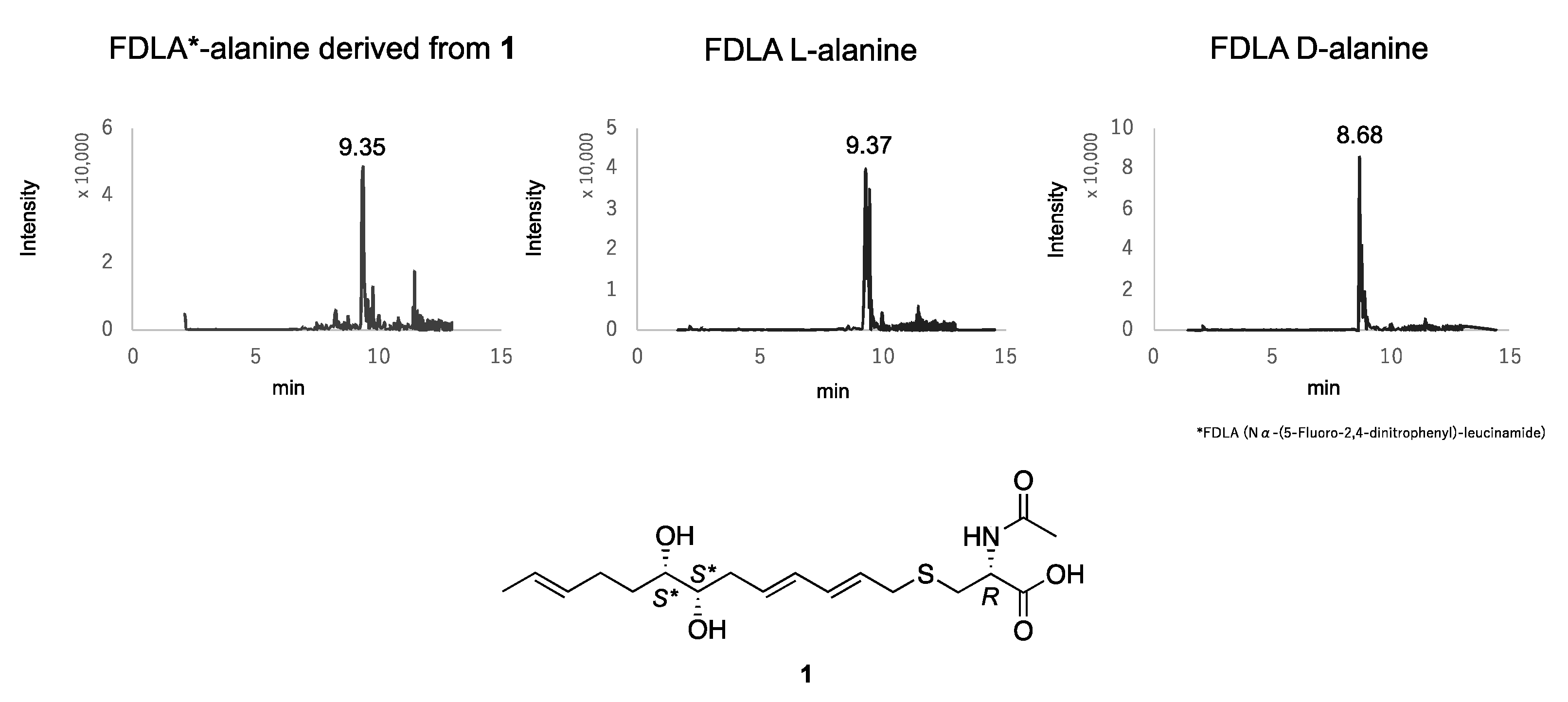
3.4. Absolute Configuration of the N-Acetylcysteine Moiety of 1
3.5. Anti-Microbial Activity of 1 and 2
4. Conclusions
Supplementary Materials
Author Contributions
Funding
Acknowledgments
Conflicts of Interest
References
- Newman, D.J.; Cragg, G.M. Natural Products as Sources of New Drugs from 1981 to 2014. J. Nat. Prod. 2016, 7, 629–661. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Kanehisa, M.; Goto, S.; Furumichi, M.; Tanabe, M.; Hirakawa, M. KEGG for representation and analysis of molecular networks involving diseases and drugs. Nucleic Acids Res. 2010, 38, D355–D360. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Ōmura, S.; Tanaka, H.; Awaya, J.; Narimatsu, Y.; Konda, Y.; Hata, T. Pyrindicin, a New Alkaloid from a Streptomyces Strain Taxonomy, Fermentation, Isolation and Biological Activity. Agric. Biol. Chem. 1974, 38, 899–906. [Google Scholar]
- Miyano, R.; Matsuo, H.; Mokudai, T.; Noguchi, Y.; Higo, M.; Nonaka, K.; Niwano, Y.; Sunazuka, T.; Shiomi, K.; Takahashi, Y.; et al. Trichothioneic acid, a new antioxidant compound produced by the fungal strain Trichoderma virens FKI-7573. J. Biosci. Bioeng. 2020. in Press. [Google Scholar] [CrossRef] [PubMed]
- Matsuo, H.; Hanamure, Y.; Miyano, R.; Takahashi, Y.; Omura, S.; Nakashima, T. Screening for Sulfur Compounds by Molybdenum-Catalyzed Oxidation Combined with Liquid Chromatography-Mass Spectrometry. Molecules 2020, 25, 240. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Freire, F.; Calderon, F.; Seco, J.M.; Fernandez-Mayoralas, A.; Quinoa, E.; Riguera, R. Relative and absolute stereochemistry of secondary/secondary diols: Low-temperature 1H NMR of their bis-MPA esters. J. Org. Chem. 2007, 72, 2297–2301. [Google Scholar] [CrossRef] [PubMed]
- Mozingo, R.; Wolf, D.E.; Harris, S.A.; Folkers, K. Hydrogenolysis of Sulfur Compounds by Raney Nickel Catalyst. J. Am. Chem. Soc. 1943, 65, 1013–1016. [Google Scholar] [CrossRef]
- Marfey, P. Determination of D-amino acids. II. Use of a bifunctional reagent, 1,5-difluoro-2,4-dinitrobenzene. Carlsberg Res. Commun. 1984, 49, 591–596. [Google Scholar] [CrossRef] [Green Version]
- Espinel-Ingroff, A.; Rodríguez-Tudela, J.L.; Martínez-Suárez, J.V. Comparison of two alternative microdilution procedures with the National Committee for Clinical Laboratory Standards reference macrodilution method M27-P for in vitro testing of fluconazole-resistant and -susceptible isolates of Candida albicans. J. Clin. Microbiol. 1995, 33, 3154–3158. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Nakashima, T.; Kimura, T.; Miyano, R.; Matsuo, H.; Hirose, T.; Kimishima, A.; Nonaka, K.; Iwatsuki, M.; Nakanishi, J.; Takahashi, Y.; et al. Nanaomycin H: A new nanaomycin analog. J. Biosci. Bioeng. 2017, 123, 765–770. [Google Scholar] [CrossRef] [PubMed]
- Newton, G.L.; Bewley, C.A.; Dwyer, T.J.; Horn, R.; Aharonowitz, Y.; Cohen, G.; Davies, J.; Faulkner, D.J.; Fahey, R.C. The structure of U17 isolated from Streptomyces clavuligerus and its properties as an antioxidant thiol. Eur. J. Biochem. 1995, 230, 821–825. [Google Scholar] [CrossRef] [PubMed]
- Gerald, L.N.; Nancy, B.; Robert, C.F. Biosynthesis and functions of mycothiol, the unique protective thiol of actinobacteria. Microbiol. Mol. Biol. Rev. 2008, 72, 471–494. [Google Scholar]





{kind=link}
{kind=link}
{kind=link}
{kind=link}
| Thioporidiol A (1) | Thioporidiol B (2) | |||
|---|---|---|---|---|
| Position | δH | δC | δH | δC |
| 1 | 1.63 (dd, 3.5, 1.5, 3H) | 18.1 | 1.63 (dd, 4.0, 1.0, 3H) | 18.1 |
| 2 | 5.46 (dd, J = 14.0, 3.5 Hz, 1H) | 126.0 | 5.45 (dd, J = 14.0, 4.0 Hz, 1H) | 126.1 |
| 3 | 5.45 (dddd, J = 14.0, 5.0, 5.0, 1.5 Hz, 1H) | 132.3 | 5.45 (dddd, J = 14.0, 5.0, 3.0, 1.5 Hz, 1H) | 131.1 |
| 4 | 2.03 (dddd, J = 14.0, 10.0, 9.0, 5.0 Hz, 1H) | 29.9 | 2.03 (dddd, J = 14.0, 8.0, 8.0, 5.0 Hz, 1H) | 30.0 |
| 2.20 (dddd, J = 14.0, 10.0, 5.0, 5.0 Hz, 1H) | 2.15 (dddd, J = 14.0, 9.0, 6.0, 3.0 Hz, 1H) | |||
| 5 | 1.41 (dddd, J = 14.0, 9.0, 9.0, 5.0 Hz, 1H) | 33.6 | 1.49 (dddd, J = 14.0, 8.0, 8.0, 6.0 Hz, 1H) | 33.9 |
| 1.68 (dddd, J = 14.0, 10.0, 7.0, 3.0 Hz, 1H) | 1.56 (dddd, J = 14.0, 9.0, 8.0, 4.0 Hz, 1H) | |||
| 6 | 3.38 (ddd, J = 9.0, 6.0, 3.0 Hz, 1H) | 74.8 | 3.41 (ddd, J = 8.0, 4.0, 4.0 Hz, 1H) | 73.9 |
| 7 | 3.40 (ddd, J = 8.0, 6.0, 3.5 Hz, 1H) | 75.8 | 3.45 (ddd, J = 8.0, 4.5, 4.0 Hz, 1H) | 75.0 |
| 8 | 2.19 (ddd, J = 14.5, 8.0, 7.0 Hz, 1H) | 37.3 | 2.23 (ddd, J = 14.0, 8.0, 7.0 Hz, 1H) | 37.5 |
| 2.42 (ddd, J = 14.5, 7.0, 3.5 Hz, 1H) | 2.34 (ddd, J = 14.0, 7.0, 4.5 Hz, 1H) | |||
| 9 | 5.77 (ddd, J = 14.0, 7.0, 7.0 Hz, 1H) | 132.3 | 5.74 (ddd, J = 14.0, 7.0, 7.0 Hz, 1H) | 132.2 |
| 10 | 6.12 (d, J = 14.0, 11.0 Hz, 1H) | 132.8 | 6.13 (d, J = 14.0, 10.0 Hz, 1H) | 132.8 |
| 11 | 6.16 (d, J = 14.0, 11.0 Hz, 1H) | 134.7 | 6.17 (d, J = 14.0, 10.0 Hz, 1H) | 134.6 |
| 12 | 5.53 (ddd, J = 14.0, 7.5, 7.0 Hz, 1H) | 128.0 | 5.54 (ddd, J = 14.0, 7.5, 7.0 Hz, 1H) | 128.1 |
| 13 | 3.19 (dd, J = 14.0, 7.0 Hz, 1H) | 34.8 | 3.19 (dd, J = 14.5, 7.0 Hz, 1H) | 34.8 |
| 3.21 (dd, J = 14.0, 7.5 Hz, 1H) | 3.21 (dd, J = 14.5, 7.5 Hz, 1H) | |||
| 1´ | 173.9 | 173.8 | ||
| 2´ | 4.57 (dd, J = 8.0, 5.0 Hz, 1H) | 53.4 | 4.57 (dd, J = 8.0, 5.0 Hz, 1H) | 53.3 |
| 3´ | 2.74 (dd, J = 14.0, 8.0 Hz, 1H) | 33.0 | 2.74 (dd, J = 14.0, 8.0 Hz, 1H) | 33.0 |
| 2.95 (dd, J = 14.0, 5.0 Hz, 1H) | 2.95 (dd, J = 14.0, 5.0 Hz, 1H) | |||
| 4´ | 173.3 | 173.3 | ||
| 5´ | 2.00 (s, 3H) | 22.4 | 2.00 (s, 3H) | 22.4 |
© 2020 by the authors. Licensee MDPI, Basel, Switzerland. This article is an open access article distributed under the terms and conditions of the Creative Commons Attribution (CC BY) license (http://creativecommons.org/licenses/by/4.0/).
Share and Cite
Matsuo, H.; Noguchi, Y.; Miyano, R.; Higo, M.; Nonaka, K.; Sunazuka, T.; Takahashi, Y.; Ōmura, S.; Nakashima, T. Thioporidiols A and B: Two New Sulfur Compounds Discovered by Molybdenum-Catalyzed Oxidation Screening from Trichoderma polypori FKI-7382. Antibiotics 2020, 9, 236. https://doi.org/10.3390/antibiotics9050236
Matsuo H, Noguchi Y, Miyano R, Higo M, Nonaka K, Sunazuka T, Takahashi Y, Ōmura S, Nakashima T. Thioporidiols A and B: Two New Sulfur Compounds Discovered by Molybdenum-Catalyzed Oxidation Screening from Trichoderma polypori FKI-7382. Antibiotics. 2020; 9(5):236. https://doi.org/10.3390/antibiotics9050236
Chicago/Turabian StyleMatsuo, Hirotaka, Yoshihiko Noguchi, Rei Miyano, Mayuka Higo, Kenichi Nonaka, Toshiaki Sunazuka, Yōko Takahashi, Satoshi Ōmura, and Takuji Nakashima. 2020. "Thioporidiols A and B: Two New Sulfur Compounds Discovered by Molybdenum-Catalyzed Oxidation Screening from Trichoderma polypori FKI-7382" Antibiotics 9, no. 5: 236. https://doi.org/10.3390/antibiotics9050236
APA StyleMatsuo, H., Noguchi, Y., Miyano, R., Higo, M., Nonaka, K., Sunazuka, T., Takahashi, Y., Ōmura, S., & Nakashima, T. (2020). Thioporidiols A and B: Two New Sulfur Compounds Discovered by Molybdenum-Catalyzed Oxidation Screening from Trichoderma polypori FKI-7382. Antibiotics, 9(5), 236. https://doi.org/10.3390/antibiotics9050236