The Basis for Natural Multiresistance to Phage in Pseudomonas aeruginosa


Abstract
:1. Introduction
2. Results
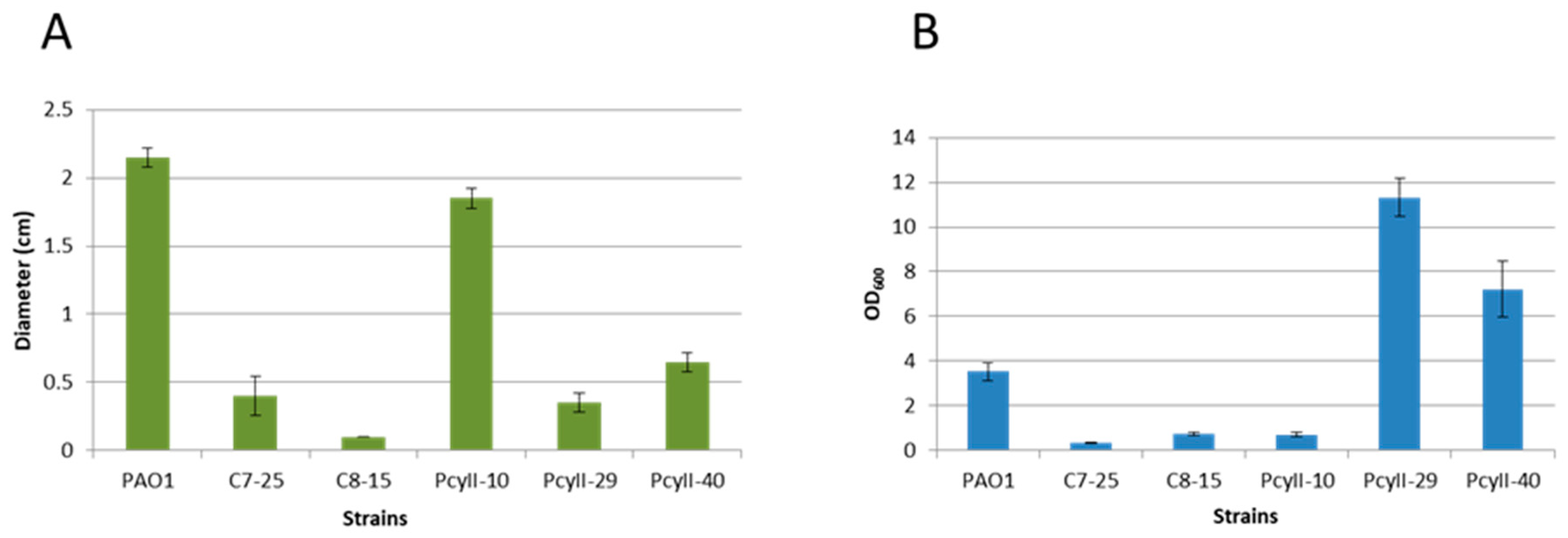
2.1. Characteristics of Multiphage-Resistant Strains
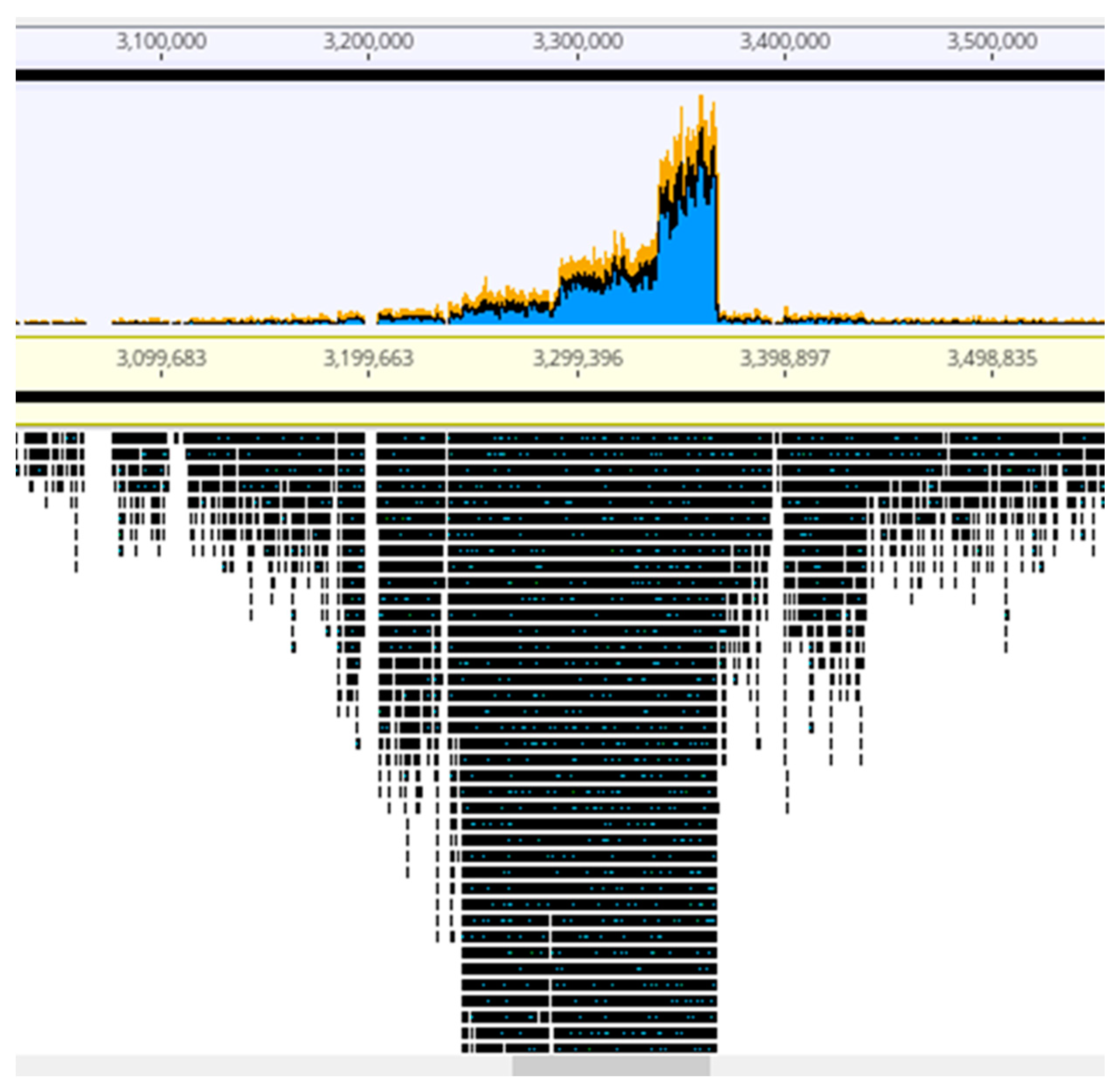
2.2. Genome Sequence Characteristics
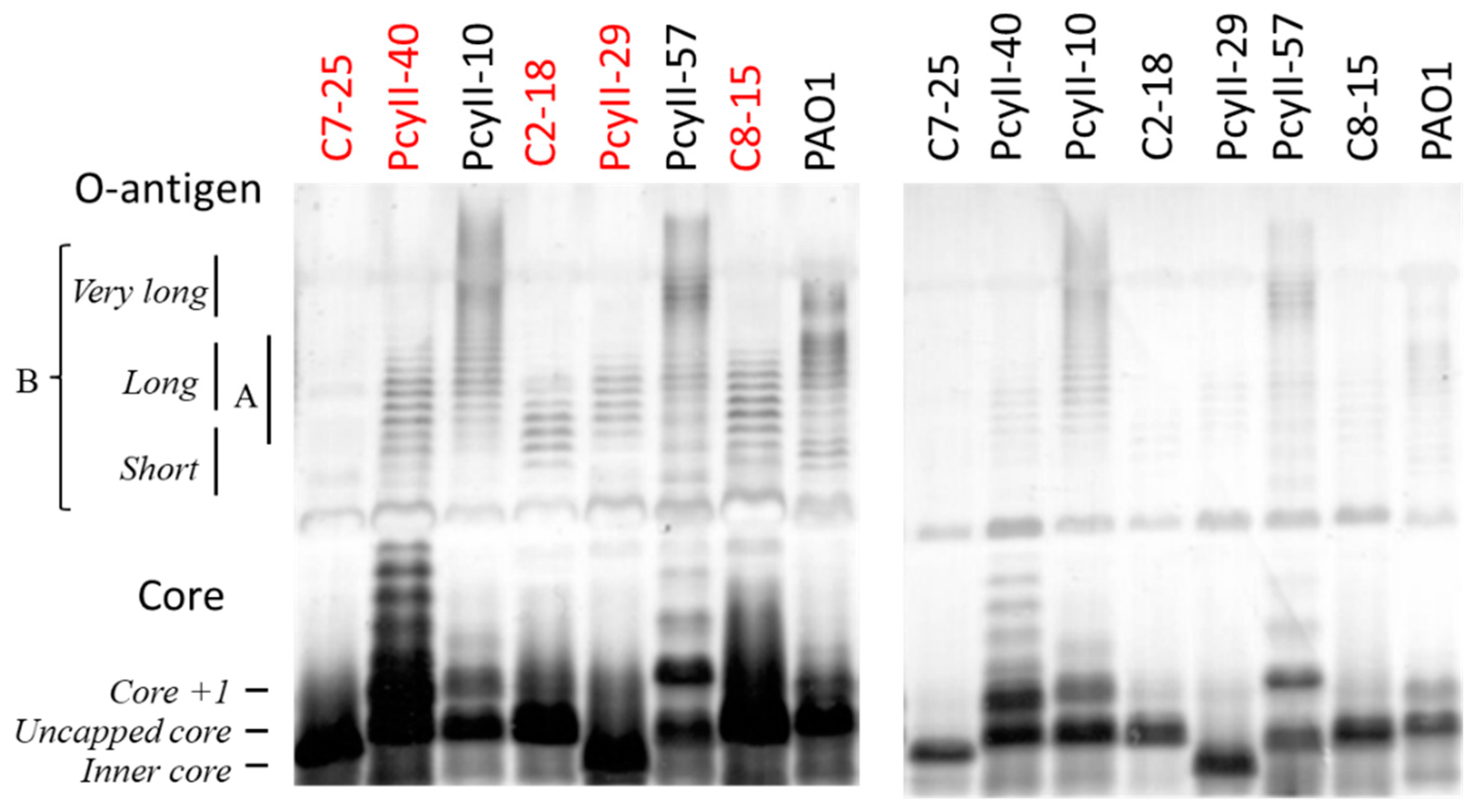
2.3. The LPS and O-antigen Biosynthesis Genes
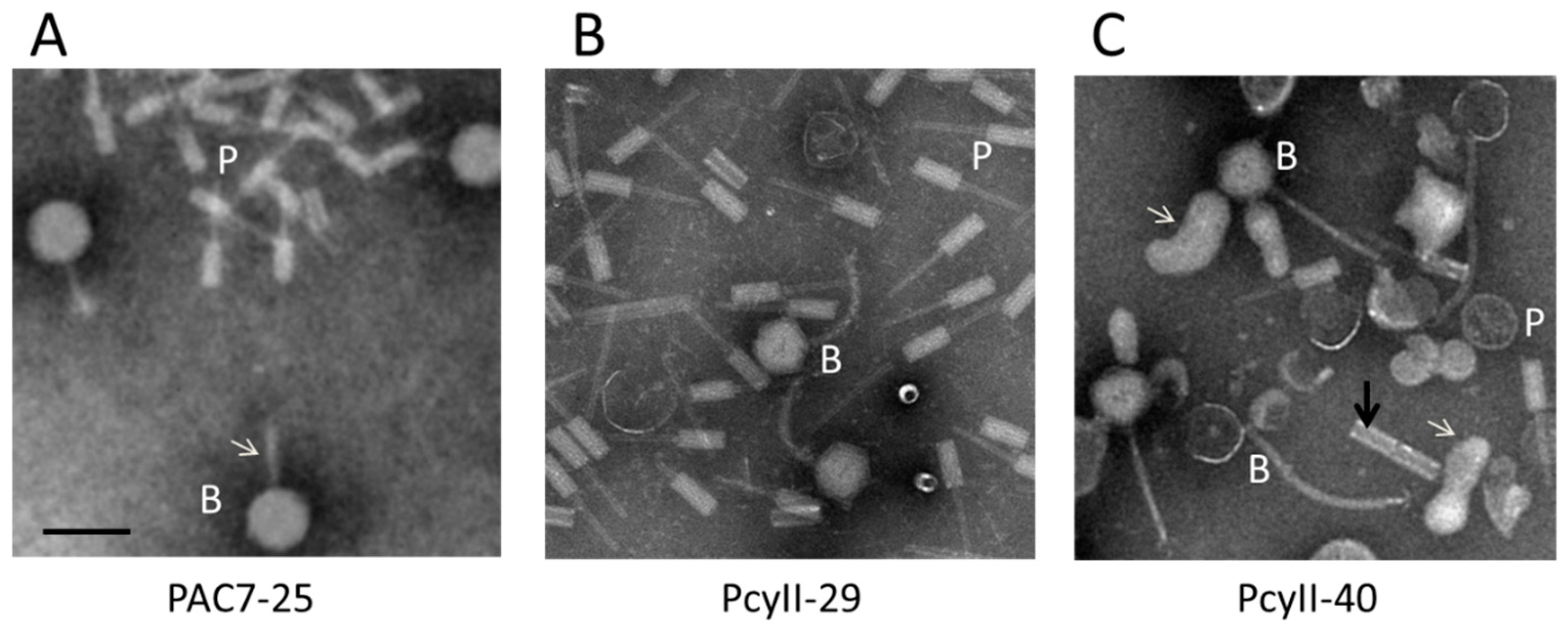
2.4. Pili and Flagella
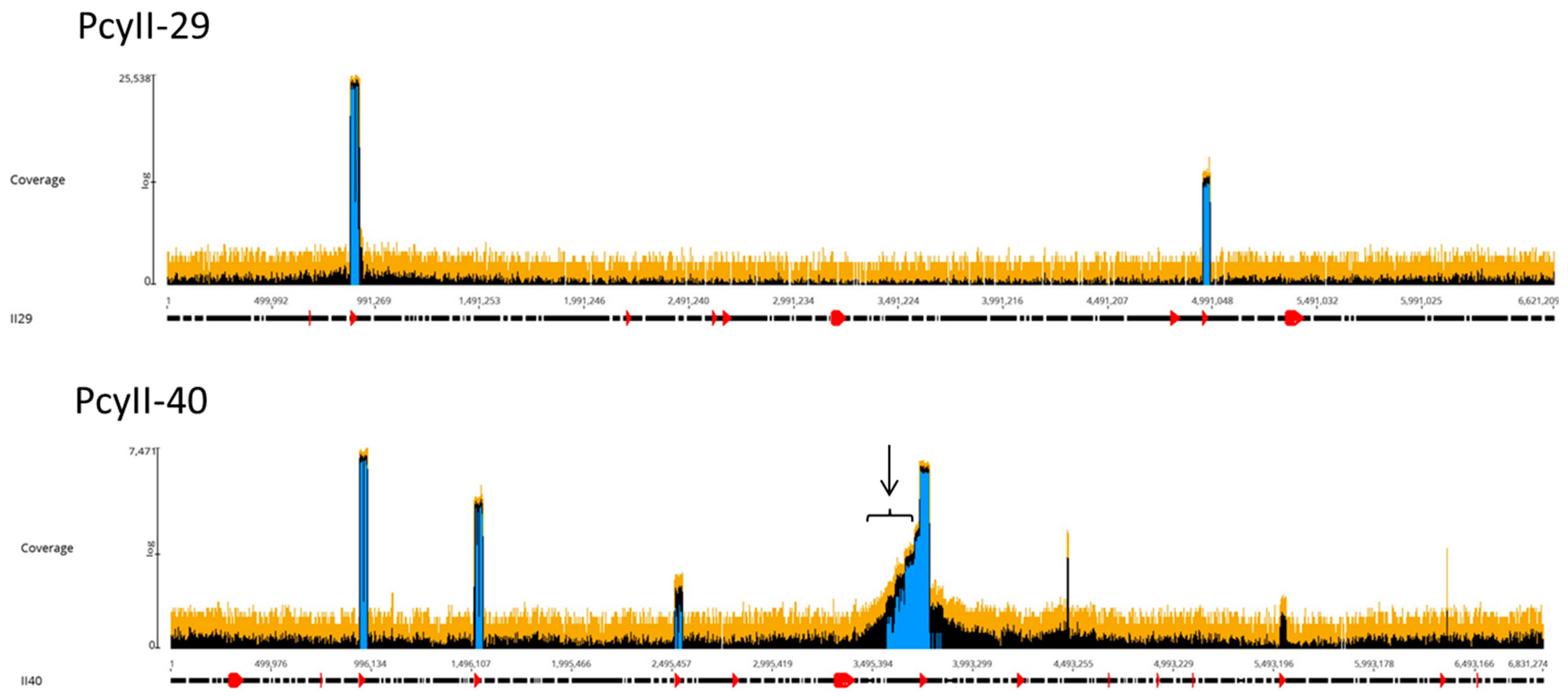
2.5. The Accessory Genome: Regions of Genome Plasticity
2.6. Prophages and Genomic Islands
2.7. CRISPR-Cas Systems
2.8. Transposases and IS
2.9. Activation of Prophages and Phage Tail Pyocins
3. Discussion
4. Materials and Methods
4.1. Ethics Statement
4.2. Strains and Media
4.3. Prophage Induction and Purification
4.4. Phenotypic Assays
4.4.1. Swarming Motility
4.4.2. Twitching Motility
4.4.3. Biofilm Formation
4.5. DNA Extraction and Analysis
4.6. Whole Genome Sequencing and Annotation
4.7. Nucleotide Sequence Accession Number
5. Conclusions
Supplementary Materials
Author Contributions
Funding
Acknowledgments
Conflicts of Interest
References
- Larche, J.; Pouillot, F.; Essoh, C.; Libisch, B.; Straut, M.; Lee, J.C.; Soler, C.; Lamarca, R.; Gleize, E.; Gabard, J.; et al. Rapid identification of international multidrug-resistant Pseudomonas aeruginosa clones by multiple-locus variable number of tandem repeats analysis and investigation of their susceptibility to lytic bacteriophages. Antimicrob. Agents Chemother. 2012, 56, 6175–6180. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Croughs, P.D.; Klaassen, C.H.W.; van Rosmalen, J.; Maghdid, D.M.; Boers, S.A.; Hays, J.P.; Goessens, W.H.F. Unexpected mechanisms of resistance in Dutch Pseudomonas aeruginosa isolates collected during 14 years of surveillance. Int. J. Antimicrob. Agents 2018, 52, 407–410. [Google Scholar] [CrossRef] [PubMed]
- Vu-Thien, H.; Corbineau, G.; Hormigos, K.; Fauroux, B.; Corvol, H.; Clement, A.; Vergnaud, G.; Pourcel, C. Multiple-locus variable-number tandem-repeat analysis for longitudinal survey of sources of Pseudomonas aeruginosa infection in cystic fibrosis patients. J. Clin. Microbiol. 2007, 45, 3175–3183. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Markussen, T.; Marvig, R.L.; Gomez-Lozano, M.; Aanaes, K.; Burleigh, A.E.; Hoiby, N.; Johansen, H.K.; Molin, S.; Jelsbak, L. Environmental heterogeneity drives within-host diversification and evolution of Pseudomonas aeruginosa. mBio 2014, 5, e01592-14. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Battle, S.E.; Rello, J.; Hauser, A.R. Genomic islands of Pseudomonas aeruginosa. Fems Microbiol. Lett. 2009, 290, 70–78. [Google Scholar] [CrossRef] [Green Version]
- Wozniak, R.A.; Waldor, M.K. Integrative and conjugative elements: Mosaic mobile genetic elements enabling dynamic lateral gene flow. Nat. Rev. Microbiol. 2010, 8, 552–863. [Google Scholar] [CrossRef] [PubMed]
- Wolfgang, M.C.; Kulasekara, B.R.; Liang, X.; Boyd, D.; Wu, K.; Yang, Q.; Miyada, C.G.; Lory, S. Conservation of genome content and virulence determinants among clinical and environmental isolates of Pseudomonas aeruginosa. Proc. Natl. Acad. Sci. USA 2003, 100, 8484–8489. [Google Scholar] [CrossRef] [Green Version]
- Kung, V.L.; Ozer, E.A.; Hauser, A.R. The accessory genome of Pseudomonas aeruginosa. Microbiol. Mol. Biol. Rev. Mmbr. 2010, 74, 621–641. [Google Scholar] [CrossRef] [Green Version]
- Pohl, S.; Klockgether, J.; Eckweiler, D.; Khaledi, A.; Schniederjans, M.; Chouvarine, P.; Tummler, B.; Haussler, S. The extensive set of accessory Pseudomonas aeruginosa genomic components. Fems Microbiol. Lett. 2014, 356, 235–241. [Google Scholar] [CrossRef] [Green Version]
- Valot, B.; Guyeux, C.; Rolland, J.Y.; Mazouzi, K.; Bertrand, X.; Hocquet, D. What It Takes to Be a Pseudomonas aeruginosa? The Core Genome of the Opportunistic Pathogen Updated. PLoS ONE 2015, 10, e0126468. [Google Scholar] [CrossRef]
- Mathee, K.; Narasimhan, G.; Valdes, C.; Qiu, X.; Matewish, J.M.; Koehrsen, M.; Rokas, A.; Yandava, C.N.; Engels, R.; Zeng, E.; et al. Dynamics of Pseudomonas aeruginosa genome evolution. Proc. Natl. Acad. Sci. USA 2008, 105, 3100–3105. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Subedi, D.; Kohli, G.S.; Vijay, A.K.; Willcox, M.; Rice, S.A. Accessory genome of the multi-drug resistant ocular isolate of Pseudomonas aeruginosa PA34. PLoS ONE 2019, 14, e0215038. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Botelho, J.; Grosso, F.; Peixe, L. Antibiotic resistance in Pseudomonas aeruginosa—Mechanisms, epidemiology and evolution. Drug Resist. Updates Rev. Comment. Antimicrob. Anticancer Chemother. 2019, 44, 100640. [Google Scholar] [CrossRef] [PubMed]
- Roy, P.H.; Tetu, S.G.; Larouche, A.; Elbourne, L.; Tremblay, S.; Ren, Q.; Dodson, R.; Harkins, D.; Shay, R.; Watkins, K.; et al. Complete genome sequence of the multiresistant taxonomic outlier Pseudomonas aeruginosa PA7. PLoS ONE 2010, 5, e8842. [Google Scholar] [CrossRef] [PubMed]
- Klockgether, J.; Cramer, N.; Wiehlmann, L.; Davenport, C.F.; Tummler, B. Pseudomonas aeruginosa Genomic Structure and Diversity. Front. Microbiol. 2011, 2, 150. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Boukerb, A.M.; Decor, A.; Ribun, S.; Tabaroni, R.; Rousset, A.; Commin, L.; Buff, S.; Doleans-Jordheim, A.; Vidal, S.; Varrot, A.; et al. Genomic Rearrangements and Functional Diversification of lecA and lecB Lectin-Coding Regions Impacting the Efficacy of Glycomimetics Directed against Pseudomonas aeruginosa. Front. Microbiol. 2016, 7, 811. [Google Scholar] [CrossRef]
- Williams, K.P. Integration sites for genetic elements in prokaryotic tRNA and tmRNA genes: Sublocation preference of integrase subfamilies. Nucleic Acids Res. 2002, 30, 866–875. [Google Scholar] [CrossRef] [Green Version]
- Winstanley, C.; Langille, M.G.; Fothergill, J.L.; Kukavica-Ibrulj, I.; Paradis-Bleau, C.; Sanschagrin, F.; Thomson, N.R.; Winsor, G.L.; Quail, M.A.; Lennard, N.; et al. Newly introduced genomic prophage islands are critical determinants of in vivo competitiveness in the Liverpool Epidemic Strain of Pseudomonas aeruginosa. Genome Res. 2009, 19, 12–23. [Google Scholar] [CrossRef] [Green Version]
- Ramirez-Diaz, M.I.; Diaz-Magana, A.; Meza-Carmen, V.; Johnstone, L.; Cervantes, C.; Rensing, C. Nucleotide sequence of Pseudomonas aeruginosa conjugative plasmid pUM505 containing virulence and heavy-metal resistance genes. Plasmid 2011, 66, 7–18. [Google Scholar] [CrossRef]
- Wagner, P.L.; Waldor, M.K. Bacteriophage control of bacterial virulence. Infect. Immun. 2002, 70, 3985–3993. [Google Scholar] [CrossRef] [Green Version]
- Sanchez, D.; Gomila, M.; Bennasar, A.; Lalucat, J.; Garcia-Valdes, E. Genome analysis of environmental and clinical P. aeruginosa isolates from sequence type-1146. PLoS ONE 2014, 9, e107754. [Google Scholar] [CrossRef] [PubMed]
- Tsao, Y.F.; Taylor, V.L.; Kala, S.; Bondy-Denomy, J.; Khan, A.N.; Bona, D.; Cattoir, V.; Lory, S.; Davidson, A.R.; Maxwell, K.L. Phage Morons Play an Important Role in Pseudomonas aeruginosa Phenotypes. J. Bacteriol. 2018, 200, e00189-18. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Bondy-Denomy, J.; Qian, J.; Westra, E.R.; Buckling, A.; Guttman, D.S.; Davidson, A.R.; Maxwell, K.L. Prophages mediate defense against phage infection through diverse mechanisms. ISME J. 2016, 10, 2854–2866. [Google Scholar] [CrossRef] [PubMed]
- De Smet, J.; Hendrix, H.; Blasdel, B.G.; Danis-Wlodarczyk, K.; Lavigne, R. Pseudomonas predators: Understanding and exploiting phage-host interactions. Nat. Rev. Microbiol. 2017, 15, 517. [Google Scholar] [CrossRef] [PubMed]
- Latino, L.; Essoh, C.; Blouin, Y.; Vu Thien, H.; Pourcel, C. A novel Pseudomonas aeruginosa Bacteriophage, Ab31, a Chimera Formed from Temperate Phage PAJU2 and P. putida Lytic Phage AF: Characteristics and Mechanism of Bacterial Resistance. PLoS ONE 2014, 9, e93777. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Le, S.; Yao, X.; Lu, S.; Tan, Y.; Rao, X.; Li, M.; Jin, X.; Wang, J.; Zhao, Y.; Wu, N.C.; et al. Chromosomal DNA deletion confers phage resistance to Pseudomonas aeruginosa. Sci. Rep. 2014, 4, 4738. [Google Scholar] [CrossRef] [Green Version]
- Latino, L.; Midoux, C.; Vergnaud, G.; Pourcel, C. Investigation of Pseudomonas aeruginosa strain PcyII-10 variants resisting infection by N4-like phage Ab09 in search for genes involved in phage adsorption. PLoS ONE 2019, 14, e0215456. [Google Scholar] [CrossRef] [Green Version]
- Samson, J.E.; Magadan, A.H.; Sabri, M.; Moineau, S. Revenge of the phages: Defeating bacterial defences. Nat. Rev. Microbiol. 2013, 11, 675–687. [Google Scholar] [CrossRef]
- Koonin, E.V.; Makarova, K.S.; Zhang, F. Diversity, classification and evolution of CRISPR-Cas systems. Curr. Opin. Microbiol. 2017, 37, 67–78. [Google Scholar] [CrossRef]
- Doron, S.; Melamed, S.; Ofir, G.; Leavitt, A.; Lopatina, A.; Keren, M.; Amitai, G.; Sorek, R. Systematic discovery of antiphage defense systems in the microbial pangenome. Science 2018, 359, eaar4120. [Google Scholar] [CrossRef] [Green Version]
- Labrie, S.J.; Samson, J.E.; Moineau, S. Bacteriophage resistance mechanisms. Nat. Rev. Microbiol. 2010, 8, 317–327. [Google Scholar] [CrossRef] [PubMed]
- Hille, F.; Charpentier, E. CRISPR-Cas: Biology, mechanisms and relevance. Philos. Trans. R. Soc. of Lond. Series B Biol. Sci. 2016, 371, 20150496. [Google Scholar] [CrossRef] [PubMed]
- Cady, K.C.; White, A.S.; Hammond, J.H.; Abendroth, M.D.; Karthikeyan, R.S.; Lalitha, P.; Zegans, M.E.; O’Toole, G.A. Prevalence, conservation and functional analysis of Yersinia and Escherichia CRISPR regions in clinical Pseudomonas aeruginosa isolates. Microbiology 2011, 157, 430–437. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Essoh, C.; Blouin, Y.; Loukou, G.; Cablanmian, A.; Lathro, S.; Kutter, E.; Thien, H.V.; Vergnaud, G.; Pourcel, C. The Susceptibility of Pseudomonas aeruginosa Strains from Cystic Fibrosis Patients to Bacteriophages. PLoS ONE 2013, 8, e60575. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Van Belkum, A.; Soriaga, L.B.; LaFave, M.C.; Akella, S.; Veyrieras, J.B.; Barbu, E.M.; Shortridge, D.; Blanc, B.; Hannum, G.; Zambardi, G.; et al. Phylogenetic Distribution of CRISPR-Cas Systems in Antibiotic-Resistant Pseudomonas aeruginosa. mBio 2015, 6, e01796-15. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Cullen, L.; Weiser, R.; Olszak, T.; Maldonado, R.F.; Moreira, A.S.; Slachmuylders, L.; Brackman, G.; Paunova-Krasteva, T.S.; Zarnowiec, P.; Czerwonka, G.; et al. Phenotypic characterization of an international Pseudomonas aeruginosa reference panel: Strains of cystic fibrosis (CF) origin show less in vivo virulence than non-CF strains. Microbiology 2015, 161, 1961–1977. [Google Scholar] [CrossRef]
- Holloway, B.W. Genetic recombination in Pseudomonas aeruginosa. J. Gen. Microbiol. 1955, 13, 572–581. [Google Scholar] [CrossRef] [Green Version]
- Latino, L.; Midoux, C.; Hauck, Y.; Vergnaud, G.; Pourcel, C. Pseudolysogeny and sequential mutations build multiresistance to virulent bacteriophages in Pseudomonas aeruginosa. Microbiology 2016, 162, 748–763. [Google Scholar] [CrossRef]
- Latino, L.; Caroff, M.; Pourcel, C. Fine structure analysis of lipopolysaccharides in bacteriophage-resistant Pseudomonas aeruginosa PAO1 mutants. Microbiology 2017, 163, 848–855. [Google Scholar] [CrossRef]
- Ormala, A.M.; Jalasvuori, M. Phage therapy: Should bacterial resistance to phages be a concern, even in the long run? Bacteriophage 2013, 3, e24219. [Google Scholar] [CrossRef] [Green Version]
- Leon, M.; Bastias, R. Virulence reduction in bacteriophage resistant bacteria. Front. Microbiol. 2015, 6, 343. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Hosseinidoust, Z.; van de Ven, T.G.; Tufenkji, N. Evolution of Pseudomonas aeruginosa virulence as a result of phage predation. Appl. Environ. Microbiol. 2013, 79, 6110–6116. [Google Scholar] [CrossRef] [Green Version]
- Hosseinidoust, Z.; Tufenkji, N.; van de Ven, T.G. Predation in homogeneous and heterogeneous phage environments affects virulence determinants of Pseudomonas aeruginosa. Appl. Environ. Microbiol. 2013, 79, 2862–2871. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Kay, M.K.; Erwin, T.C.; McLean, R.J.; Aron, G.M. Bacteriophage ecology in Escherichia coli and Pseudomonas aeruginosa mixed-biofilm communities. Appl. Environ. Microbiol. 2011, 77, 821–829. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Meaden, S.; Koskella, B. Exploring the risks of phage application in the environment. Front. Microbiol. 2013, 4, 358. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Oechslin, F. Resistance Development to Bacteriophages Occurring during Bacteriophage Therapy. Viruses 2018, 10, 351. [Google Scholar] [CrossRef] [Green Version]
- Wright, R.C.T.; Friman, V.P.; Smith, M.C.M.; Brockhurst, M.A. Resistance Evolution against Phage Combinations Depends on the Timing and Order of Exposure. mBio 2019, 10, e01652-19. [Google Scholar] [CrossRef] [Green Version]
- Pourcel, C.; Midoux, C.; Vergnaud, G.; Latino, L. A carrier state is established in Pseudomonas aeruginosa by phage LeviOr01, a newly isolated ssRNA levivirus. J. Gen. Virol. 2017, 98, 2181–2189. [Google Scholar] [CrossRef]
- Krylov, V.; Pleteneva, E.; Bourkaltseva, M.; Shaburova, O.; Volckaert, G.; Sykilinda, N.; Kurochkina, L.; Mesyanzhinov, V. Myoviridae bacteriophages of Pseudomonas aeruginosa: A long and complex evolutionary pathway. Res. Microbiol. 2003, 154, 269–275. [Google Scholar] [CrossRef]
- Islam, S.T.; Huszczynski, S.M.; Nugent, T.; Gold, A.C.; Lam, J.S. Conserved-residue mutations in Wzy affect O-antigen polymerization and Wzz-mediated chain-length regulation in Pseudomonas aeruginosa PAO1. Sci. Rep. 2013, 3, 3441. [Google Scholar] [CrossRef] [Green Version]
- Klausen, M.; Heydorn, A.; Ragas, P.; Lambertsen, L.; Aaes-Jorgensen, A.; Molin, S.; Tolker-Nielsen, T. Biofilm formation by Pseudomonas aeruginosa wild type, flagella and type IV pili mutants. Mol. Microbiol. 2003, 48, 1511–1524. [Google Scholar] [CrossRef] [PubMed]
- O’Toole, G.A.; Kolter, R. Flagellar and twitching motility are necessary for Pseudomonas aeruginosa biofilm development. Mol. Microbiol. 1998, 30, 295–304. [Google Scholar] [CrossRef] [PubMed]
- Kohler, T.; Curty, L.K.; Barja, F.; van Delden, C.; Pechere, J.C. Swarming of Pseudomonas aeruginosa is dependent on cell-to-cell signaling and requires flagella and pili. J. Bacteriol. 2000, 182, 5990–5996. [Google Scholar] [CrossRef] [Green Version]
- Darling, A.C.; Mau, B.; Blattner, F.R.; Perna, N.T. Mauve: Multiple alignment of conserved genomic sequence with rearrangements. Genome Res. 2004, 14, 1394–1403. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Llanes, C.; Pourcel, C.; Richardot, C.; Plesiat, P.; Fichant, G.; Cavallo, J.D.; Merens, A. Diversity of beta-lactam resistance mechanisms in cystic fibrosis isolates of Pseudomonas aeruginosa: A French multicentre study. J. Antimicrob. Chemother. 2013, 68, 1763–1771. [Google Scholar] [CrossRef] [Green Version]
- King, J.D.; Kocincova, D.; Westman, E.L.; Lam, J.S. Review: Lipopolysaccharide biosynthesis in Pseudomonas aeruginosa. Innate Immun. 2009, 15, 261–312. [Google Scholar] [CrossRef]
- Raymond, C.K.; Sims, E.H.; Kas, A.; Spencer, D.H.; Kutyavin, T.V.; Ivey, R.G.; Zhou, Y.; Kaul, R.; Clendenning, J.B.; Olson, M.V. Genetic variation at the O-antigen biosynthetic locus in Pseudomonas aeruginosa. J. Bacteriol. 2002, 184, 3614–3622. [Google Scholar] [CrossRef] [Green Version]
- Dean, C.R.; Franklund, C.V.; Retief, J.D.; Coyne, M.J.; Jr Hatano, K.; Evans, D.J.; Pier, G.B.; Goldberg, J.B. Characterization of the serogroup O11 O-antigen locus of Pseudomonas aeruginosa PA103. J. Bacteriol. 1999, 181, 4275–4284. [Google Scholar] [CrossRef] [Green Version]
- Kintz, E.; Scarff, J.M.; DiGiandomenico, A.; Goldberg, J.B. Lipopolysaccharide O-antigen chain length regulation in Pseudomonas aeruginosa serogroup O11 strain PA103. J. Bacteriol. 2008, 190, 2709–2716. [Google Scholar] [CrossRef] [Green Version]
- Hao, Y.; Murphy, K.; Lo, R.Y.; Khursigara, C.M.; Lam, J.S. Single-Nucleotide Polymorphisms Found in the migA and wbpX Glycosyltransferase Genes Account for the Intrinsic Lipopolysaccharide Defects Exhibited by Pseudomonas aeruginosa PA14. J. Bacteriol. 2015, 197, 2780–2791. [Google Scholar] [CrossRef] [Green Version]
- Liebens, V.; Defraine, V.; Van der Leyden, A.; De Groote, V.N.; Fierro, C.; Beullens, S.; Verstraeten, N.; Kint, C.; Jans, A.; Frangipani, E.; et al. A putative de-N-acetylase of the PIG-L superfamily affects fluoroquinolone tolerance in Pseudomonas aeruginosa. Pathog. Dis. 2014, 71, 39–54. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Mattick, J.S. Type IV pili and twitching motility. Annu. Rev. Microbiol. 2002, 56, 289–314. [Google Scholar] [CrossRef] [PubMed]
- Burrows, L.L. Pseudomonas aeruginosa twitching motility: Type IV pili in action. Annu. Rev. Microbiol. 2012, 66, 493–520. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Leighton, T.L.; Buensuceso, R.N.; Howell, P.L.; Burrows, L.L. Biogenesis of Pseudomonas aeruginosa type IV pili and regulation of their function. Environ. Microbiol. 2015, 17, 4148–4163. [Google Scholar] [CrossRef]
- Tammam, S.; Sampaleanu, L.M.; Koo, J.; Manoharan, K.; Daubaras, M.; Burrows, L.L.; Howell, P.L. PilMNOPQ from the Pseudomonas aeruginosa type IV pilus system form a transenvelope protein interaction network that interacts with PilA. J. Bacteriol. 2013, 195, 2126–2135. [Google Scholar] [CrossRef] [Green Version]
- Smyth, C.J.; Marron, M.B.; Twohig, J.M.; Smith, S.G. Fimbrial adhesins: Similarities and variations in structure and biogenesis. FEMS Immunol. Med. Microbiol. 1996, 16, 127–139. [Google Scholar] [CrossRef]
- Leech, A.J.; Mattick, J.S. Effect of site-specific mutations in different phosphotransfer domains of the chemosensory protein ChpA on Pseudomonas aeruginosa motility. J. Bacteriol. 2006, 188, 8479–8486. [Google Scholar] [CrossRef] [Green Version]
- Nakayama, K.; Takashima, K.; Ishihara, H.; Shinomiya, T.; Kageyama, M.; Kanaya, S.; Ohnishi, M.; Murata, T.; Mori, H.; Hayashi, T. The R-type pyocin of Pseudomonas aeruginosa is related to P2 phage, and the F-type is related to lambda phage. Mol. Microbiol. 2000, 38, 213–231. [Google Scholar] [CrossRef] [PubMed]
- Michel-Briand, Y.; Baysse, C. The pyocins of Pseudomonas aeruginosa. Biochimie 2002, 84, 499–510. [Google Scholar] [CrossRef]
- Essar, D.W.; Eberly, L.; Han, C.Y.; Crawford, I.P. DNA sequences and characterization of four early genes of the tryptophan pathway in Pseudomonas aeruginosa. J. Bacteriol. 1990, 172, 853–866. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Ceyssens, P.J.; Lavigne, R. Bacteriophages of Pseudomonas. Future Microbiol. 2010, 5, 1041–1055. [Google Scholar] [CrossRef] [PubMed]
- Kwan, T.; Liu, J.; Dubow, M.; Gros, P.; Pelletier, J. Comparative genomic analysis of 18 Pseudomonas aeruginosa bacteriophages. J. Bacteriol. 2006, 188, 1184–1187. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Li, G.; Shen, M.; Lu, S.; Le, S.; Tan, Y.; Wang, J.; Zhao, X.; Shen, W.; Guo, K.; Yang, Y.; et al. Identification and Characterization of the HicAB Toxin-Antitoxin System in the Opportunistic Pathogen Pseudomonas aeruginosa. Toxins 2016, 8, 113. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Pourcel, C.; Midoux, C.; Hauck, Y.; Vergnaud, G.; Latino, L. Large Preferred Region for Packaging of Bacterial DNA by phiC725A, a Novel Pseudomonas aeruginosa F116-Like Bacteriophage. PLoS ONE 2017, 12, e0169684. [Google Scholar] [CrossRef] [PubMed]
- Nowak-Thompson, B.; Chaney, N.; Wing, J.S.; Gould, S.J.; Loper, J.E. Characterization of the pyoluteorin biosynthetic gene cluster of Pseudomonas fluorescens Pf-5. J. Bacteriol. 1999, 181, 2166–2174. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Huang, X.; Yan, A.; Zhang, X.; Xu, Y. Identification and characterization of a putative ABC transporter PltHIJKN required for pyoluteorin production in Pseudomonas sp. M18. Gene 2006, 376, 68–78. [Google Scholar] [CrossRef] [PubMed]
- Klockgether, J.; Reva, O.; Larbig, K.; Tummler, B. Sequence analysis of the mobile genome island pKLC102 of Pseudomonas aeruginosa C. J. Bacteriol. 2004, 186, 518–534. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Wurdemann, D.; Tummler, B. In silico comparison of pKLC102-like genomic islands of Pseudomonas aeruginosa. FEMS Microbiol. Lett. 2007, 275, 244–249. [Google Scholar] [CrossRef] [Green Version]
- Kiewitz, C.; Larbig, K.; Klockgether, J.; Weinel, C.; Tummler, B. Monitoring genome evolution ex vivo: Reversible chromosomal integration of a 106 kb plasmid at two tRNA(Lys) gene loci in sequential Pseudomonas aeruginosa airway isolates. Microbiology 2000, 146 Pt 10, 2365–2373. [Google Scholar] [CrossRef] [Green Version]
- Vergnaud, G.; Midoux, C.; Blouin, Y.; Bourkaltseva, M.; Krylov, V.; Pourcel, C. Transposition Behavior Revealed by High-Resolution Description of Pseudomonas aeruginosa Saltovirus Integration Sites. Viruses 2018, 10, 245. [Google Scholar] [CrossRef] [Green Version]
- Smith, K.; Martin, L.; Rinaldi, A.; Rajendran, R.; Ramage, G.; Walker, D. Activity of pyocin S2 against Pseudomonas aeruginosa biofilms. Antimicrob. Agents Chemother. 2012, 56, 1599–1601. [Google Scholar] [CrossRef] [Green Version]
- Couvin, D.; Bernheim, A.; Toffano-Nioche, C.; Touchon, M.; Michalik, J.; Neron, B.; Rocha, E.P.; Vergnaud, G.; Gautheret, D.; Pourcel, C. CRISPRCasFinder, an update of CRISRFinder, includes a portable version, enhanced performance and integrates search for Cas proteins. Nucleic Acids Res. 2018, 46, W246–W251. [Google Scholar] [CrossRef] [Green Version]
- Gertman, E.; White, B.N.; Berry, D.; Kropinski, A.M. IS222, a new insertion element associated with the genome of Pseudomonas aeruginosa. J. Bacteriol. 1986, 166, 1134–1136. [Google Scholar] [CrossRef] [Green Version]
- Kropinski, A.M.; Farinha, M.A.; Jansons, I. Nucleotide sequence of the Pseudomonas aeruginosa insertion sequence IS222: Another member of the IS3 family. Plasmid 1994, 31, 222–228. [Google Scholar] [CrossRef] [PubMed]
- Turnbull, L.; Toyofuku, M.; Hynen, A.L.; Kurosawa, M.; Pessi, G.; Petty, N.K.; Osvath, S.R.; Carcamo-Oyarce, G.; Gloag, E.S.; Shimoni, R.; et al. Explosive cell lysis as a mechanism for the biogenesis of bacterial membrane vesicles and biofilms. Nat. Commun. 2016, 7, 11220. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Chen, J.; Ram, G.; Penades, J.R.; Brown, S.; Novick, R.P. Pathogenicity island-directed transfer of unlinked chromosomal virulence genes. Mol. Cell. 2015, 57, 138–149. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Freschi, L.; Bertelli, C.; Jeukens, J.; Moore, M.P.; Kukavica-Ibrulj, I.; Emond-Rheault, J.G.; Hamel, J.; Fothergill, J.L.; Tucker, N.P.; McClean, S.; et al. Genomic characterisation of an international Pseudomonas aeruginosa reference panel indicates that the two major groups draw upon distinct mobile gene pools. FEMS Microbiol. Lett. 2018, 365, fny120. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Johansson, E.; Welinder-Olsson, C.; Gilljam, M.; Pourcel, C.; Lindblad, A. Genotyping of Pseudomonas aeruginosa reveals high diversity, stability over time and good outcome of eradication. J. Cyst. Fibros. 2015, 14, 353–360. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Loenen, W.A.; Dryden, D.T.; Raleigh, E.A.; Wilson, G.G. Type I restriction enzymes and their relatives. Nucleic Acids Res. 2014, 42, 20–44. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Tock, M.R.; Dryden, D.T. The biology of restriction and anti-restriction. Curr. Opin. Microbiol. 2005, 8, 466–472. [Google Scholar] [CrossRef] [PubMed]
- Pleska, M.; Lang, M.; Refardt, D.; Levin, B.R.; Guet, C.C. Phage-host population dynamics promotes prophage acquisition in bacteria with innate immunity. Nat. Ecol. Evol. 2018, 2, 359–366. [Google Scholar] [CrossRef] [PubMed]
- Chen, J.; Quiles-Puchalt, N.; Chiang, Y.N.; Bacigalupe, R.; Fillol-Salom, A.; Chee, M.S.J.; Fitzgerald, J.R.; Penades, J.R. Genome hypermobility by lateral transduction. Science 2018, 362, 207–212. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Chiang, Y.N.; Penades, J.R.; Chen, J. Genetic transduction by phages and chromosomal islands: The new and noncanonical. PLoS Pathog. 2019, 15, e1007878. [Google Scholar] [CrossRef] [Green Version]
- Heussler, G.E.; O’Toole, G.A. Friendly Fire: Biological Functions and Consequences of Chromosomal Targeting by CRISPR-Cas Systems. J. Bacteriol. 2016, 198, 1481–1486. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Miller, R.V.; Rubero, V.J. Mucoid conversion by phages of Pseudomonas aeruginosa strains from patients with cystic fibrosis. J. Clin. Microbiol. 1984, 19, 717–719. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Holloway, B.W. Genetics of Pseudomonas. Bacteriol. Rev. 1969, 33, 419–443. [Google Scholar] [CrossRef] [Green Version]
- Dedrick, R.M.; Jacobs-Sera, D.; Bustamante, C.A.; Garlena, R.A.; Mavrich, T.N.; Pope, W.H.; Reyes, J.C.; Russell, D.A.; Adair, T.; Alvey, R.; et al. Prophage-mediated defence against viral attack and viral counter-defence. Nat. Microbiol. 2017, 2, 16251. [Google Scholar] [CrossRef]
- Chung, I.Y.; Jang, H.J.; Bae, H.W.; Cho, Y.H. A phage protein that inhibits the bacterial ATPase required for type IV pilus assembly. Proc. Natl. Acad. Sci. USA 2014, 111, 11503–11508. [Google Scholar] [CrossRef] [Green Version]
- Davies, E.V.; James, C.E.; Williams, D.; O’Brien, S.; Fothergill, J.L.; Haldenby, S.; Paterson, S.; Winstanley, C.; Brockhurst, M.A. Temperate phages both mediate and drive adaptive evolution in pathogen biofilms. Proc. Natl. Acad. Sci. USA 2016, 113, 8266–8271. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Davies, E.V.; James, C.E.; Kukavica-Ibrulj, I.; Levesque, R.C.; Brockhurst, M.A.; Winstanley, C. Temperate phages enhance pathogen fitness in chronic lung infection. ISME J. 2016, 10, 2553–2555. [Google Scholar] [CrossRef] [PubMed]
- Burns, N.; James, C.E.; Harrison, E. Polylysogeny magnifies competitiveness of a bacterial pathogen in vivo. Evol. Appl. 2015, 8, 346–351. [Google Scholar] [CrossRef] [PubMed]
- Craig, L.; Pique, M.E.; Tainer, J.A. Type IV pilus structure and bacterial pathogenicity. Nat. Rev. Microbiol. 2004, 2, 363–378. [Google Scholar] [CrossRef] [PubMed]
- Tang, H.; Kays, M.; Prince, A. Role of Pseudomonas aeruginosa pili in acute pulmonary infection. Infect. Immun. 1995, 63, 1278–1285. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Heo, Y.J.; Chung, I.Y.; Choi, K.B.; Cho, Y.H. R-type pyocin is required for competitive growth advantage between Pseudomonas aeruginosa strains. J. Microbiol. Biotechnol. 2007, 17, 180–185. [Google Scholar] [PubMed]
- James, C.E.; Davies, E.V.; Fothergill, J.L.; Walshaw, M.J.; Beale, C.M.; Brockhurst, M.A.; Winstanley, C. Lytic activity by temperate phages of Pseudomonas aeruginosa in long-term cystic fibrosis chronic lung infections. ISME J. 2015, 9, 1391–1398. [Google Scholar] [CrossRef] [Green Version]
- Essoh, C.; Latino, L.; Midoux, C.; Blouin, Y.; Loukou, G.; Nguetta, S.P.; Lathro, S.; Cablanmian, A.; Kouassi, A.K.; Vergnaud, G.; et al. Investigation of a Large Collection of Pseudomonas aeruginosa Bacteriophages Collected from a Single Environmental Source in Abidjan, Cote d’Ivoire. PLoS ONE 2015, 10, e0130548. [Google Scholar] [CrossRef] [Green Version]
- Krylov, V.N.; Smirnova, T.A.; Minenkova, I.B.; Plotnikova, T.G.; Zhazikov, I.Z.; Khrenova, E.A. Pseudomonas bacteriophage phi KZ contains an inner body in its capsid. Can. J. Microbiol. 1984, 30, 758–762. [Google Scholar] [CrossRef]
- Hitchcock, P.J.; Brown, T.M. Morphological heterogeneity among Salmonella lipopolysaccharide chemotypes in silver-stained polyacrylamide gels. J. Bacteriol. 1983, 154, 269–277. [Google Scholar] [CrossRef] [Green Version]
- Fomsgaard, A.; Freudenberg, M.A.; Galanos, C. Modification of the silver staining technique to detect lipopolysaccharide in polyacrylamide gels. J. Clin. Microbiol. 1990, 28, 2627–2631. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Chikhi, R.; Medvedev, P. Informed and automated k-mer size selection for genome assembly. Bioinformatics 2014, 30, 31–37. [Google Scholar] [CrossRef] [PubMed]
- Boetzer, M.; Pirovano, W. SSPACE-LongRead: Scaffolding bacterial draft genomes using long read sequence information. BMC Bioinform. 2014, 15, 211. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Chaisson, M.J.; Tesler, G. Mapping single molecule sequencing reads using basic local alignment with successive refinement (BLASR): Application and theory. BMC Bioinform. 2012, 13, 238. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Boetzer, M.; Pirovano, W. Toward almost closed genomes with GapFiller. Genome Biol. 2012, 13, R56. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Vallenet, D.; Engelen, S.; Mornico, D.; Cruveiller, S.; Fleury, L.; Lajus, A.; Rouy, Z.; Roche, D.; Salvignol, G.; Scarpelli, C.; et al. MicroScope: A platform for microbial genome annotation and comparative genomics. Database J. Biol. Databases Curation 2009, 2009, bap021. [Google Scholar] [CrossRef] [PubMed]
- Aziz, R.K.; Bartels, D.; Best, A.A.; DeJongh, M.; Disz, T.; Edwards, R.A.; Formsma, K.; Gerdes, S.; Glass, E.M.; Kubal, M.; et al. The RAST Server: Rapid annotations using subsystems technology. BMC Genom. 2008, 9, 75. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Van Domselaar, G.H.; Stothard, P.; Shrivastava, S.; Cruz, J.A.; Guo, A.; Dong, X.; Lu, P.; Szafron, D.; Greiner, R.; Wishart, D.S. BASys: A web server for automated bacterial genome annotation. Nucleic Acids Res. 2005, 33, W455–W459. [Google Scholar] [CrossRef] [Green Version]
- Arndt, D.; Grant, J.R.; Marcu, A.; Sajed, T.; Pon, A.; Liang, Y.; Wishart, D.S. PHASTER: A better, faster version of the PHAST phage search tool. Nucleic Acids Res. 2016, 44, W16–W21. [Google Scholar] [CrossRef] [Green Version]
- Zhou, Y.; Liang, Y.; Lynch, K.H.; Dennis, J.J.; Wishart, D.S. PHAST: A fast phage search tool. Nucleic Acids Res. 2011, 39, W347–W352. [Google Scholar] [CrossRef]
- Ozer, E.A.; Allen, J.P.; Hauser, A.R. Characterization of the core and accessory genomes of Pseudomonas aeruginosa using bioinformatic tools Spine and AGEnt. BMC Genom. 2014, 15, 737. [Google Scholar] [CrossRef] [Green Version]
- Dhillon, B.K.; Laird, M.R.; Shay, J.A.; Winsor, G.L.; Lo, R.; Nizam, F.; Pereira, S.K.; Waglechner, N.; McArthur, A.G.; Langille, M.G.; et al. IslandViewer 3: More flexible, interactive genomic island discovery, visualization and analysis. Nucleic Acids Res. 2015, 43, W104–W108. [Google Scholar] [CrossRef]
- Romero-Calle, D.; Guimaraes Benevides, R.; Goes-Neto, A.; Billington, C. Bacteriophages as Alternatives to Antibiotics in Clinical Care. Antibiotics 2019, 8, 138. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Pires, D.P.; Melo, L.; Vilas Boas, D.; Sillankorva, S.; Azeredo, J. Phage therapy as an alternative or complementary strategy to prevent and control biofilm-related infections. Curr. Opin. Microbiol. 2017, 39, 48–56. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Sulakvelidze, A.; Alavidze, Z.; Morris, J.G., Jr. Bacteriophage therapy. Antimicrob. Agents Chemother. 2001, 45, 649–659. [Google Scholar] [CrossRef] [PubMed] [Green Version]






{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
| Strain | Genome ID | Size (bp) | %GC | CDS * | tRNA * | Serotype | ST * | Closely Related Genome ** |
|---|---|---|---|---|---|---|---|---|
| PcyII-10 | LT673656 | 6,288,645 | 66.50% | 5787 | 64 | O6 | 1233 | LESB58 (NC_011770) |
| PAC7-25 | LR739071 | 6,479,881 | 66.30% | 5955 | 65 | O6 | 1710 | YL84 (CP007147) |
| PcyII-29 | LR739068 | 6,621,209 | 66.30% | 6156 | 65 | O4 | 175 | F22031 (CP007399) |
| PcyII-40 | LR739069 | 6,831,274 | 66.10% | 6395 | 66 | O11 | 309 | UCBPP-PA14 (CP000438) |
© 2020 by the authors. Licensee MDPI, Basel, Switzerland. This article is an open access article distributed under the terms and conditions of the Creative Commons Attribution (CC BY) license (http://creativecommons.org/licenses/by/4.0/).
Share and Cite
Pourcel, C.; Midoux, C.; Vergnaud, G.; Latino, L. The Basis for Natural Multiresistance to Phage in Pseudomonas aeruginosa. Antibiotics 2020, 9, 339. https://doi.org/10.3390/antibiotics9060339
Pourcel C, Midoux C, Vergnaud G, Latino L. The Basis for Natural Multiresistance to Phage in Pseudomonas aeruginosa. Antibiotics. 2020; 9(6):339. https://doi.org/10.3390/antibiotics9060339
Chicago/Turabian StylePourcel, Christine, Cédric Midoux, Gilles Vergnaud, and Libera Latino. 2020. "The Basis for Natural Multiresistance to Phage in Pseudomonas aeruginosa" Antibiotics 9, no. 6: 339. https://doi.org/10.3390/antibiotics9060339
APA StylePourcel, C., Midoux, C., Vergnaud, G., & Latino, L. (2020). The Basis for Natural Multiresistance to Phage in Pseudomonas aeruginosa. Antibiotics, 9(6), 339. https://doi.org/10.3390/antibiotics9060339