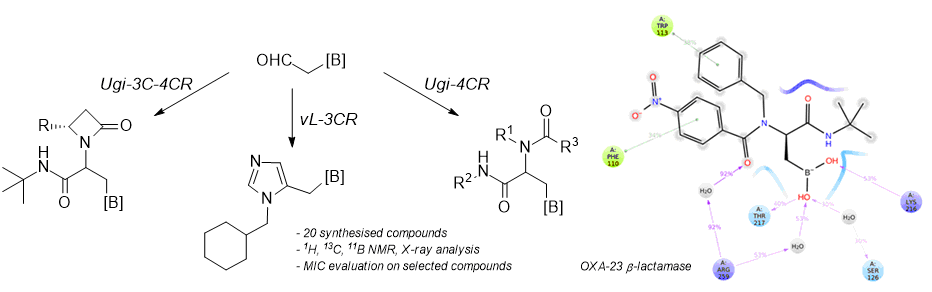
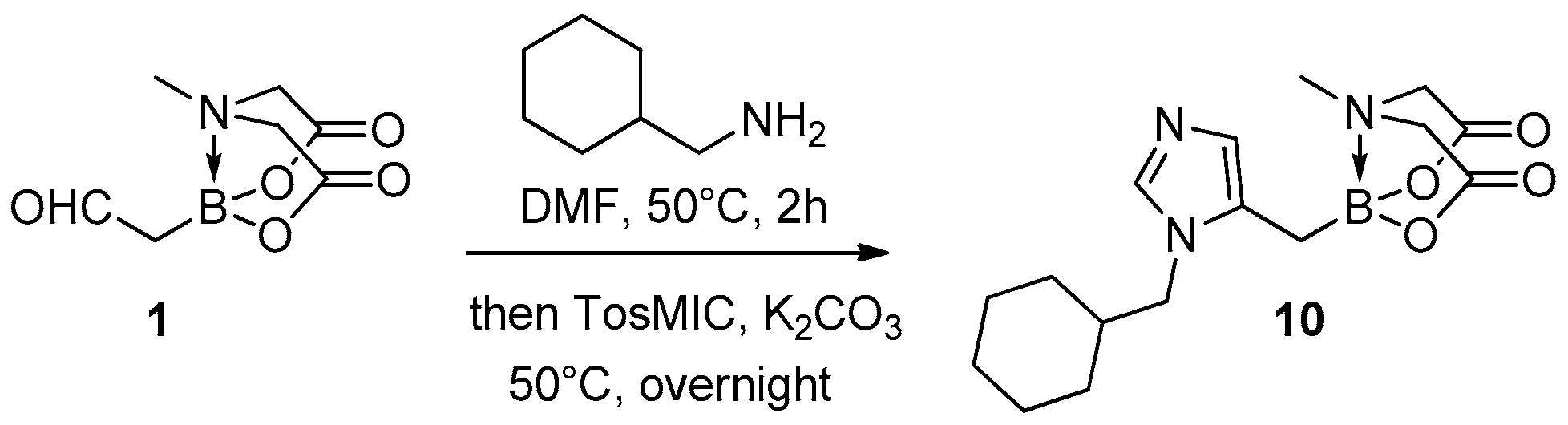
4.5. Procedure for the VL-3CR Reaction (GP-D)
A test tube for carousel was equipped with a magnetic stir bar, and it was charged with compound 1 (100 mg, 0.5 mmol) and dry DMF (5 mL). Cyclohexylmethanamine (0.08 mL, 0.6 mmol), was added and the resulting mixture was kept under stirring for 2 h at 50 °C under an atmosphere of nitrogen. Then, potassium carbonate (104 mg, 0.75 mmol) and TosMIC (148 mg, 0.75 mmol) were added in portions and the reaction was left overnight at 50 °C under stirring. The resulting mixture was concentrated in vacuo, and then it was partitioned between ethyl acetate/water. The aqueous phase was extracted with ethyl acetate (×3) and the organic phase was washed with brine (×5), dried over Na2SO4, and concentrated in vacuo, to give a residue that was purified by silica gravimetric chromatography column (eluent: DCM/MeOH = 99/1 to 90/10). The purified product was obtained with 32% yield as a brown solid.
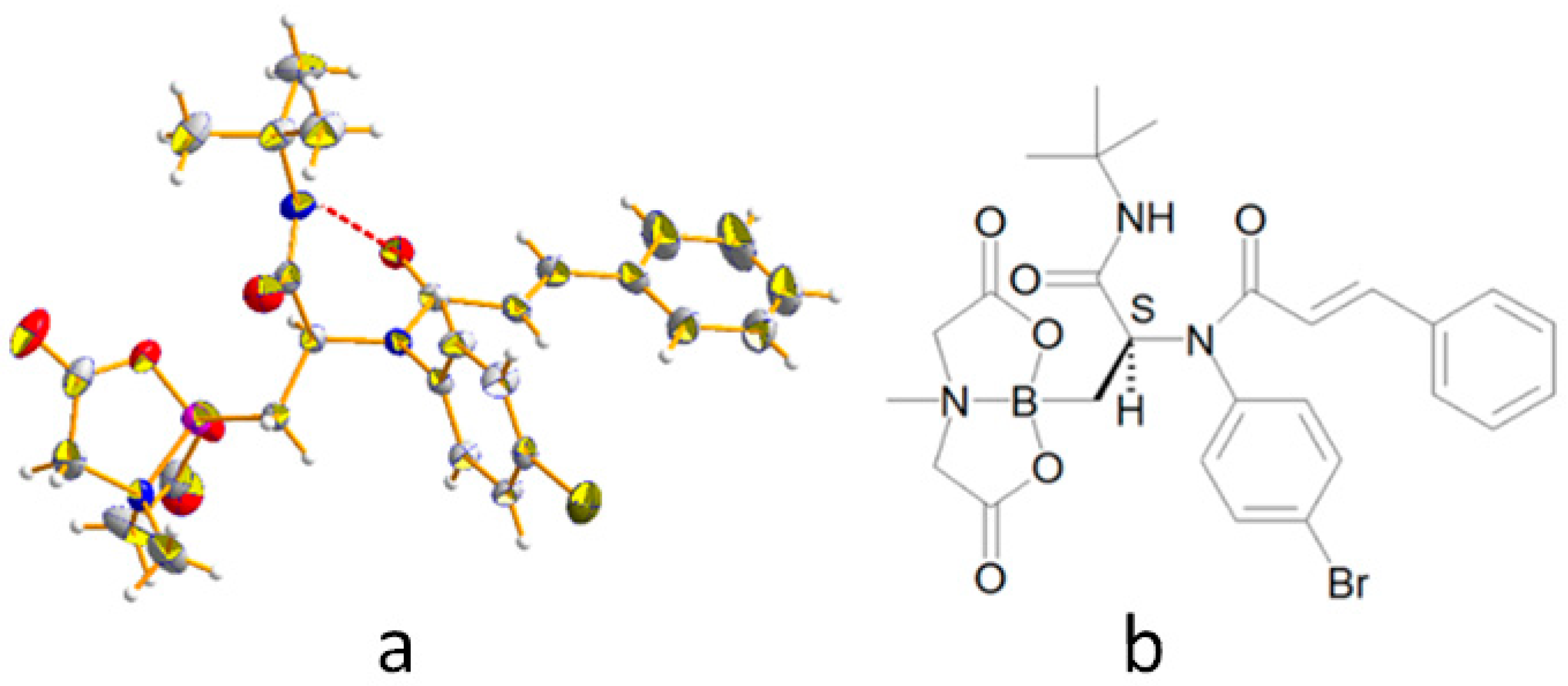
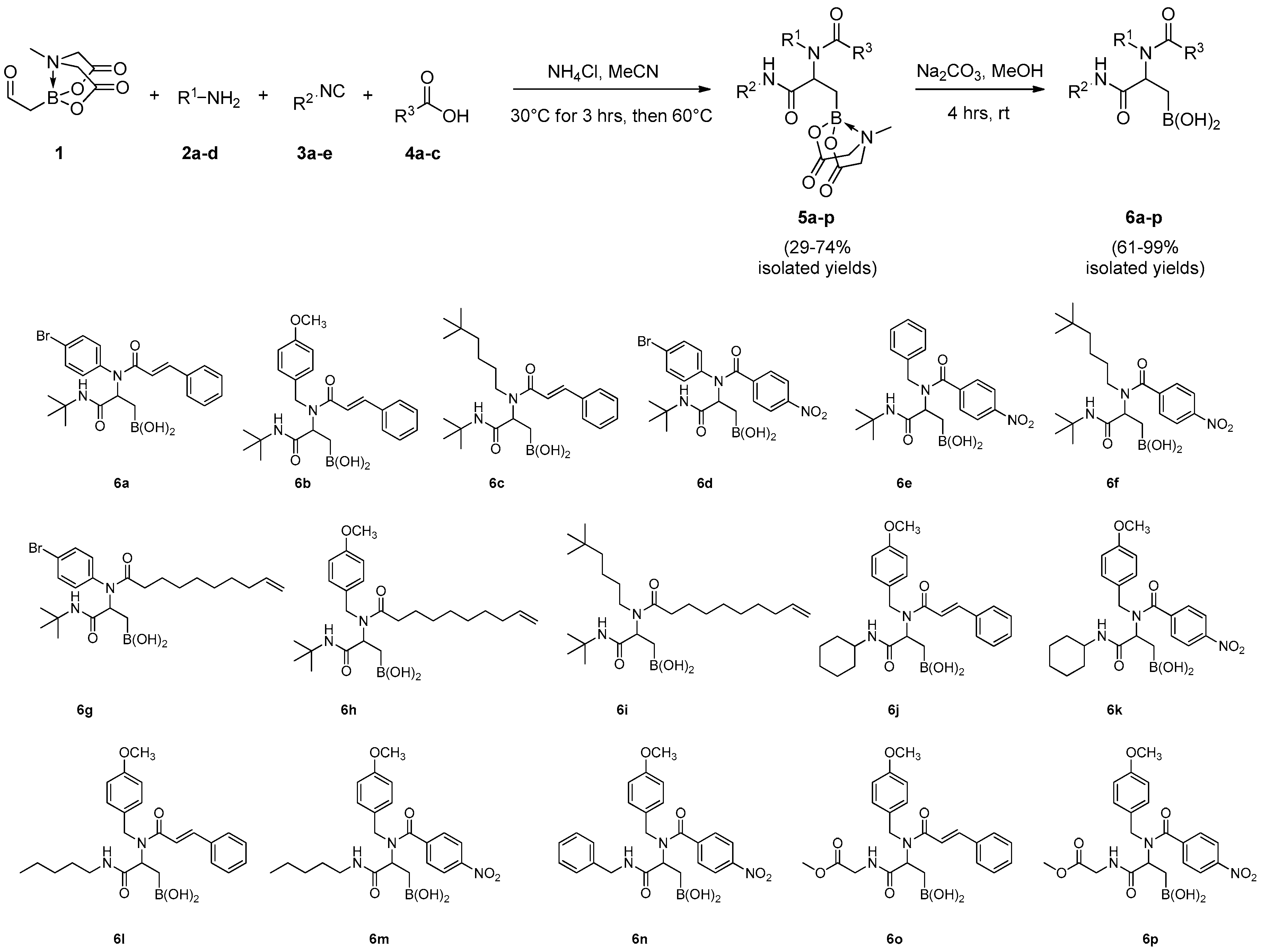
N-(4-bromophenyl)-N-(1-(tert-butylamino)-3-(4-methyl-2,6-dioxotetrahydro-2H-4λ4,8λ4-[1,3,2]oxazaborolo[2,3-b][1,3,2]oxazaborol-8-yl)-1-oxopropan-2-yl)cinnamamide (5a): Prepared according to GP-A using α-borylaldehyde 1, 4-bromoaniline 2a, trans cinnamic acid 4a, and tert-butyl isocyanide 3a. Obtained as a white solid (yield = 74%); 1H-NMR (400 MHz, CD3OD) δ 7.68–7.52 (m, 3H), 7.35–7.25 (m, 7H), 6.22 (d, J = 15.6 Hz, 1H), 5.38–5.21 (m, 1H), 4.17 (d, J = 17.1 Hz, 1H), 4.13 (d, J = 16.8 Hz, 1H), 4.00 (d, J = 17.1 Hz, 1H), 3.96 (d, J = 16.8 Hz, 1H), 2.99 (s, 3H), 1.36 (s, 9H), 1.18 (d, J = 14.4 Hz, 1H), 0.96 (dd, J = 14.4, 4.4 Hz, 1H), (NH missed); 13C-NMR (101 MHz, CD3OD) δ 171.3, 170.0, 169.4, 167.6, 143.2, 138.9, 135.4, 132.9, 132.8, 132.7 (2C), 132.4, 130.4, 129.3 (2C), 128.2 (2C), 123.0, 119.2, 62.6, 62.5, 58.1, 51.6, 46.0, 28.2 (3C); HR-MS (ESI) m/z: [M + Na]+ calcd for C27H31BBrN3NaO6 606.1387; found 606.1399.
N-(1-(tert-Butylamino)-3-(4-methyl-2,6-dioxotetrahydro-2H-4λ4,8λ4-[1,3,2]oxazaborolo[2,3-b][1,3,2]oxazaborol-8-yl)-1-oxopropan-2-yl)-N-(4-methoxybenzyl)cinnamamide (5b): Prepared according to GP-A using α-borylaldehyde 1, 4-methoxybenzylamine 2b, trans cinnamic acid 4a and tert-butyl isocyanide 3a. Obtained as a white solid (yield = 51%); 1H-NMR (400 MHz, 115 °C, DMSO-d6) δ 7.54 (m, 3H), 7.37 (d, J = 7.2 Hz, 2H), 7.25 (d, J = 8.1 Hz, 2H), 7.00 (d, J = 16.0 Hz, 1H), 6.87 (d, J = 8.1 Hz, 2H), 6.75 (s, 1H), 6.48 (d, J = 16.0 Hz, 1H), 4.88–4.81 (m, 2H), 4.54 (d, J = 16.2 Hz, 1H), 4.14 (d, J = 16.9 Hz, 1H), 4.10 (d, J = 16.8 Hz, 1H), 3.96 (d, J = 16.9 Hz, 1H), 3.94 (d, J = 16.8 Hz, 1H), 3.74 (s, 3H), 2.95 (s, 3H), 1.34–1.27 (m, 1H), 1.17 (s, 9H), 0.95–0.85 (m, 1H); 13C-NMR (101 MHz, 25 °C, DMSO-d6, 6:4 rotameric mixture) δ 170.4–166.4 (4C), 158.5, 144.6, 141.7 and 141.5 (1C), 135.9 and 135.5 (1C), 131.8, 129.3 and 129.2 (2C), 128.6, 128.4, 128.2 and 128.1 (1C), 120.0, 114.2 (2C), 62.3–62.0 (2C), 55.5, 55.4, 50.6 and 50.5 (1C), 46.8, 46.3 and 46.1 (1C), 29.4, 28.6 and 28.5 (1C); HR-MS (ESI) m/z: [M + Na]+ calcd for C29H36BN3NaO7 572.2544; found 572.2524.
N-(1-(tert-Butylamino)-3-(4-methyl-2,6-dioxotetrahydro-2H-4λ4,8λ4-[1,3,2]oxazaborolo[2,3-b][1,3,2]oxazaborol-8-yl)-1-oxopropan-2-yl)-N-(5,5-dimethylhexyl)cinnamamide (5c): Prepared according to GP-A using α-borylaldehyde 1, 5,5-dimethylhexan-1-amine 2c, trans cinnamic acid 4a and tert-butyl isocyanide 3a. Obtained as a white solid (yield = 29%); 1H-NMR (400 MHz, CD3OD) δ 7.75 (d, J = 15.4 Hz, 1H), 7.71–7.61 (m, 5H), 7.06 (d, J = 15.4 Hz, 1H), 5.02 (m, J = 7.6 Hz, 0.7H), 4.50 (m, 0.3H), 4.17 (d, J = 17.1 Hz, 1H), 4.13 (d, J = 16.8 Hz, 1H), 4.02 (d, J = 17.1 Hz, 1H), 4.00 (d, J = 16.8 Hz, 1H), 3.67–3.53 (m, 2H), 3.08 (s, 2.1H), 3.01 (s, 0.9H), 1.73–1.58 (m, 4H), 1.45 (dd, J = 14.5, 8.8 Hz, 1H), 1.43–1.15 (m, 11H), 1.08 (dd, J = 14.5, 6.5 Hz, 1H), 0.90 (s, 9H), (NH missed); 13C-NMR (101 MHz, CD3OD) δ 171.5, 169.4, 168.8, 168.2, 142.7, 135.2, 130.2 and 129.6 (1C), 128.6 (2C), 128.0–127.6 (2C), 118.3, 61.8 (2C), 57.3, 55.9, 50.7, 45.3, 43.6, 31.4 (2C), 30.5, 28.4 (3C), 27.5 and 27.3 (3C), 21.9 (1C); HR-MS (ESI) m/z: [M + Na]+ calcd for C29H44BN3NaO6 564.3221; found 564.3244.
N-(4-Bromophenyl)-N-(1-(tert-butylamino)-3-(4-methyl-2,6-dioxotetrahydro-2H-4λ4,8λ4-[1,3,2]oxazaborolo[2,3-b][1,3,2]oxazaborol-8-yl)-1-oxopropan-2-yl)-4-nitrobenzamide (5d): Prepared according to GP-A using α-borylaldehyde 1, 4-bromoaniline 2a, 4-nitrobenzoic acid 4b and tert-butyl isocyanide 3a. Obtained as a white solid (yield = 57%); 1H-NMR (400 MHz, CD3OD) δ 8.08 (d, J = 8.2 Hz, 2H), 7.58 (d, J = 8.2 Hz, 2H), 7.38 (d, J = 8.1 Hz, 2H), 7.21 (d, J = 8.1 Hz, 2H), 5.25 (dd, J = 7.0 Hz, 1H), 4.22 (d, J = 17.1 Hz, 1H), 4.18 (d, J = 16.8 Hz, 1H), 4.03 (d, J = 17.1 Hz, 1H), 4.01 (d, J = 16.8 Hz, 1H), 3.02 (s, 3H), 1.38 (s, 9H), 1.26 (dd, J = 7.0, 4.2 Hz, 2H), (NH missed); 13C-NMR (75 MHz, CD3OD, 2:1 rotameric mixture) δ 174.0–169.6 (4C), 149.7 and 149.6 (1C), 144.4 and 144.1 (1C), 140.9 and 140.2 (1C), 133.7–133.5 (4C), 130.6 (2C), 124.5 and 124.4 (2C), 123.5 and 123.3 (1C), 63.5, 63.4, 61.2 and 60.7 (1C), 58.7, 46.9, 29.2 (3C), (B-Cα missed); 11B-NMR (128 MHz, CD3CN) δ 12.26; HR-MS (ESI) m/z: [M + Na]+ calcd for C25H28BBrN4NaO8 625.1081; found 625.1092.
N-Benzyl-N-(1-(tert-butylamino)-3-(4-methyl-2,6-dioxotetrahydro-2H-4λ4,8λ4-[1,3,2]oxazaborolo[2,3-b][1,3,2]oxazaborol-8-yl)-1-oxopropan-2-yl)-4-nitrobenzamide (5e): Prepared according to GP-A using α-borylaldehyde 1, benzylamine 2d, 4-nitrobenzoic acid 4b and tert-butyl isocyanide 3a. Obtained as a white solid (yield = 51%); 1H-NMR (400 MHz, CD3OD) δ 8.24 (d, J = 7.5 Hz, 2H), 7.75 (d, J = 7.5 Hz, 2H), 7.35–7.05 (m, 5H), 4.80 (d, J = 15.8 Hz, 1H), 4.71 (m, 1H), 4.48 (d, J = 15.8 Hz, 1H), 4.24 (d, J = 16.8 Hz, 1H), 4.22 (d, J = 16.4 Hz, 1H), 4.14–3.98 (m, 2H), 3.04 (s, 3H), 1.58 (d, J = 8.0 Hz, 1H), 1.35 (d, J = 8.0 Hz, 1H), 1.29 (s, 9H), (NH missed); 13C-NMR (101 MHz, CD3OD) δ 171.4–168.8 (4C), 148.2, 142.6, 137.0, 128.5 (2C), 128.2, 127.7 (2C), 127.4, 127.2, 123.3 (2C), 61.7, 61.5, 58.2, 57.3, 52.0, 45.1, 42.5, 27.4 (3C); 11B NMR (128 MHz, CD3CN) δ 12.33; HR-MS (ESI) m/z: [M + Na]+ calcd for C26H31BN4NaO8 561.2133; found 561.2109.
N-(1-(tert-Butylamino)-3-(4-methyl-2,6-dioxotetrahydro-2H-4λ4,8λ4-[1,3,2]oxazaborolo[2,3-b][1,3,2]oxazaborol-8-yl)-1-oxopropan-2-yl)-N-(5,5-dimethylhexyl)-4-nitrobenzamide (5f): Prepared according to GP-A using α-borylaldehyde 1, 5,5-dimethylhexan-1-amine 2c, 4-nitrobenzoic acid 4b and tert-butyl isocyanide 3a. Obtained as a white solid (yield = 40%); 1H-NMR (400 MHz, CD3OD) δ 8.36 (d, J = 8.5 Hz, 2H), 7.83 (d, J = 8.5 Hz, 2H), 4.74 (dd, J = 9.6, 5.9 Hz, 1H), 4.21 (d, J = 17.1 Hz, 1H), 4.19 (d, J = 16.9 Hz, 1H), 4.05 (d, J = 17.1 Hz, 1H), 4.03 (d, J = 16.9 Hz, 1H), 3.42–3.21 (m, 2H), 3.07 (s, 3H), 1.53 (dd, J = 14.0, 9.6 Hz, 1H), 1.48–1.21 (m, 12H), 0.92 (m, 4H), 0.78 (s, 9H), (NH missed); 13C-NMR (101 MHz, CD3OD) δ 171.8, 171.3, 169.4, 168.9, 148.4, 142.8, 127.8 (2C), 123.5 (2C), 61.7, 61.6, 58.1, 57.1, 50.8, 45.2 (2C), 43.1, 30.0, 28.2 (3C), 27.5 (3C), 21.5 (2C); HR-MS (ESI) m/z: [M + Na]+ calcd for C27H41BN4NaO8 583.2915; found 583.2931.
N-(4-Bromophenyl)-N-(1-(tert-butylamino)-3-(4-methyl-2,6-dioxotetrahydro-2H-4λ4,8λ4-[1,3,2]oxazaborolo[2,3-b][1,3,2]oxazaborol-8-yl)-1-oxopropan-2-yl)dec-9-enamide (5g): Prepared according to GP-A using α-borylaldehyde 1, 4-bromoaniline 2a, 9-decenoic acid 4c and tert-butyl isocyanide 3a. Obtained as a white solid (yield = 50%); 1H-NMR (400 MHz, CD3OD) δ 7.62 (d, J = 8.3 Hz, 2H), 7.23 (d, J = 8.3 Hz, 2H), 5.88–5.73 (m, J = 13.0 Hz, 1H), 5.20 (m, 1H), 4.95 (dd, J =13.0 Hz, 2H), 4.14–4.12 (m, 1H), 4.08 (d, J = 16.7 Hz, 1H), 3.97 (d, J = 17.6 Hz, 1H), 3.92 (d, J = 16.7 Hz, 1H), 2.95 (s, 3H), 2.34–2.26 (m, 1H), 2.09–1.97 (m, 4H), 1.65–1.48 (m, 3H), 1.36 (s, 9H), 1.27–1.13 (m, 6H), 1.04 (dd, J = 14.4, 10.5 Hz, 1H), 0.84 (dd, J = 14.4, 4.8 Hz, 1H), (NH missed); 13C-NMR (75 MHz, CD3OD) δ 176.1, 172.7, 171.0, 170.3, 140.4, 140.0, 133.9, 133.7, 133.5, 133.2, 123.9, 114.9, 63.6, 63.4, 58.79, 58.6, 46.9, 36.1 (2C), 35.1, 30.4 (3C), 29.2 (3C), 26.6 (2C); HR-MS (ESI) m/z: [M + Na]+ calcd for C28H41BBrN3NaO6 628.2169; found 628.2154.
N-(1-(tert-Butylamino)-3-(4-methyl-2,6-dioxotetrahydro-2H-4λ4,8λ4-[1,3,2]oxazaborolo[2,3-b][1,3,2]oxazaborol-8-yl)-1-oxopropan-2-yl)-N-(4-methoxybenzyl)dec-9-enamide (5h): Prepared according to GP-A using α-borylaldehyde 1, 4-methoxybenzylamine 2b, 9-decenoic acid 4c and tert-butyl isocyanide 3a. Obtained as a white solid (yield = 61%); 1H-NMR (400 MHz, 115 °C, DMSO-d6) δ 7.18 (d, J = 8.1 Hz, 2H), 6.85 (d, J = 8.1 Hz, 2H), 6.69 (s, 1H), 5.90–5.72 (m, 1H), 4.97 (dd, J = 14.4 Hz, 2H), 4.74–4.55 (m, 2H), 4.39 (d, J = 16.4 Hz, 1H), 4.16–4.03 (m, 2H), 4.00–3.88 (m, 2H), 3.75 (s, 3H), 2.93 (s, 3H), 2.40–2.17 (m, 2H), 2.09–1.95 (m, 2H), 1.54 (m, 2H), 1.44–1.23 (m, 8H), 1.18 (s, 9H), 0.93–0.81 (m, 2H); 13C-NMR (101 MHz, 25 °C, DMSO-d6, 1:1 rotameric mixture) δ 174.6–169.4 (4C), 159.2 and 158.8 (1C), 139.9, 133.0 and 132.0 (1C), 129.4, 128.6, 115.7, 114.8, 114.2, 63.0 and 62.6 (2C), 58.4 (0.5C), 56.2, 55.8 (0.5C), 51.3 and 51.1 (1C), 47.8, 46.9 and 46.8 (1C), 34.2 and 33.5 (2C), 30.1 and 29.5 (4C), 29.2 (3C), 25.9 and 25.7 (2C); HR-MS (ESI) m/z: [M + Na]+ calcd for C30H46BN3NaO7 594.3327; found 594.3320.
N-(1-(tert-Butylamino)-3-(4-methyl-2,6-dioxotetrahydro-2H-4λ4,8λ4-[1,3,2]oxazaborolo[2,3-b][1,3,2]oxazaborol-8-yl)-1-oxopropan-2-yl)-N-(5,5-dimethylhexyl)dec-9-enamide (5i): Prepared according to GP-A using α-borylaldehyde 1, 5,5-dimethylhexan-1-amine 2c, 9-decenoic acid 4c and tert-butyl isocyanide 3a. Obtained as a white solid (yield = 38%); 1H-NMR (400 MHz, DMSO-d6, 6:4 rotameric mixture) δ 7.48 (s, 0.4H), 7.04 (s, 0.6H), 5.92–5.68 (m, 1H), 4.96 (dd, J =14.2 Hz, 2H), 4.74 (t, J = 7.1 Hz, 0.6H), 4.28 (dd, J = 8.6, 5.0 Hz, 0.4H), 4.21–4.06 (m, 2H), 4.04–3.87 (m, 2H), 3.29–3.19 (m, 1.2H), 3.17–3.04 (m, 0.8H), 2.96 (s, 1.8H), 2.90 (s, 1.2H), 2.33–2.22 (m, 2H), 2.03–1.93 (m, 2H), 1.59–1.39 (m, 4H), 1.37–1.32 (m, 2.4H), 1.30–1.07 (m, 19.8H), 0.85 (s, 5.5H), 0.84 (s, 3.5H), 0.80 (d, J = 7.1 Hz, 0.3H), 0.67 (dd, J = 14.4, 5.0 Hz, 0.5H); 13C-NMR (101 MHz, DMSO-d6, 6:4 rotameric mixture) δ 174.0, 172.8 and 172.1 (1C), 170.9, 170.0 and 169.6 (1C), 139.9, 115.7, 62.8–62.5(2C), 57.9 and 55.7 (1C), 51.4 and 51.0 (2C), 46.8 and 46.7 (1C), 44.7 and 44.5 (1C), 34.3- 31.2 (6C), 30.3 (3C), 29.4 (3C), 26.1 and 25.9 (2C), 23.2 and 22.9 (2C); 11B-NMR (128 MHz, CD3CN) δ 12.56; HR-MS (ESI) m/z: [M + Na]+ calcd for C30H54BN3NaO6 586.4003; found 586.4024.
N-(1-(Cyclohexylamino)-3-(4-methyl-2,6-dioxotetrahydro-2H-4λ4,8λ4-[1,3,2]oxazaborolo[2,3-b][1,3,2]oxazaborol-8-yl)-1-oxopropan-2-yl)-N-(4-methoxybenzyl)cinnamamide (5j): Prepared according to GP-A using α-borylaldehyde 1, 4-methoxybenzylamine 2b, trans cinnamic acid 4a and cyclohexyl isocyanide 3b. Obtained as a white solid (yield = 49%); 1H-NMR (400 MHz, CD3OD) δ 7.67 (d, J = 6.9 Hz, 1H), 7.61 (d, J = 15.5 Hz, 1H), 7.46 (d, J = 3.6 Hz, 1H), 7.40 (t, J = 3.6, 6.9 Hz, 2H), 7.35 (d, J = 3.6 Hz, 1H), 7.24 (d, J = 8.6 Hz, 2H), 6.95 (d, J = 15.5 Hz, 1H), 6.90 (d, J = 8.6 Hz, 2H), 5.13–5.05 (m, 1H), 4.95 (d, J = 17.1 Hz, 1H), 4.74 (d, J = 17.1 Hz, 1H), 4.17 (d, J = 17.1 Hz, 1H), 4.13 (d, J = 17.3 Hz, 1H), 3.99 (d, J = 17.1 Hz, 1H), 3.97 (d, J = 17.3 Hz, 1H), 3.77 (s, 3H), 3.52–3.44 (m, 1H), 3.03 (s, 2H), 2.98 (s, 1H), 1.89–1.80 (m 1H), 1.79–1.73 (m, 1H), 1.73–1.64 (m, 2H), 1.62–1.52 (m, 2H), 1.47–1.20 (m, 5H), 1.16–1.09 (m, 1H), (NH missed); 13C-NMR (75 MHz, CD3OD) δ 172.4–169.7 (4C), 160.8, 144.6, 136.7, 131.91, 131.3, 130.2 (2C), 129.5 (2C), 129.3 (2C), 120.2, 115.5 (2C), 63.5 (2C), 57.5, 56.1, 52.3, 47.0, 43.3, 33.7 (2C), 31.0, 26.9, 26.2 (2C); HR-MS (ESI) m/z: [M + Na]+ calcd for C31H38BN3NaO7 598.2701; found 598.2689.
N-(1-(Cyclohexylamino)-3-(4-methyl-2,6-dioxotetrahydro-2H-4λ4,8λ4-[1,3,2]oxazaborolo[2,3-b][1,3,2]oxazaborol-8-yl)-1-oxopropan-2-yl)-N-(4-methoxybenzyl)-4-nitrobenzamide (5k): Prepared according to GP-A using α-borylaldehyde 1, 4-methoxybenzylamine 2b, 4-nitrobenzoic acid 4b, and cyclohexyl isocyanide 3b. Obtained as a white solid (yield = 53%); 1H-NMR (400 MHz, CD3OD, 6:4 rotameric mixture) δ 8.27 (d, J = 8.1 Hz, 2H), 7.77 (d, J = 8.1 Hz, 2H), 7.04 (d, J = 7.9 Hz, 2H), 6.82 (d, J = 7.9 Hz, 2H), 4.74–4.62 (m, 1.4H), 4.57 (s, 0.2H), 4.42 (d, J = 15.5 Hz, 0.6H), 4.25 (d, J = 17.1 Hz, 1H), 4.23 (d, J = 16.7 Hz, 1H), 4.13 (d, J = 17.1 Hz, 1H), 4.11 (d, J = 16.7 Hz, 1H), 4.06 (m, 0.8H), 3.77 (s, 3H), 3.64–3.47 (m, 1H), 3.04 (s, 3H), 1.87–1.55 (m, 6H), 1.46–1.22 (m, 6H), (NH missed); 13C-NMR (101 MHz, CD3OD) δ 170.9–168.0 (4C), 159.5, 148.3, 148.2, 129.2 (2C), 128.6, 127.8 (2C), 123.4 (2C), 113.9 (2C), 61.7, 61.6, 57.4, 54.4, 51.7, 45.2, 41.7, 32.1 (2C), 29.3, 25.2 (2C), 24.5; HR-MS (ESI) m/z: [M + Na]+ calcd for C29H35BN4NaO9 617.2395; found 617.2382.
N-(4-Methoxybenzyl)-N-(3-(4-methyl-2,6-dioxotetrahydro-2H-4λ4,8λ4-[1,3,2]oxazaborolo[2,3-b][1,3,2]oxazaborol-8-yl)-1-oxo-1-(pentylamino)propan-2-yl)cinnamamide (5l): Prepared according to GP-A using α-borylaldehyde 2, 4-methoxybenzylamine 3b, trans cinnamic acid 4a, and 1-pentyl isocyanide 5e. Obtained as a white solid (yield = 40%); 1H-NMR (400 MHz, CD3OD, rotameric mixture) δ 7.80–7.51 (m, 2H), 7.49–7.31 (m, 4H), 7.29–7.21 (m, 2H), 7.13–6.79 (m, 3H), 5.14 (m, 0.5H), 5.00–4.91 (m, 0.6H), 4.71 (m, 1.9H), 4.18 (d, J = 16.8 Hz, 1H), 4.14 (d, J = 16.0 Hz, 1H), 4.00 (d, J = 16.8 Hz, 1H), 3.95–3.90 (m, 1H), 3.81–3.73 (m, 3H), 3.24–3.15 (m, 0.5H), 3.14–2.78 (m, 4.5H), 1.76–1.05 (m, 7H), 1.07–0.73 (m, 4H), (NH missed); 13C-NMR (101 MHz, CD3OD, rotameric mixture) δ 172.4–168.6 (4C), 159.7, 144.9 and 143.5 (1C), 135.9 and 135.7 (1C), 131.2 and 130.8 (1C) 130.7–129.7 (2C), 129.3 and 129.2 (2C), 128.5 and 128.4 (2C), 128.2, 119.2, and 117.8 (1C), 114.6 and 114.4 (2C), 62.5, 62.5, 56.3, 55.0, 51.3, 46.0, 40.7 and 39.9 (1C), 29.5, 29.1 (2C), 22.7, 13.6; HR-MS (ESI) m/z: [M + Na]+ calcd for C30H38BN3NaO7 586.2701; found 586.2719.
N-(4-Methoxybenzyl)-N-(3-(4-methyl-2,6-dioxotetrahydro-2H-4λ4,8λ4-[1,3,2]oxazaborolo[2,3-b][1,3,2]oxazaborol-8-yl)-1-oxo-1-(pentylamino)propan-2-yl)-4-nitrobenzamide (5m): Prepared according to GP-A using α-borylaldehyde 1, 4-methoxybenzylamine 2b, 4-nitrobenzoic acid 4b, and 1-pentyl isocyanide 3c. Obtained as a white solid (yield = 53%); 1H-NMR (400 MHz, 115 °C, DMSO-d6, 9:1 rotameric mixture, only the major rotamer reported here) δ 8.17 (d, J = 7.8 Hz, 2H), 7.66 (d, J = 7.8 Hz, 2H), 7.24 (s, 1H), 7.11 (s, 2H), 6.78 (d, J = 7.8 Hz, 2H), 4.66–4.51 (m, 2H), 4.46 (d, J = 15.6 Hz, 1H), 4.15 (d, J = 15.3 Hz, 1H), 4.12 (d, J = 15.6 Hz, 1H), 3.97 (d, J = 15.3 Hz, 1H), 3.93 (d, J = 15.6 Hz, 1H), 3.73 (s, 3H), 3.01 (d, J = 6.0 Hz, 2H), 2.91 (s, 3H), 1.47–1.21 (m, 8H), 0.89 (t, J = 6.0 Hz, 3H); 13C-NMR (101 MHz, 25 °C, DMSO-d6, 7:3 rotameric mixture) δ 170.9–168.8 (4C), 158.6 and 158.3 (1C), 148.0 and 147.7 (1C), 144.3 and 143.7 (1C), 131.2 and 129.9 (1C), 129.1 (2C), 128.8, 128.0, 124.0, 123.7, 113.9, 113.7, 62.1 (2C), 55.5, 55.4, 49.9, 46.2, 39.3, 29.4, 29.1 (2C), 22.3, 14.4; HR-MS (ESI) m/z: [M + Na]+ calcd for C28H35BN4NaO9 605.2395; found 605.2380.
N-(1-(Benzylamino)-3-(4-methyl-2,6-dioxotetrahydro-2H-4λ4,8λ4-[1,3,2]oxazaborolo[2,3-b][1,3,2]oxazaborol-8-yl)-1-oxopropan-2-yl)-N-(4-methoxybenzyl)-4-nitrobenzamide (5n): Prepared according to GP-A using α-borylaldehyde 1, 4-methoxybenzylamine 2b, 4-nitrobenzoic acid 4b, and benzyl isocyanide 3d. Obtained as a white solid (yield = 43%); 1H-NMR (400 MHz, CD3OD) δ 8.24 (d, J = 7.7 Hz, 2H), 7.75 (d, J = 7.7 Hz, 2H), 7.40–7.17 (m, 5H), 6.99 (d, J = 7.2 Hz, 2H), 6.69 (d, J = 7.2 Hz, 2H), 4.77–4.69 (m, 1H), 4.66 (d, J = 15.3 Hz, 1H), 4.41 (d, J = 15.3 Hz, 1H), 4.33 (s, 1.6H), 4.25 (d, J = 17.1 Hz, 1H), 4.23 (d, J = 16.2 Hz, 1H), 4.15 (d, J = 17.1 Hz, 0.2H), 4.13 (d, J = 17.1 Hz, 1H), 4.07 (d, J = 16.2 Hz, 1H), 3.97 (d, J = 17.1 Hz, 0.2H), 3.73 (s, 3H), 3.03 (s, 3H), 1.42–1.24 (m, 2H), (NH missed); 13C-NMR (101 MHz, CD3OD) δ 171.9, 171.0, 169.4, 168.9, 168.0, 159.3, 148.1, 142.8, 138.3, 129.3 (2C), 128.2 (2C), 127.7 (2C), 127.3, 126.8 (2C), 123.3 (2C), 113.7 (2C), 61.7, 61.5, 57.1, 54.3, 51.9, 45.2, 43.0, 29.3; HR-MS (ESI) m/z: [M + Na]+ calcd for C30H31BN4NaO9 625.2082; found 625.2069.
Methyl (2-(N-(4-methoxybenzyl)cinnamamido)-3-(4-methyl-2,6-dioxotetrahydro-2H-4λ4,8λ4-[1,3,2]oxazaborolo[2,3-b][1,3,2]oxazaborol-8-yl)propanoyl)glycinate (5o): Prepared according to GP-A using α-borylaldehyde 1, 4-methoxybenzylamine 2b, trans cinnamic acid 4a, and methyl isocyanoacetate 3e. Obtained as a brown solid (yield = 38%); 1H-NMR (300 MHz, CD3OD, 8:2 rotameric mixture) δ 7.73–7.51 (m, 2H), 7.48–7.18 (m, 6H), 7.02–6.78 (m, 3H), 5.23 (m, 0.8H), 4.92 (m, 1.2H), 4.72 (d, J = 17.2 Hz, 0.8H), 4.62 (d, J = 14.8 Hz, 0.2H), 4.14 (d, J = 17.1 Hz, 1H), 4.11 (d, J = 16.7 Hz, 1H), 3.98 (d, J = 17.1 Hz, 1H), 3.96 (d, J = 16.7 Hz, 1H), 3.83 (d, J = 12.0 Hz, 2H), 3.74 (s, 3H), 3.69 (s, 3H), 2.98 (s, 2.4H), 2.88 (s, 0.6H), 1.70–1.53 (m, 0.2H), 1.44 (dd, J = 14.4, 8.3 Hz, 0.8H), 1.14 (dd, J = 14.4, 6.6 Hz, 0.8H), 1.05–0.93 (m, 0.2H), (NH missed); 13C-NMR (75 MHz, CD3OD, rotameric mixture) δ 177.4–171.8 (5C), 163.3 and 162.9 (1C), 148.8 and 146.8 (1C), 139.0 and 138.5 (1C), 134.1, 133.9, 133.5, 132.6 and 132.4 (2C), 131.9 and 131.8 (2C), 131.5, 122.4 and 120.7 (1C), 117.9 and 117.7 (2C), 65.9, 65.8, 63.0, 60.9, 58.3, 55.2, 45.6, 44.7, 33.3; HR-MS (ESI) m/z: [M + Na]+ calcd for C28H32BN3NaO9 588.2129; found 588.2114.
Methyl (2-(N-(4-Methoxybenzyl)-4-nitrobenzamido)-3-(4-methyl-2,6-dioxotetrahydro-2H-4λ4,8λ4-[1,3,2]oxazaborolo[2,3-b][1,3,2]oxazaborol-8-yl)propanoyl)glycinate (5p): Prepared according to GP-A using α-borylaldehyde 1, 4-methoxybenzylamine 2b, 4-nitrobenzoic acid 4b, and methyl isocyanoacetate 3d. Obtained as a brown solid (yield = 42%); 1H-NMR (300 MHz, 115 °C, DMSO-d6) δ 8.19 (d, J = 8.2 Hz, 2H), 7.78 (s, 1H), 7.71 (d, J = 8.2 Hz, 2H), 7.25–7.05 (m, 2H), 6.79 (d, J = 7.9 Hz, 2H), 4.82–4.56 (m, 2H), 4.46 (d, J = 15.8 Hz, 1H), 4.17 (d, J = 17.0 Hz, 1H), 4.14 (d, J = 17.1 Hz, 1H), 3.97 (d, J = 17.0 Hz, 1H), 3.95 (d, J = 17.1 Hz, 1H), 3.83 (dd, J = 10.1, 5.5 Hz, 2H), 3.75 (s, 3H), 3.70 (s, 3H), 2.91 (s, 3H), 1.46–1.12 (m, 2H); 13C-NMR (75 MHz, 25 °C, DMSO-d6, rotameric mixture) δ 171.3–168.4 (5C), 158.2 and 157.9 (1C), 147.7 and 147.3 (1C), 143.8 and 143.1 (1C), 130.8, 128.9 (2C), 128.4, 127.6, 123.5 and 123.4 (2C), 113.5 and 113.3 (2C), 61.6 (2C), 59.3, 55.1, 51.8, 49.5, 45.8, 40.9, (B-Cα missed); 11B-NMR (128 MHz, CD3CN) δ 12.17; HR-MS (ESI) m/z: [M + Na]+ calcd for C26H29BN4NaO11 607.1824; found 607.1828.
(2-(N-(4-Bromophenyl)cinnamamido)-3-(tert-butylamino)-3-oxopropyl)boronic acid (6a): Prepared according to GP-B starting from 5a. Obtained as a white solid (yield = 97%); 1H-NMR (400 MHz, 115 °C, DMSO-d6) δ 7.63 (d, J = 8.4 Hz, 2H), 7.51 (d, J = 15.5 Hz, 1H), 7.40–7.28 (m, 7H), 6.92 (s, 1H), 6.26 (d, J = 15.5 Hz, 1H), 5.20 (dd, J = 9.4, 6.1 Hz, 1H), 1.29 (s, 9H), 0.98 (dd, J = 15.2, 9.4 Hz, 1H), 0.78 (dd, J = 15.2, 6.1 Hz, 1H), (BOH missed); 13C-NMR (75 MHz, 25 °C, DMSO-d6, 6:4 rotameric mixture) δ 172.7 and 171.5 (1C), 164.9, 141.6 and 141.1 (1C), 138.6, 134.5, 132.6, 132.0 (2C), 129.9 and 129.8 (1C), 129.0 (3C), 127.7 (2C), 121.5, 119.5 and 119.0 (1C), 58.8 and 56.9 (1C), 51.2 and 50.1 (1C), 28.5 and 28.3 (3C), 19.3; 11B-NMR (128 MHz, CD3CN) δ 32.01; HR-MS (ESI) m/z: [C22H24BrN2O2B(OMe)2 + Na]+ calcd for C24H30BBrN2NaO4 523.1380; found 523.1391.
(3-(tert-butylamino)-2-(N-(4-methoxybenzyl)cinnamamido)-3-oxopropyl)boronic acid (6b): Prepared according to GP-B starting from 5b. Obtained as a white solid (yield = 78%); 1H-NMR (400 MHz, CD3OD) δ 7.74 (d, J = 15.4 Hz, 1H), 7.60–7.55 (m, 2H), 7.41–7.37 (m, 3H), 7.27 (d, J = 8.5 Hz, 2H), 7.11 (d, J = 15.4 Hz, 1H), 6.94 (d, J = 8.5 Hz, 2H), 5.02 (d, J = 16.7 Hz, 1H), 4.70–4.62 (m, 2H), 3.79 (s, 3H), 1.37–1.27 (m, 9H), 1.18–1.14 (m, 1H), 0.88–0.80 (m, 1H), (NH, BOH missed); 13C-NMR (75 MHz, CD3OD) δ 176.0, 169.8, 161.2, 146.1, 136.4, 131.8, 130.5, 130.3 (2C), 129.9 (2C), 129.6 (2C), 118.7, 115.6 (2C), 62.9, 56.1, 53.8, 52.9, 31.1, 28.9 (3C); 11B-NMR (128 MHz, CD3CN) δ 31.55; HR-MS (ESI) m/z: [C24H29N2O3B(OMe)2 + Na]+ calcd for C26H35BN2NaO5 489.2537; found 489.2549.
(3-(tert-Butylamino)-2-(N-(5,5-dimethylhexyl)cinnamamido)-3-oxopropyl)boronic acid (6c): Prepared according to GP-B starting from 5c. Obtained as a white solid (yield = 61%); 1H-NMR (400 MHz, CD3OD) δ 7.75 (d, J = 15.4 Hz, 1H), 7.71–7.59 (m, 2H), 7.44 (m, 3H), 7.11 (d, J = 15.4 Hz, 1H), 4.50 (m, 1H), 3.99–3.72 (m, 1H), 3.53–3.35 (m, 1H), 1.78–1.48 (m, 2H), 1.48–1.11 (m, 15H), 0.90 (s, 9H), (NH, BOH missed); 13C-NMR (75 MHz, CD3OD) δ 171.2, 170.2, 146.2, 134.1, 131.9, 130.4 (2C), 129.7 (2C), 118.2, 63.4, 54.3, 52.3, 45.3, 32.1, 31.0, 30.0 (3C), 29.0 (3C), 23.2, (B-Cα missed); 11B-NMR (128 MHz, CD3CN) δ 30.99; HR-MS (ESI) m/z: [C24H37N2O2B(OMe)2 + Na]+ calcd for C26H43BN2NaO4 481.3214; found 481.3234.
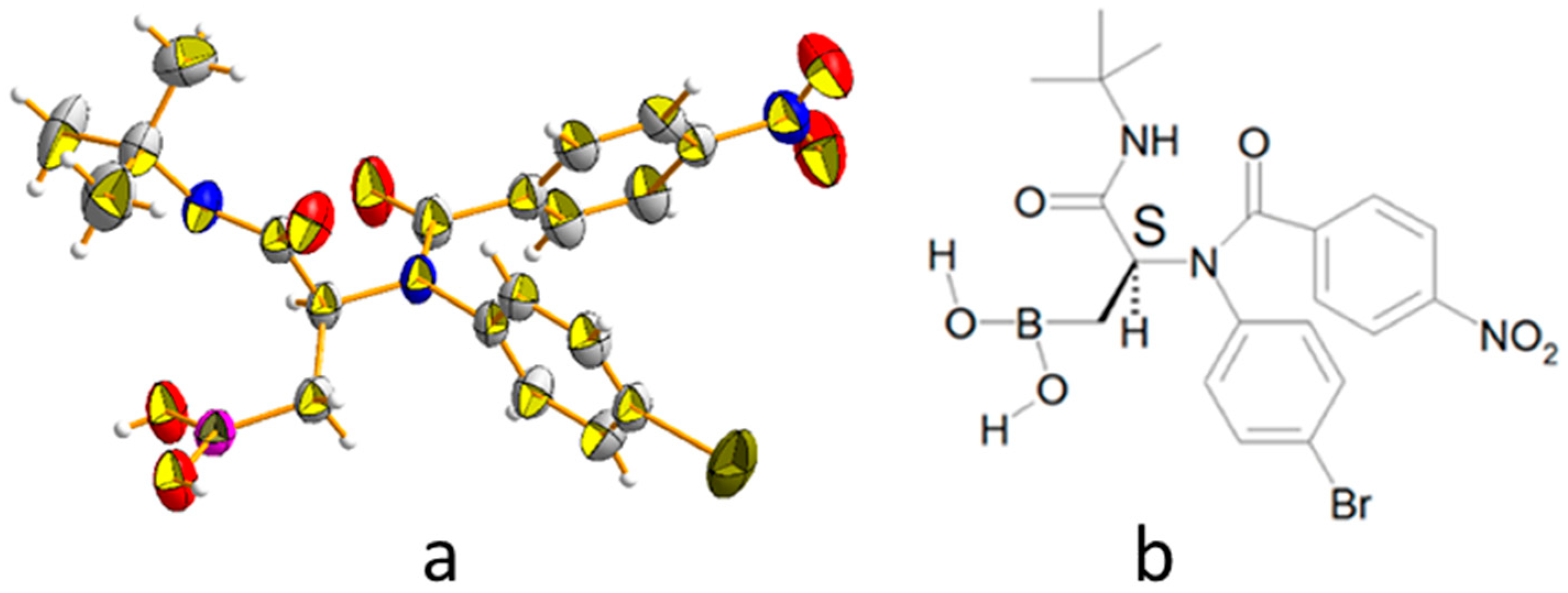
(2-(N-(4-Bromophenyl)-4-nitrobenzamido)-3-(tert-butylamino)-3-oxopropyl)boronic acid (6d): Prepared according to GP-B starting from 5d. Obtained as a white solid (yield = 99%); 1H-NMR (400 MHz, 115 °C, DMSO-d6, 1:1 rotameric mixture) δ 8.12–7.96 (m, 2H), 7.53 (d, J = 8.5 Hz, 1H), 7.48 (d, J = 8.5 Hz, 1H), 7.45–7.38 (m, 2H), 7.28–7.18 (m, 2H), 6.91 (s, 1H), 5.29–5.13 (m, 1H), 1.32 (s, 9H), 1.10 (dd, J = 15.5, 9.0 Hz, 0.5H), 1.04 (dd, J = 14.1, 8.1 Hz, 0.5H), 0.97 (dd, J = 8.1, 5.1 Hz, 0.5H), 0.91 (dd, J = 15.5, 6.8 Hz, 0.5H), (BOH missed); 13C-NMR (75 MHz, 25 °C, DMSO-d6, 2:1 rotameric mixture) δ 170.9, 167.7, 147.4 and 147.2 (1C), 143.4, 138.8, 132.5 (2C), 131.9 and 131.8 (1C), 131.5 (2C), 128.9, 123.1 (2C), 120.8 and 120.7 (1C), 57.7, 50.3, 28.5 and 28.2 (3C), (B-Cα missed); 11B-NMR (128 MHz, CD3CN) δ 31.06; HRMS (ESI) m/z: [C20H21BrN3O4B(OMe)2 + Na]+ calcd for C22H27BBrN3NaO6 542.1074; found 542.1061.
(2-(N-Benzyl-4-nitrobenzamido)-3-(tert-butylamino)-3-oxopropyl)boronic acid (6e): Prepared according to GP-B starting from 5e. Obtained as a white solid (yield = 89%); 1H-NMR (400 MHz, 115 °C, DMSO-d6, 7:3 rotameric mixture) δ 8.27–8.16 (m, 2H), 7.82 (s, 0.3H), 7.67 (d, J = 8.6 Hz, 0.6H), 7.61 (d, J = 8.6 Hz, 1.4H), 7.30–7.16 (m, 5H), 6.59 (s, 0.7H), 4.77 (dd, J = 16.3, 9.2 Hz, 1H), 4.72–4.55 (m, 1H), 4.51 (dd, J = 16.3, 9.2 Hz, 1H), 1.30 (dd, J = 15.5, 6.8 Hz, 1H), 1.24 (s, 9H), 1.13 (dd, J = 15.5, 6.8 Hz, 1H), (BOH missed); 13C-NMR (75 MHz, 25 °C, DMSO-d6, rotameric mixture) δ 171.2–170.2 (2C), 147.6, 143.6, 139.0, 128.0 (2C), 127.7 (2C), 126.8 (2C), 126.3, 123.7 (2C), 59.9 and 59.4 (1C), 50.3 and 50.0 (1C), 46.5, 28.3 (3C), 18.5; HR-MS (ESI) m/z: [C21H24N3O4B(OMe)2 + Na]+ calcd for C23H30BN3NaO6 478.2125; found 478.2104. Anal. calcd for C21H26BN3O6: C, 59.03; H, 6.13; B, 2.53; N, 9.83; O, 22.47; found: C, 59.33; H, 6.25; N, 9.56.
(3-(tert-Butylamino)-2-(N-(5,5-dimethylhexyl)-4-nitrobenzamido)-3-oxopropyl)boronic acid (6f): Prepared according to GP-B starting from 5f. Obtained as a white solid (yield = 95%); 1H-NMR (400 MHz, CD3OD) δ 8.38 (d, J = 8.1 Hz, 2H), 7.69 (d, J = 8.1 Hz, 2H), 4.75–4.63 (m, 1H), 3.43–3.24 (m, 2H), 1.69–1.52 (m, 2H), 1.50–1.23 (m, 12H), 1.14–0.99 (m, 3H), 0.81 (s, 9H), (NH, BOH missed); 13C-NMR (75 MHz, CD3OD) δ 172.0, 171.8, 150.3, 143.7, 129.3 (2C), 125.3 (2C), 61.3, 51.2, 45.1 (2C), 31.9, 31.2, 29.9 (3C), 29.0 (3C), 23.3 (2C); 11B NMR (128 MHz, CD3CN) δ 31.22; HR-MS (ESI) m/z: [C22H34N3O4B(OMe)2 + Na]+ calcd for C24H40BN3NaO6 500.2908; found 500.2924.
(2-(N-(4-Bromophenyl)dec-9-enamido)-3-(tert-butylamino)-3-oxopropyl)boronic acid (6g): Prepared according to GP-B starting from 5g. Obtained as a white solid (yield = 99%); 1H-NMR (400 MHz, 115 °C, DMSO-d6) δ 7.59 (d, J = 8.2 Hz, 2H), 7.25 (d, J = 8.2 Hz, 2H), 7.12 (s, 1H), 5.87–5.73 (m, 1H), 5.09 (dd, J = 9.9, 5.8 Hz, 1H), 4.96 (dd, J = 21.4, 13.7 Hz, 2H), 2.08–1.89 (m, 4H), 1.51–1.42 (m, 2H), 1.37–1.14 (m, 17H), 0.86 (dd, J = 14.9, 9.9 Hz, 1H), 0.66 (dd, J = 14.9, 5.8 Hz, 1H), (BOH missed); 13C-NMR (75 MHz, 25 °C, DMSO-d6, rotameric mixture) δ 172.7 and 172.2 (1C), 172.0 and 171.5 (1C), 139.1, 138.8, 132.4 (2C), 131.9 (2C), 121.3 and 121.1 (1C), 114.6, 56.2, 50.0, 34.2, 33.1, 28.5, 28.4 (3C), 28.3, 28.2, 28.1, 24.8, 17.2; 11B-NMR (128 MHz, CD3CN) δ 31.21; HR-MS (ESI) m/z: [C23H34BrN2O2B(OMe)2 + Na]+ calcd for C25H40BBrN2NaO4 545.2162; found 545.2150.
(3-(tert-Butylamino)-2-(N-(4-methoxybenzyl)dec-9-enamido)-3-oxopropyl)boronic acid (6h): Prepared according to GP-B starting from 5h. Obtained as a white solid (yield = 74%); 1H-NMR (400 MHz, CD3OD, 8:2 rotameric mixture) δ 7.22 (d, J = 8.2 Hz, 2H), 6.95 (d, J = 8.2 Hz, 2H), 5.91–5.73 (m, 1H), 5.06–4.88 (m, 2H), 4.76 (d, J = 16.7 Hz, 1H), 4.57–4.45 (m, 2H), 3.80 (s, 3H), 2.61–2.41 (m, 2H), 2.12–1.96 (m, 2H), 1.79–1.52 (m, 2H), 1.52–1.18 (m, 17H), 1.10 (dd, J = 14.0 Hz, 1H), 0.79 (dd, J = 14.0 Hz, 1H), (NH, BOH missed); 13C-NMR (75 MHz, CD3OD) δ 179.5, 177.9, 163.4, 142.6, 134.8, 132.2, and 131.91 (2C), 117.9 (2C), 117.3, 64.4, 58.3, 55.7, 55.0, 37.3(2C), 33.3–32.5 (4C), 31.1 (3C), 28.9 (2C); 11B-NMR (128 MHz, CD3CN) δ 31.54; HR-MS (ESI) m/z: [C25H39N2O3B(OMe)2 + Na]+ calcd for C27H45BN2NaO5 511.3319; found 511.3334.
(3-(tert-Butylamino)-2-(N-(5,5-dimethylhexyl)dec-9-enamido)-3-oxopropyl)boronic acid (6i): Prepared according to GP-B starting from 5i. Obtained as a white solid (yield = 99%); 1H-NMR (400 MHz, 115 °C, DMSO-d6) δ 7.11 (s, 1H), 5.90–5.73 (m, 1H), 4.97 (dd, J = 23.7, 13.7 Hz, 2H), 4.80–4.53 (m, 1H), 3.34–3.17 (m, 2H), 2.32 (t, J = 6.6 Hz, 2H), 2.04 (d, J = 6.3 Hz, 2H), 1.63–1.43 (m, 6H), 1.43–1.11 (m, 20H), 0.88 (s, 9H), 0.79 (m, 1H), (BOH missed); 13C-NMR (75 MHz, 25 °C, CD3OD) δ 173.1, 172.3, 138.9, 114.7, 57.6, and 55.1 (1C), 44.6, 43.3 (2C), 33.2 (2C), 32.6 (2C), 30.7 (1C) 30.0 (1C), 29.2 (3C), 28.7 (2C), 28.3 (3C), 25.1 and 24.8 (2C), 22.1 and 21.7 (1C); 11B-NMR (128 MHz, CD3CN) δ 31.19; HR-MS (ESI) m/z: [C25H47N2O2B(OMe)2 + Na]+ calcd for C27H53BN2NaO4 503.3996; found 503.4006.
(3-(Cyclohexylamino)-2-(N-(4-methoxybenzyl)cinnamamido)-3-oxopropyl)boronic acid (6j): Prepared according to GP-B starting from 5j. Obtained as a white solid (yield = 83%); 1H-NMR (400 MHz, 115 °C, DMSO-d6, 6:4 rotameric mixture) δ 7.65 (d, J = 7.1 Hz, 1H), 7.58–7.46 (m, 3H), 7.43 (d, J = 7.1 Hz, 1H), 7.39–7.29 (m, 2H), 7.24–7.13 (m, 2H), 6.96–6.79 (m, 2H), 5.18–5.12 (m, 0.4H), 4.84 (d, J = 17.6 Hz, 0.6H), 4.77–4.69 (m, 0.6H), 4.62 (d, J = 15.2 Hz, 0.4H), 4.56 (d, J = 17.6 Hz, 0.6H), 4.49 (d, J = 15.2 Hz, 0.4H), 3.70 (s, 3H), 3.48–3.37 (m, 1H), 1.64–1.44 (m, 4H), 1.32 (dd, J = 15.2 Hz, 1H), 1.25–1.00 (m, 6H), 0.85 (dd, J = 15.2, 5.9 Hz, 0.5H), 0.77 (dd, J = 15.2, 5.9 Hz, 0.5H), (NH, BOH missed); 13C-NMR (75 MHz, 25 °C, DMSO-d6, 6:4 rotameric mixture) δ 170.5 and 169.9 (1C), 166.5 and 166.2 (1C), 158.1 and 158.0 (1C), 141.4, 135.0, and 135.0 (1C), 131.3 and 131.2 (2C), 129.6, 128.8 (3C), 127.8 (2C), 127.6, 119.6, 113.8, 113.5, 55.5, 55.0, 47.8, and 47.6 (1C), 47.1, 32.1 (2C), 29.0, 25.2, 24.3 (2C); 11B-NMR (128 MHz, CD3CN) δ 31.40; HR-MS (ESI) m/z: [C26H32N2O4B(OMe) + Na]+ calcd for C27H35BN2NaO5 501.2537; found 501.2549.
(3-(Cyclohexylamino)-2-(N-(4-methoxybenzyl)-4-nitrobenzamido)-3-oxopropyl)boronic acid (6k): Prepared according to GP-B starting from 5k. Obtained as a white solid (yield = 66%); 1H-NMR (400 MHz, CD3OD, 6:4 rotameric mixture) δ 8.45–8.25 (m, 2H), 7.70 (d, J = 8.0 Hz, 2H), 7.31 (d, J = 7.4 Hz, 1H), 7.09 (d, J = 7.4 Hz, 1H), 6.94–6.76 (m, 2H), 5.27 (d, J = 16.2 Hz, 0.4H), 4.79–4.64 (m, 0.6H), 4.64–4.50 (m, 1.2H), 4.46 (d, J = 16.2 Hz, 0.4H), 4.41–4.29 (m, 0.4H), 3.81 (s, 1.2H), 3.78 (s, 1.8H), 3.71–3.48 (m, 1H), 1.93–1.70 (m, 4H), 1.70–1.54 (m, 2H), 1.47–1.05 (m, 5H), 1.05–0.74 (m, 1H), (NH, BOH missed); 13C NMR (75 MHz, CD3OD, 6:4 rotameric mixture) δ 174.0 and 173.6 (1C), 169.6 and 169.5 (1C), 161.3 and 160.7 (1C), 150.2, 144.4 and 143.6 (1C), 132.2, 130.5, 130.1, 129.5, 129.1, 125.2 (2C), 115.5, 115.2, 61.4, and 60.8 (1C), 56.0, 54.2, 51.6, and 50.6 (1C), 33.8 (2C), 31.0, 26.8, 26.3 (2C); 11B-NMR (128 MHz, CD3CN) δ 31.58; HR-MS (ESI) m/z: [C24H28N3O5B(OMe)2 + Na]+ calcd for C26H34BN3NaO7 534.2388; found 534.2373.
(2-(N-(4-Methoxybenzyl)cinnamamido)-3-oxo-3-(pentylamino)propyl)boronic acid (6l): Prepared according to GP-B starting from 5l. Obtained as a white solid (yield = 99%); 1H-NMR (400 MHz, DMSO-d6, rotameric mixture) δ 7.62–7.26 (m, 7H), 7.18 (d, J = 7.7 Hz, 2H), 6.88 (m, 3H), 5.29–5.07 (m, 0.6H), 4.82 (d, J = 17.2 Hz, 0.6H), 4.79–4.73 (m, 0.4H), 4.70 (d, J = 17.2 Hz, 0.2H), 4.55 (t, J = 8.1 Hz, 1.2H), 3.72 (s, 3H), 3.16–2.85 (m, 2H), 1.53–0.99 (m, 6H), 0.99–0.61 (m, 5H), (BOH missed); 13C-NMR (75 MHz, DMSO-d6, rotameric mixture) δ 171.3 and 170.9 (1C), 166.4 and 165.1 (1C), 158.5 and 158.1 (1C), 141.8 and 141.1 (1C), 135.3 and 135.0 (1C), 131.1 and 130.3 (1C), 129.6, 128.8 (2C), 128.4, 128.0 and 127.8 (2C), 127.5, 119.6 and 118.7 (1C), 113.9 and 113.4 (2C), 55.6, 55.0, 47.3, 28.6 (3C), 21.9, 15.7, 13.9; 11B NMR (128 MHz, CD3CN) δ 31.20; HR-MS (ESI) m/z: [C25H31N2O3B(OMe)2 + Na]+ calcd for C27H37BN2NaO5 503.2693; found 503.2704.
(2-(N-(4-Methoxybenzyl)-4-nitrobenzamido)-3-oxo-3-(pentylamino)propyl)boronic acid (6m): Prepared according to GP-B starting from 5m. Obtained as a white solid (yield = 64%); 1H-NMR (400 MHz, CD3OD, 6:4 rotameric mixture) δ 8.41–8.26 (m, 2H), 7.74 (d, J = 7.2 Hz, 2H), 7.31 (d, J = 7.2 Hz, 1H), 7.12 (d, J = 7.2 Hz, 1H), 6.94–6.82 (m, 2H), 5.26 (d, J = 15.7 Hz, 0.4H), 4.73–4.65 (m, 0.5H), 4.55 (dd, J = 15.7 Hz, 1.2H), 4.45–4.41 (m, 0.5H), 4.37 (d, J = 15.8 Hz, 0.4H), 3.81 (s, 1.3H), 3.78 (s, 1.7H), 3.28–3.21 (m, 1.1H), 3.19–3.08 (m, 0.9H), 1.61–1.42 (m, 2H), 1.42–1.22 (m, 6H), 0.96–0.90 (m, 3H), (NH, BOH missed); 13C-NMR (101 MHz, CD3OD, rotameric mixture) δ 176.1, 173.4, 162.5, 149.2, 143.2, 136.0, 129.2 (2C), 128.3 (2C), 124.0 (2C), 114.4 (2C), 61.6 and 61.0 (1C), 55.1, 54.5, 40.3, 29.9, 29.4, 29.2, 22.6. (B-Cα missed); 11B-NMR (128 MHz, CD3CN) δ 31.59; HR-MS (ESI) m/z: [C23H28N3O5B(OMe)2 + Na]+ calcd for C25H34BN3NaO7 522.2388; found 522.2401.
(3-(Benzylamino)-2-(N-(4-methoxybenzyl)-4-nitrobenzamido)-3-oxopropyl)boronic acid (6n): Prepared according to GP-B starting from 5n. Obtained as a white solid (yield = 97%); 1H-NMR (400 MHz, CD3OD) δ 8.31–8.17 (m, 2H), 7.64 (d, J = 5.9 Hz, 2H), 7.41–7.23 (m, 6H), 7.06 (d, J = 7.3 Hz, 1H), 6.91 (d, J = 7.7 Hz, 1H), 6.81 (d, J = 7.3 Hz, 1H), 4.62–4.21 (m, 5H), 3.81 (s, 3H), 1.44–1.19 (m, 2H), (NH, BOH missed); 13C-NMR (75 MHz, CD3OD) δ 169.8, 169.4, 167.0, 158.9, 150.1, 142.8, 138.2, 130.3, 130.2, 129.9 (2C), 129.1 (2C), 128.7 (2C), 125.1 (3C), 115.4, 115.3, 61.6, 56.1, 53.7, 44.6, 31.0; 11B-NMR (128 MHz, CD3CN) δ 30.99; HR-MS (ESI) m/z: [C25H24N3O5B(OMe)2 + Na]+ calcd for C27H30BN3NaO7 542.2075; found 542.2058.
(3-((2-Methoxy-2-oxoethyl)amino)-2-(N-(4-methoxybenzyl)cinnamamido)-3-oxopropyl)boronic acid (6o): Prepared according to GP-B starting from 5o. Obtained as a brown solid (yield = 99%); 1H-NMR (400 MHz, CD3OD) δ 7.79 (d, J = 15.3 Hz, 1H), 7.59–7.53 (m, 2H), 7.43–7.34 (m, 3H), 7.26 (d, J = 8.2 Hz, 2H), 7.08 (d, J = 15.3 Hz, 1H), 6.94 (d, J = 8.2 Hz, 2H), 5.03–4.93 (m, 2H), 4.74 (d, J = 17.2 Hz, 1H), 3.95 (d, J = 9.4 Hz, 2H), 3.79 (s, 3H), 3.75 (s, 3H), 1.03–0.86 (m, 2H), (NH, BOH missed); 13C-NMR (75 MHz, CD3OD) δ 175.2, 171.9, 170.5, 161.1, 146.5, 136.3, 131.8, 130.3 (2C), 129.6 (2C), 129.5 (2C), 128.6, 118.6, 115.6 (2C), 60.6, 56.0, 52.9, 51.9, 42.4, 31.0; 11B-NMR (128 MHz, CD3CN) δ 31.50; HR-MS (ESI) m/z: [C23H25N2O5B(OMe)2 + Na]+ calcd for C25H31BN2NaO7 505.2122; found 505.2135.
(3-((2-Methoxy-2-oxoethyl)amino)-2-(N-(4-methoxybenzyl)-4-nitrobenzamido)-3-oxopropyl)boronic acid (6p): Prepared according to GP-B starting from 5p. Obtained as a brown solid (yield = 91%); 1H-NMR (400 MHz, CD3OD, rotameric mixture) δ 8.44–8.22 (m, 2H), 7.91–7.64 (m, 2H), 7.46–6.75 (m, 4H), 5.38 (d, J = 14.8 Hz, 0.3H), 5.22–4.97 (m, 0.4H), 4.74–4.64 (m, 0.3H), 4.64–4.44 (m, 1.3H), 4.41–4.31 (m, 0.7H), 4.02–3.89 (m, 2H), 3.78 (s, 3H), 3.77 (s, 3H), 1.71–1.48 (m, 1H), 1.48–1.25 (m, 1H), (NH, BOH missed); 13C-NMR (101 MHz, CD3OD, rotameric mixture) δ 172.6, 171.8, 170.9, 159.7, 149.1, 143.2, and 142.9 (1C), 131.2, 129.3, and 129.1 (2C), 128.4 (2C), 124.0 (2C), 114.2 (2C), 60.6, 55.0, 52.0, 42.6, 41.2, 30.0; 11B NMR (128 MHz, CD3CN) δ 31.57; HR-MS (ESI) m/z: [C21H22N3O7B(OMe)2 + Na]+ calcd for C23H28BN3NaO9 524.1816; found 524.1827.
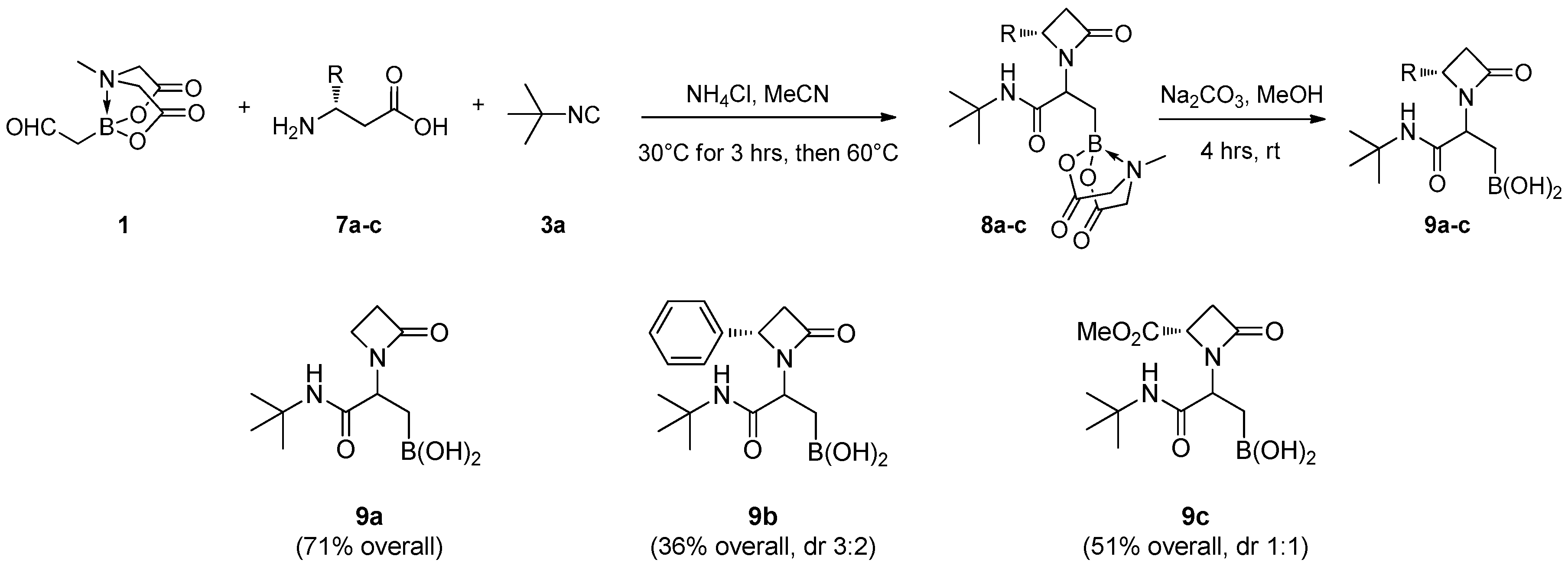
N-(tert-Butyl)-3-(4-methyl-2,6-dioxotetrahydro-2H-4λ4,8λ4-[1,3,2]oxazaborolo[2,3-b][1,3,2]oxazaborol-8-yl)-2-(2-oxoazetidin-1-yl)propenamide (8a): Prepared according to GP-C using α-borylaldehyde 1, β-alanine 7a and tert-butyl isocyanide 3a. Obtained as a white solid (yield = 72%); 1H-NMR (400 MHz, CD3OD) δ 4.35 (dd, J = 9.1, 6.2 Hz, 1H)), 4.18 (d, J = 17.1 Hz, 1H), 4.17 (d, J = 16.9 Hz, 1H), 4.02 (d, J = 17.1 Hz, 1H), 3.99 (d, J = 16.9 Hz, 1H), 3.47 (dd, J = 8.4, 4.9 Hz, 1H), 3.41 (dd, J = 8.4, 4.9 Hz, 1H), 3.01 (s, 3H), 2.94–2.86 (m, 2H), 1.36 (s, 9H), 1.22 (dd, J = 14.4, 9.1 Hz, 1H), 1.13 (dd, J = 14.4, 6.2 Hz, 1H), (NH missed); 13C-NMR (101 MHz, CD3OD) δ 171.8 and 171.7 (1C), 169.9, 169.5 (2C), 62.3 and 62.2 (2C), 54.3, 53.8, 51.5, 38.4, 35.3, 30.0, 28.1 (3C); 11B-NMR (128 MHz, CD3CN) δ 12.15; HR-MS (ESI) m/z: [M + Na]+ calcd for C15H24BN3NaO6 376.1656; found 376.1647.
N-(tert-Butyl)-3-(4-methyl-2,6-dioxotetrahydro-2H-4λ4,8λ4-[1,3,2]oxazaborolo[2,3-b][1,3,2]oxazaborol-8-yl)-2-((S)-2-oxo-4-phenylazetidin-1-yl)propenamide (8b): Prepared according to GP-C using α-borylaldehyde 1, S-β-phenylalanine 7b and tert-butyl isocyanide 3a. Obtained as a white solid (yield = 36%); 1H-NMR (400 MHz, CD3OD, 3:2 diasteroisomeric mixture, presence of rotamers) δ 7.58–7.15 (m, 5H), 4.86 (dd, J = 5.3, 2.3 Hz, 0.1H), 4.78 (dd, J = 5.3, 2.3 Hz, 0.3H), 4.70 (ddd, J = 10.8, 5.3, 2.3 Hz, 0.6H), 4.32–4.28 (m, 0.2H), 4.18–4.11 (m, 2.6H), 4.02–3.94 (m, 2H), 3.87–3.83 (m, 0.2H), 3.44–3.35 (m, 1H), 3.00 (s, 1.8H), 2.99 (s, 1.2H), 2.83–2.75 (m, 1H), 1.55–1.06 (m, 11H), (NH missed); 13C-NMR (75 MHz, CD3OD, 3:2 diasteroisomeric mixture, presence of rotamers) δ 173.2–170.5 (4C), 140.6 and 140.4 (1C), 130.2 (2C), 129.9 and 129.7 (1C), 128.3 (2C), 63.5, 63.4, and 63.3 (1C), 57.3–57.0 (1C), 47.3 and 46.2 (1C), 46.9 and 46.7 (1C), 32.1, 31.0, 29.1, and 29.0 (3C), 24.0; HR-MS (ESI) m/z: [M + Na]+ calcd for C21H28BN3NaO6 452.1969; found 452.1985.
Methyl (2S)-1-(1-(tert-butylamino)-3-(4-methyl-2,6-dioxotetrahydro -2H-4λ4,8λ4-[1,3,2]oxazaborolo[2,3-b][1,3,2]oxazaborol-8-yl)-1-oxopropan-2-yl)-4-oxoazetidine-2-carboxylate (8c): Prepared according to GP-C using α-borylaldehyde1, (3S)-3-ammonio-4-methoxy-4- oxobutanoate 7c and tert-butyl isocyanide 3a. Obtained as a white solid (yield = 52%); 1H-NMR (400 MHz, 115 °C, DMSO-d6, 1:1 diasteroisomeric mixture) δ 7.71 (s, 0.5H), 7.56 (s, 0.5H), 4.38–4.33 (m, 0.5H), 4.21–4.13 (m, 3H), 4.06–3.94 (m, 2.5H), 3.72 (s, 1.5H), 3.61 (s, 1.5H), 3.12 (dd, J = 14.2, 5.8 Hz, 1H), 2.91 (s, 1.5H), 2.89 (s, 1.5H), 2.78 (dd, J = 14.2, 2.0 Hz, 1H), 1.26 (s, 4.5H), 1.22 (s, 4.5H), 1.15 (d, J = 8.1 Hz, 0.5H), 1.10–0.99 (m, 1H), 0.93 (dd, J = 14.6, 8.1 Hz, 0.5H); 13C-NMR (75 MHz, 25 °C, DMSO-d6, 1:1 diasteroisomeric mixture) δ 172.1 and 171.3 (1C), 169.0, 168.8, 168.6, 165.8, and 165.0 (1C), 61.8 (2C), 55.2 and 52.3 (1C), 52.1 and 51.7 (1C), 51.0 and 49.0 (1C), 50.2 and 50.1 (1C), 45.7, 41.4, and 40.8 (1C), 29.0, 28.3 (3C); HR-MS (ESI) m/z: [M + Na]+ calcd for C17H26BN3NaO8 434.1711; found 434.1723.
(3-(tert-Butylamino)-3-oxo-2-(2-oxoazetidin-1-yl)propyl)boronic acid (9a): Prepared according to GP-B starting from 8a. Obtained as a white solid (yield = 99%); 1H-NMR (400 MHz, CD3OD) δ 4.35 (t, J = 7.8 Hz, 1H, CH), 3.41–3.30 (m, 2H, CH2-N), 2.92–2.86 (m, 2H, CH2-CO), 1.35 (s, 9H, tert-Bu), 1.21 (dd, J = 15.7, 7.8 Hz, 2H, CH2-B), (NH, BOH missed); 13C-NMR (101 MHz, CD3OD) δ 172.1 (CO), 169.3 (CO), 54.3 (CH-N), 51.7 ((CH3)3C-N), 38.1 (CH2-N), 35.3 (CH2-CO), 30.0 (CH2-B), 28.0 (3C, (CH3)3C); 11B-NMR (128 MHz, CD3OD) δ 28.84; HR-MS (ESI) m/z: [M + Na]+ calcd for C10H19BN2NaO4 265.1336; found 265.1357.
(3-(tert-Butylamino)-3-oxo-2-((S)-2-oxo-4-phenylazetidin-1-yl)propyl)boronic acid (9b): Prepared according to GP-B starting from 8b. Obtained as a white solid (yield = 99%); 1H-NMR (400 MHz, 115 °C, DMSO-d6, 6:4 diastereoisomeric mixture) δ 7.48–7.26 (m, 5H, Ph), 6.87 (s, 0.4H, NH), 6.71 (s, 0.6H, NH), 4.83–4.75 (m, 0.4H, CH-Ph), 4.72–4.62 (m, 0.6H, CH-Ph), 4.12–3.99 (m, 1H, CH), 3.28 (dd, J = 14.5, 5.4 Hz, 1H, CH-CO), 2.74–2.63 (m, 1H, CH-CO), 1.44–1.11 (m, 10H, tert-Bu, CH-B), 0.98 (d, J = 7.4 Hz, 1H, CH-B), (BOH missed); 13C-NMR (75 MHz, 25 °C, DMSO-d6, 6:4 diastereoisomeric mixture) δ 170.6 and 169.9 (1C, CO), 167.5 and 167.1 (1C, CO), 140.3 and 139.6 (1C, C(Ph)), 128.6 (2C, CH(Ph)), 127.9 (CH(Ph)), 126.6 (2C, CH(Ph)), 54.8–52.9 (1C, CH-N), 50.1 ((CH3)3C-N), 45.7 (CH2-CO), 31.6–29.5 (1C, CH-Ph), 29.0 (CH2-B), 28.4 and 28.2 (3C, (CH3)3C); 11B-NMR (128 MHz, CD3CN) δ 31.09; HR-MS (ESI) m/z: [C16H21N2O2B(OMe)2 + Na]+ calcd for C18H27BN2NaO4 369.1962; found 369.1970.
(3-(tert-Butylamino)-2-((S)-2-(methoxycarbonyl)-4-oxoazetidin-1-yl)-3-oxopropyl)boronic acid (9c): Prepared according to GP-B starting from 8c. Obtained as a white solid (yield = 99%); 1H-NMR (400 MHz, CD3OD, 1:2 diastereoisomeric mixture) δ 4.42-4.21 (m, 2H, CH-N, CH-CO2), 3.83 and 3.80 (s, 3H, OCH3), 3.23 (dd, J = 14.4, 5.6 Hz, 1H, CH-CO), 2.94 (q, J = 14.4, 11.8 Hz, 1H, CH-CO), 1.47–1.21 (m, 11H, tert-Bu, CH2-B), (NH, BOH missed); 13C-NMR (75 MHz, CD3OD, 1:2 diastereoisomeric mixture) δ 173.6 (CO2), 172.8 (CO), 169.5 (CO), 57.4 and 55.9 (1C, CH-N), 53.4 and 53.2 (1C, CH-CO2), 52.8 ((CH3)3C-N), 51.8 (CH3O), 42.3 (CH2-CO), 31.0 (CH2-B), 29.0 (3C, (CH3)3C); 11B-NMR (128 MHz, CD3CN) δ 31.24; HR-MS (ESI) m/z: [C12H19N2O4B(OMe)2+Na]+ calcd for C14H25BN2NaO6 351.1703; found 351.1724.
8-((1-(Cyclohexylmethyl)-1H-imidazol-5-yl) methyl)-4-methyldihydro-4λ4,8λ4-[1,3,2]oxazaborolo[2,3-b][1,3,2]oxazaborole-2,6(3H,5H)-dione (10): Prepared according to GP-D. Obtained as a brown solid (yield = 32%); 1H-NMR (300 MHz, (CD3)2CO) δ 7.65 (s, 1H, CH(Im)), 6.81 (s, 1H, CH(Im)), 4.27 (d, J = 16.9 Hz, 2H, CH2-N(MIDA)), 4.05 (d, J = 16.9 Hz, 2H, CH2-N(MIDA)), 3.88 (d, J = 7.4 Hz, 2H, CH2-N(Im)), 3.18 (s, 3H, N-CH3), 1.88–1.52 (m, 9H, CH and 4 CH2(Cy)), 1.26–1.15 (m, 2H, CH2(Cy)), 1.12–0.93 (m, 2H, CH2-B); 13C-NMR (101 MHz, CD3OD) δ 169.4 (2C, CO2), 130.7 (CH(Im)), 129.3 (C(Im)), 118.1 (CH(Im)), 61.5 (2C, CH2-N (MIDA)), 52.4 and 52.1 (1C, CH2-N), 42.2 (CH3-N (MIDA)), 38.5 (CH(Cy)), 30.5 (2C, CH2(Cy)), 30.0 (CH2(Cy)), 26.5 (CH2(Cy)), 25.9 (2C, CH2(Cy), CH2-B); HR-MS (ESI) m/z: [M + H]+ calcd for C16H25BN3O4 334.1938; found 334.1927.
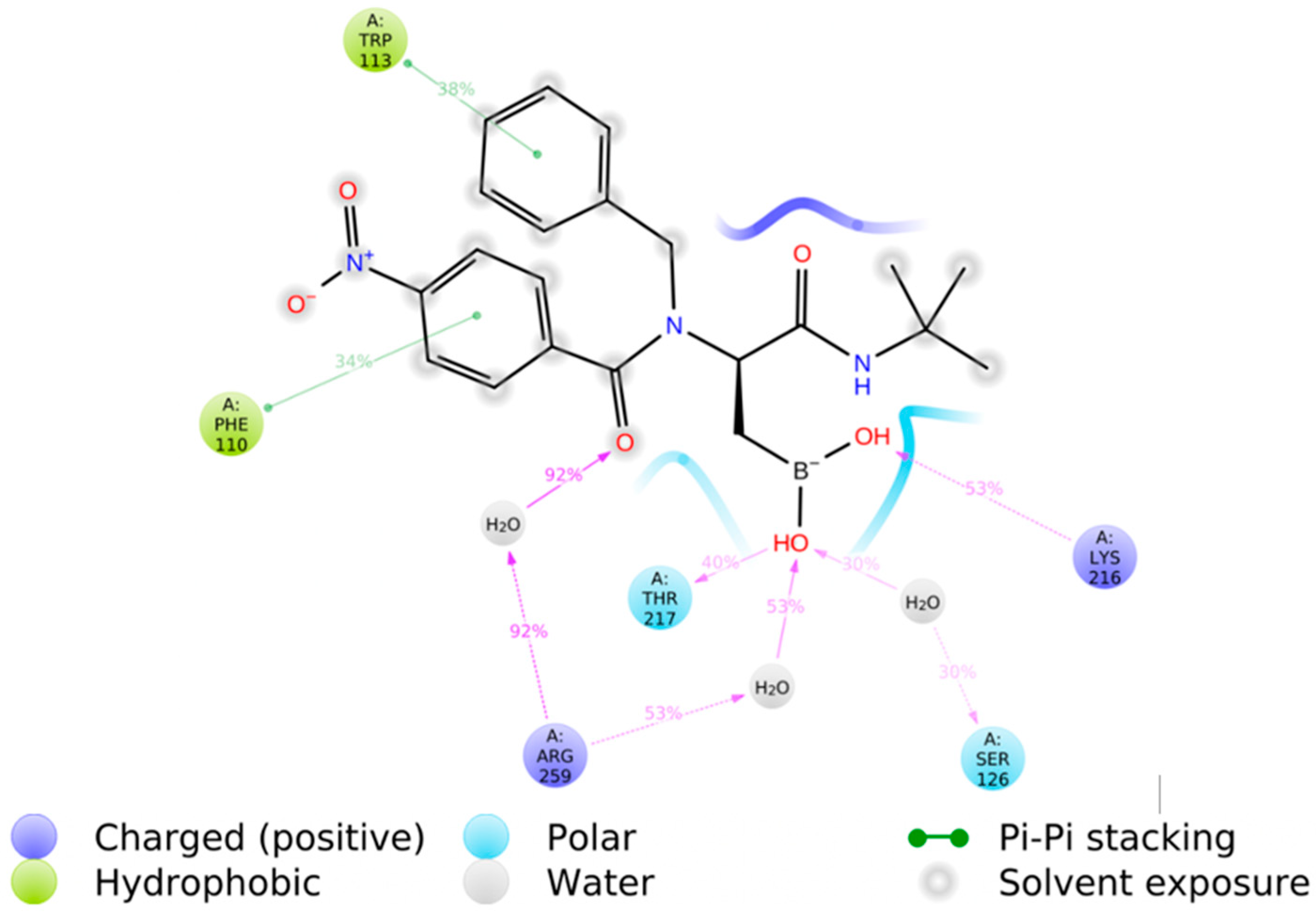
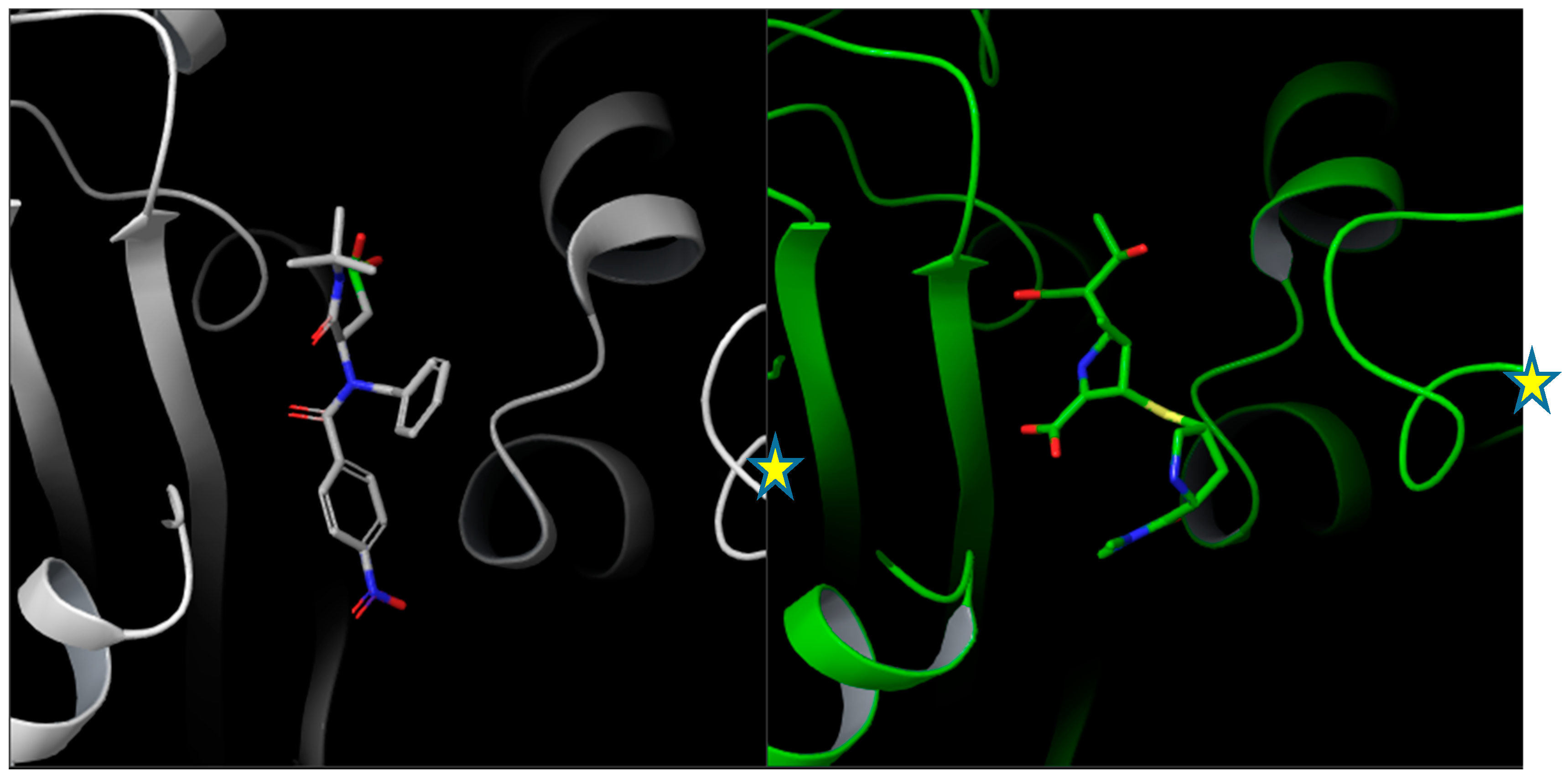
Single crystal X-ray diffraction analysis of compounds
5a and
6d: Single crystals of
5a and
6d were obtained by slow evaporation from the mother liquor. All X-ray data collections were performed at room temperature with a Bruker AXS Smart 3-circle diffractometer equipped with an APEX-II CCD detector. Graphite–monochromated Mo Kα radiation (λ = 0.71073 Å) at a nominal power of 50 kV × 30 mA of the sealed X-ray tube was employed. Highly-redundant ω-scans (Δω = 0.25 deg) at variable φ angles were performed, resulting in 100% complete spheres of data up to 2θ = 46.5 deg (
5a) and 2θ = 52.7 deg (
6d). Diffraction patterns were corrected by absorption and beam anisotropy using SADABS [
31], and then phased by direct methods with Shelx [
32]. Both compounds crystallized in centrosymmetric space groups (
5a: P2
1/c;
6d: P
) as 1:1 racemates. The interested reader can find full details of the diffraction analysis in the
Supporting Information.

 ,
, 











{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}