Short Chain Fatty Acids Modulate the Growth and Virulence of Pathosymbiont Escherichia coli and Host Response

Abstract
:1. Introduction
2. Results
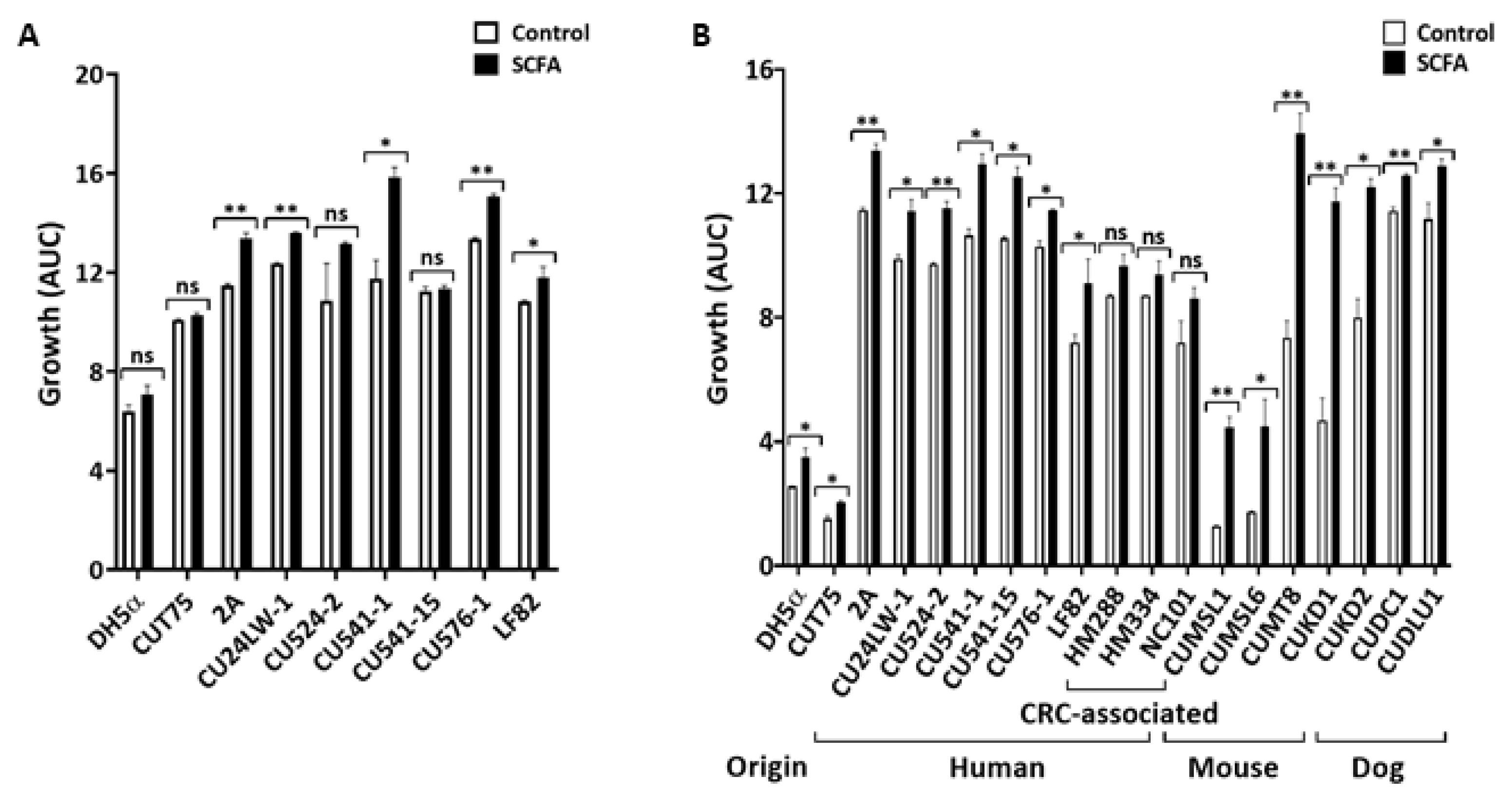
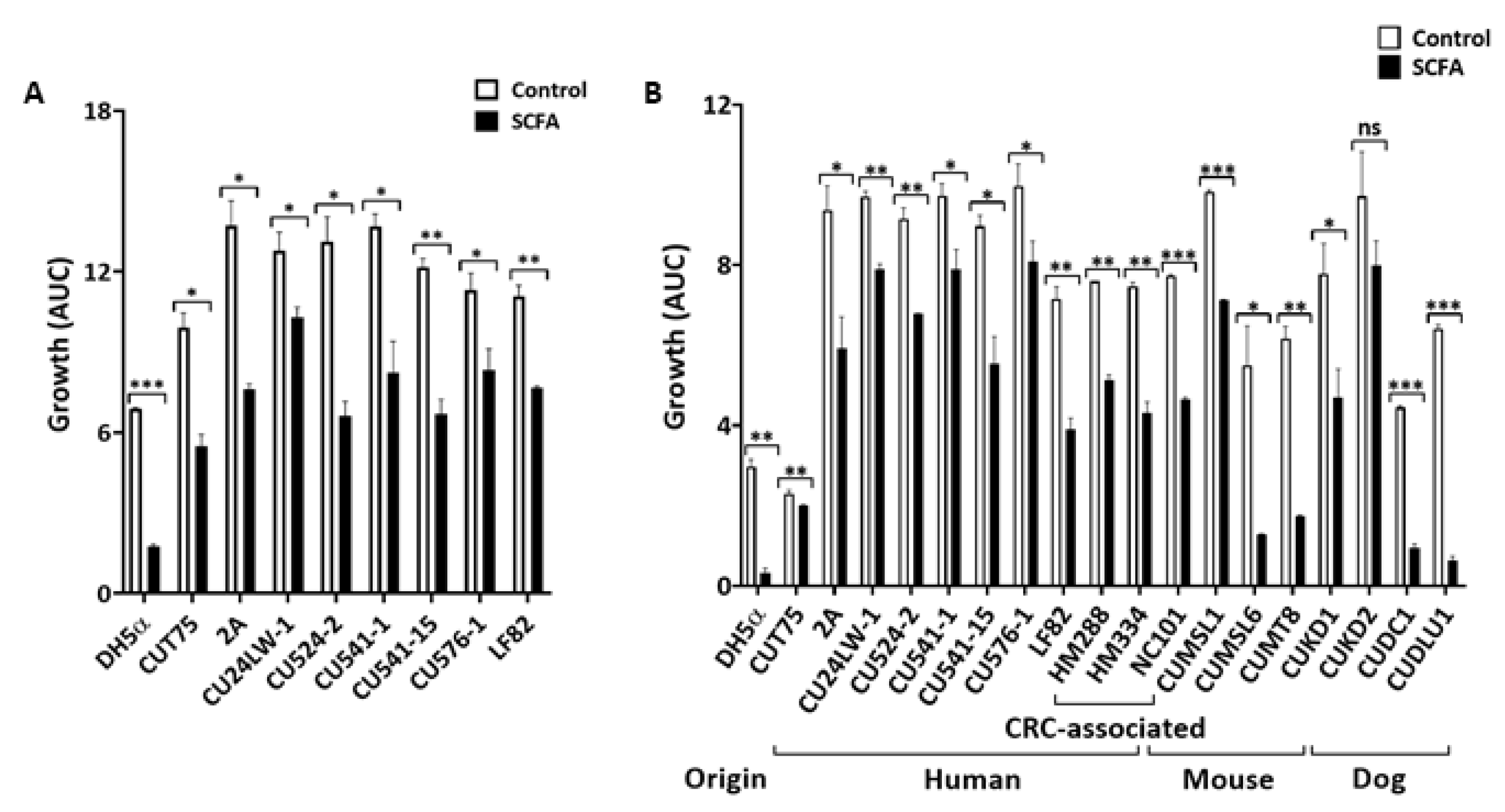
2.1. SCFA Modulate E. coli Growth In Vitro
2.1.1. Ileal SCFA (i-SCFA) Promote E. coli Growth
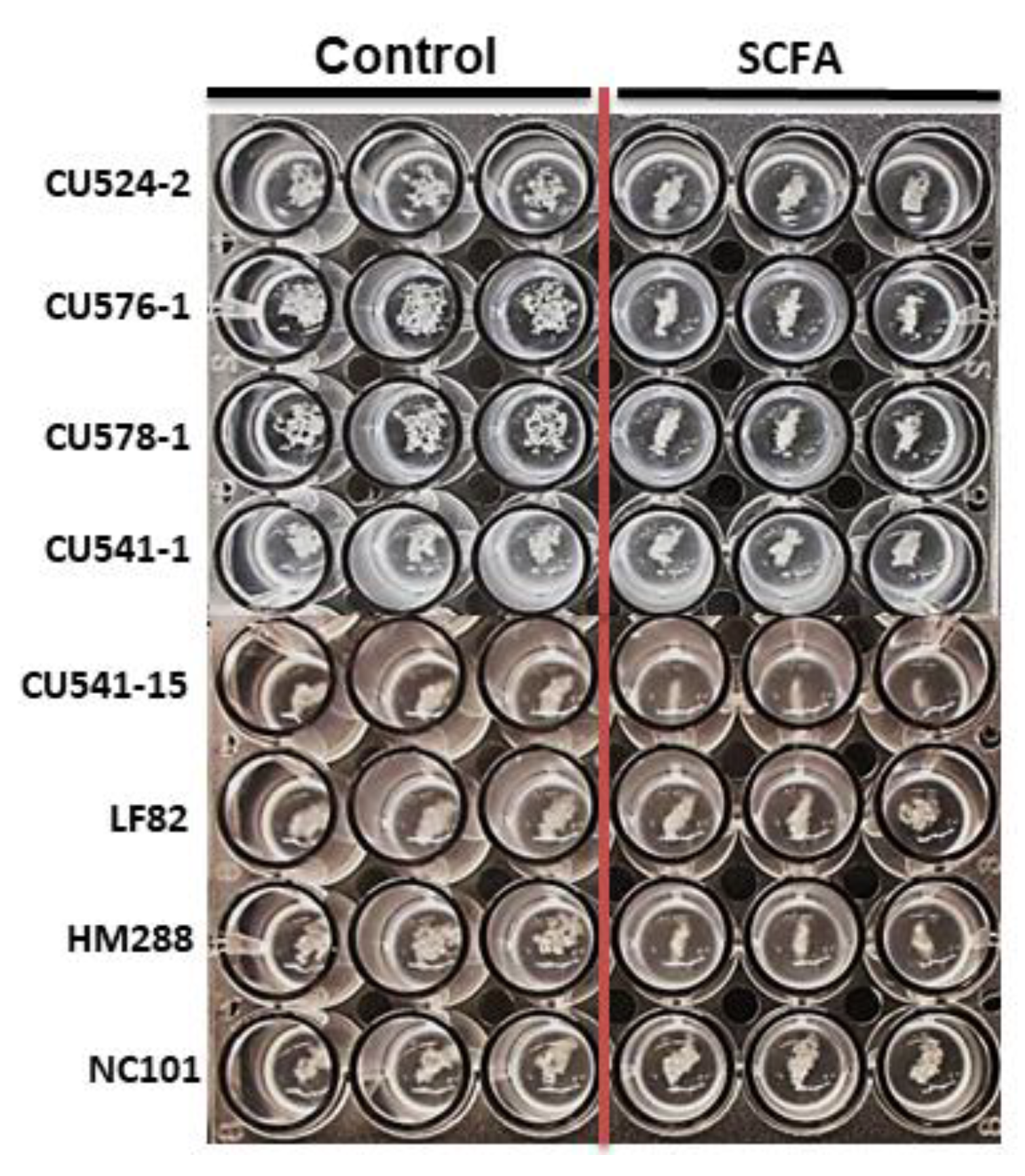
2.1.2. Colonic SCFA (c-SCFA) inhibit E. coli Growth
2.1.3. Inhibition of Growth by c-SCFA is pH-Dependent
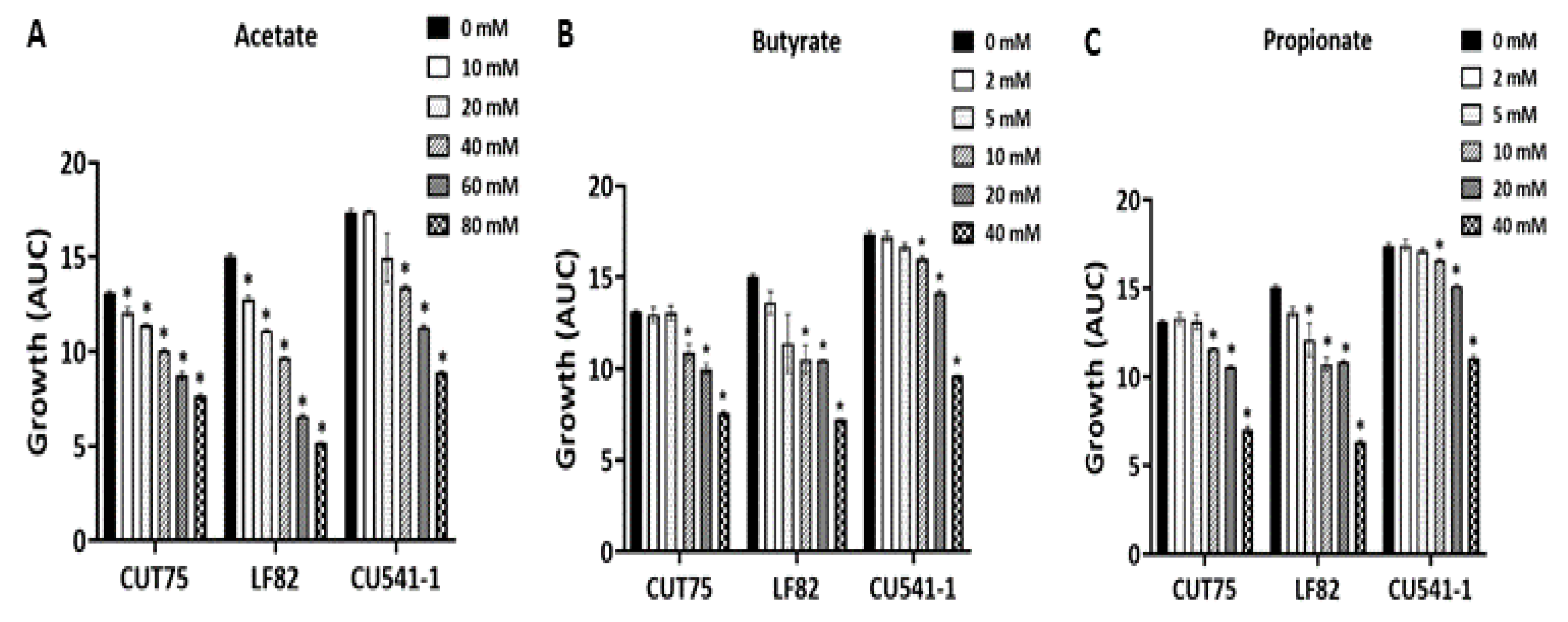
2.1.4. Inhibition of E. coli Growth by Acetate, Propionate, and Butyrate
2.2. c-SCFA Inhibit Virulence Gene Expression in E. coli
2.3. SCFA Modulate E. coli Motility
2.4. c-SCFA Inhibit Type 1 Pili FimH-Mediated Yeast Agglutination
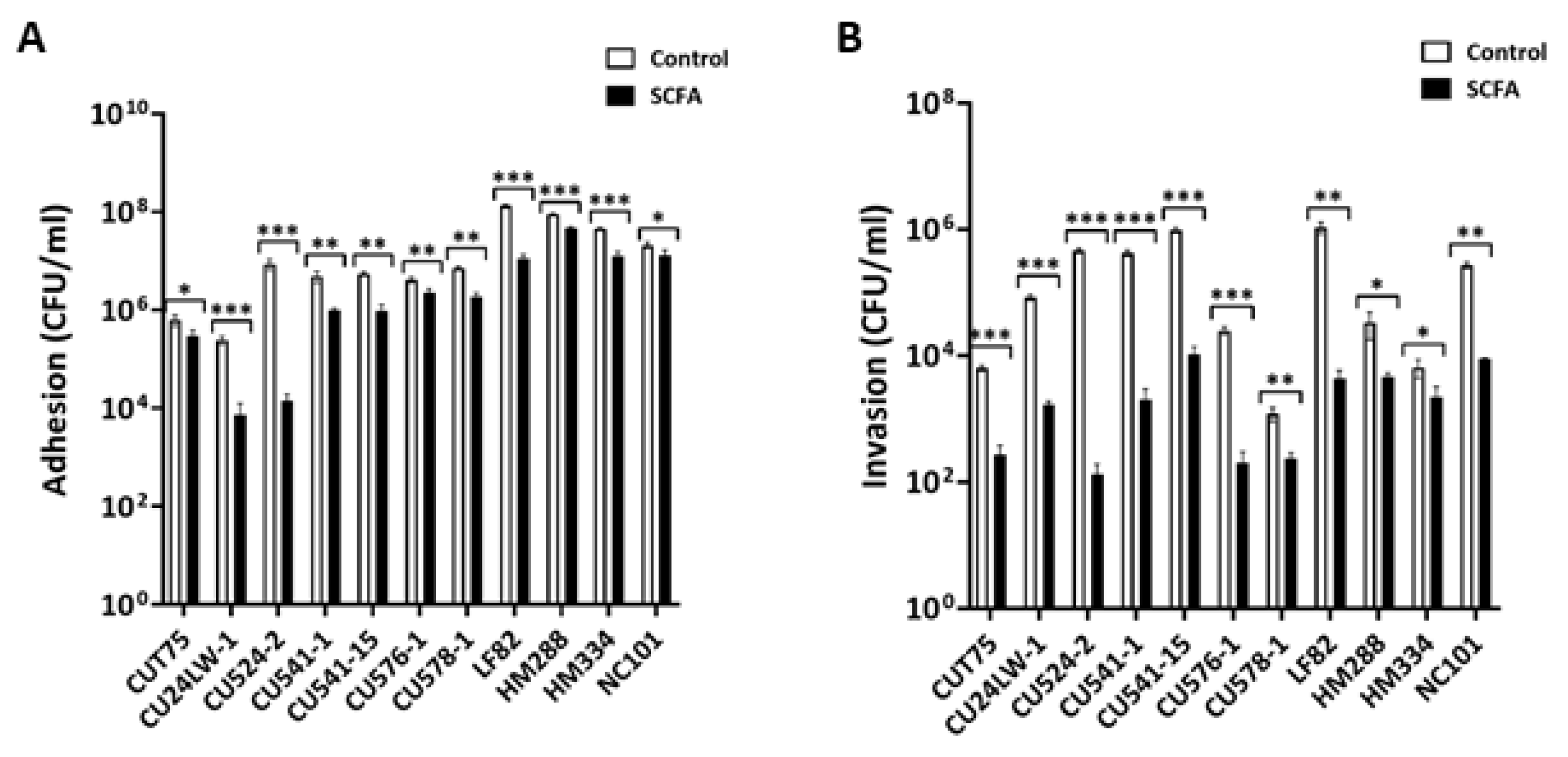
2.5. c-SCFA Inhibit E. coli Adhesion and Invasion of Intestinal Epithelial Cells
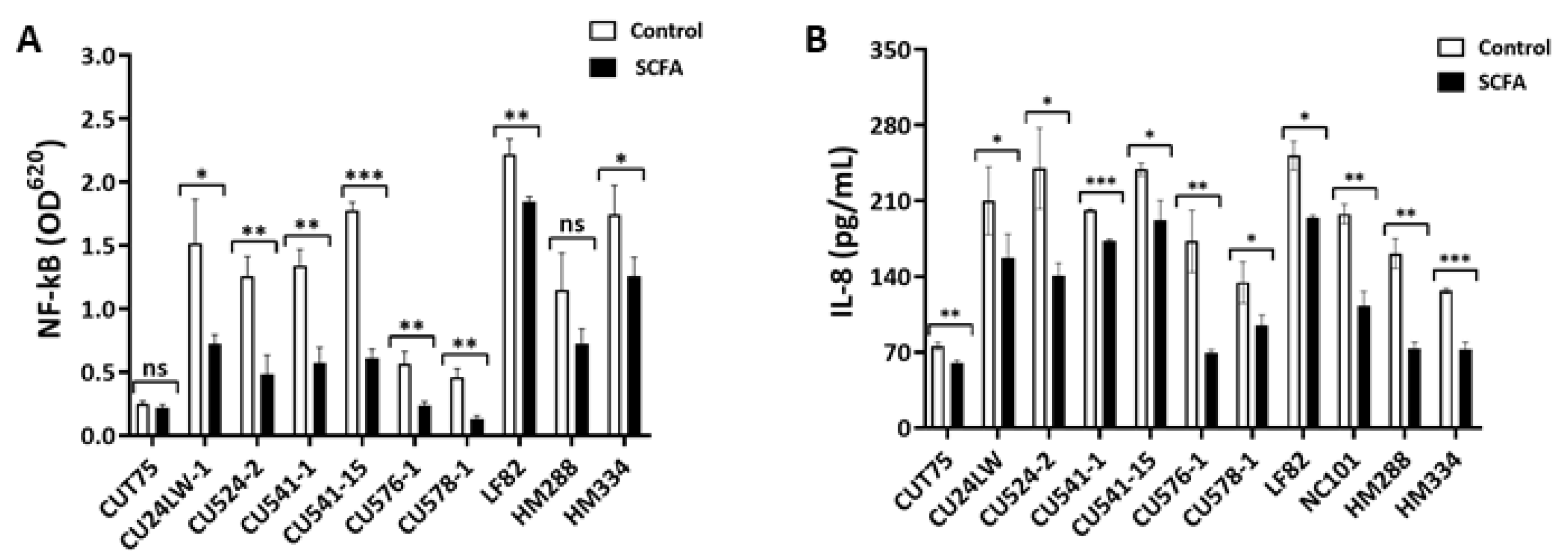
2.6. c-SCFA Inhibit Host Proinflammatory Responses
2.6.1. c-SCFA Inhibit NF-kB Signal Transduction
2.6.2. c-SCFA Inhibit IL-8 Secretion by Epithelial Cells
3. Discussion
4. Materials and Methods
4.1. Bacterial Strains
4.2. Bacterial Culture
4.3. Chemicals and Stock Solutions
4.4. Standardized Growth Analysis
4.5. Transcriptional Analysis of Virulence Genes
4.6. Motility Assay
4.7. Mammalian Cell Culture Conditions
4.8. Yeast Agglutination Assay
4.9. E. coli Adhesion and Invasion of Cultured Epithelial Cells
4.10. NF-ĸB Activation Assay
4.11. Proinflammatory Cytokine IL-8 Secretion
4.12. Statistical Analysis
5. Conclusions
Author Contributions
Funding
Acknowledgments
Conflicts of Interest
References
- Morrison, D.J.; Preston, T. Formation of short chain fatty acids by the gut microbiota and their impact on human metabolism. Gut Microbes 2016, 7, 189–200. [Google Scholar] [CrossRef] [Green Version]
- Bishehsari, F.; Engen, P.A.; Preite, N.Z.; Tuncil, Y.E.; Naqib, A.; Shaikh, M.; Rossi, M.; Wilber, S.; Green, S.; Hamaker, B.; et al. Dietary fiber treatment corrects the composition of gut microbiota, promotes SCFA production, and suppresses colon carcinogenesis. Genes 2018, 9, 102. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Rahat-Rozenbloom, S.; Fernandes, J.; Gloor, G.B.; Wolever, T.M.S. Evidence for greater production of colonic short-chain fatty acids in overweight than lean humans. Int. J. Obes. 2014, 38, 1525–1531. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Louis, P.; Flint, H.J. Formation of propionate and butyrate by the human colonic microbiota. Environ. Microbiol. 2017, 29–41. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Agus, A.; Denizot, J.; Thévenot, J.; Martinez-Medina, M.; Massier, S.; Sauvanet, P.; Bernalier-Donadille, A.; Denis, S.; Hofman, P.; Bonnet, R.; et al. Western diet induces a shift in microbiota composition enhancing susceptibility to Adherent-Invasive, E. coli infection and intestinal inflammation. Sci. Rep. 2016, 6, 19032. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Flint, H.J.; Duncan, S.H.; Scott, K.P.; Louis, P. Links between diet, gut microbiota composition and gut metabolism. Proc. Nutr. Soc. 2015, 74, 13–22. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Louis, P.; Flint, H.J. Diversity, metabolism and microbial ecology of butyrate-producing bacteria from the human large intestine. FEMS Microbiol. Lett. 2009, 294, 1–8. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Ríos-Covián, D.; Ruas-Madiedo, P.; Margolles, A.; Gueimonde, M.; De los Reyes-Gavilán, C.G.; Salazar, N. Intestinal short chain fatty acids and their link with diet and human health. Front. Microbiol. 2016, 7, 185. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Macfarlane, G.T.; Gibson, G.R.; Cummings, J.H. Comparison of fermentation reactions in different regions of the human colon. J. Appl. Microbiol. 1992, 72, 57–64. [Google Scholar] [CrossRef]
- Cummings, J.H.; Pomare, E.W.; Branch, H.W.J.; Naylor, C.P.E.; MacFarlane, G.T. Short chain fatty acids in human large intestine, portal, hepatic and venous blood. Gut 1987, 28, 1221–1227. [Google Scholar] [CrossRef] [Green Version]
- Venegas, D.P.; De La Fuente, M.K.; Landskron, G.; González, M.J.; Quera, R.; Dijkstra, G.; Harmsen, H.; Faber, K.; Hermoso, M. Short chain fatty acids (SCFAs)mediated gut epithelial and immune regulation and its relevance for inflammatory bowel diseases. Front. Immunol. 2019, 10, 277. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Bultman, S.J. Interplay between diet, gut microbiota, epigenetic events, and colorectal cancer. Mol. Nutr. Food Res. 2017, 61, 10. [Google Scholar] [CrossRef] [Green Version]
- Kim, C.H.; Park, J.; Kim, M. Gut Microbiota-Derived Short-Chain Fatty Acids, T Cells, and Inflammation. Immune Netw. 2014, 14, 277. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Mekonnen, S.A.; Merenstein, D.; Fraser, C.M.; Marco, M.L. Molecular mechanisms of probiotic prevention of antibiotic-associated diarrhea. Curr. Opin. Biotechnol. 2020, 61, 226–234. [Google Scholar] [CrossRef]
- Lackraj, T.; Kim, J.I.; Tran, S.L.; Barnett Foster, D.E. Differential modulation of flagella expression in enterohaemorrhagic Escherichia coli O157: H7 by intestinal short-chain fatty acid mixes. Microbiology (UK) 2016, 162, 1761–1772. [Google Scholar] [CrossRef] [PubMed]
- Lawhon, S.D. Genetic and Environmental Regulation of Virulence Genes in Salmonella enterica Serovar Typhimurium. Ph.D. Thesis, North Carolina State University, Raleigh, NC, USA, 2003. [Google Scholar]
- Sorbara, M.T.; Dubin, K.; Littmann, E.R.; Moody, T.U.; Fontana, E.; Seok, R.; Leiner, I.; Taur, Y.; Peled, J.; Van Den Brink, M.; et al. Inhibiting antibiotic-resistant Enterobacteriaceae by microbiota-mediated intracellular acidification. J. Exp. Med. 2019, 216, 84–98. [Google Scholar] [CrossRef] [PubMed]
- Pascal, V.; Pozuelo, M.; Borruel, N.; Casellas, F.; Campos, D.; Santiago, A.; Martinez, X.; Varela, E.; Sarrabayrouse, G.; Machiels, K.; et al. A microbial signature for Crohn’s disease. Gut 2017, 66, 813–822. [Google Scholar] [CrossRef] [PubMed]
- Gao, Z.; Guo, B.; Gao, R.; Zhu, Q.; Qin, H. Microbiota disbiosis is associated with colorectal cancer. Front. Microbiol. 2015, 6. [Google Scholar] [CrossRef] [PubMed]
- Wang, T.; Cai, G.; Qiu, Y.; Fei, N.; Zhang, M.; Pang, X.; Jia, W.; Cai, S.; Zhao, L. Structural segregation of gut microbiota between colorectal cancer patients and healthy volunteers. ISME J. 2012, 6, 320–329. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Machiels, K.; Joossens, M.; Sabino, J.; De Preter, V.; Arijs, I.; Eeckhaut, V.; Ballet, V.; Claes, K.; Van Immerseel, F.; Verbeke, K.; et al. A decrease of the butyrate-producing species roseburia hominis and faecalibacterium prausnitzii defines dysbiosis in patients with ulcerative colitis. Gut 2014, 63, 1275–1283. [Google Scholar] [CrossRef] [PubMed]
- Darfeuille-Michaud, A. Adherent-invasive Escherichia coli: A putative new E. coli pathotype associated with Crohn’s disease. Int. J. Med. Microbiol. 2002, 292, 185–193. [Google Scholar] [CrossRef] [PubMed]
- Martinez-Medina, M.; Garcia-Gil, L.J. Escherichia coli in chronic inflammatory bowel diseases: An update on adherent invasive Escherichia coli pathogenicity. World, J. Gastrointest Pathophysiol. 2014, 5, 213–227. [Google Scholar] [CrossRef] [PubMed]
- Simpson, K.W.; Dogan, B.; Rishniw, M.; Goldstein, R.E.; Klaessig, S.; McDonough, P.L.; German, A.; Yates, R.; Russell, D.; Johnson, S.; et al. Adherent and invasive Escherichia coli is associated with granulomatous colitis in Boxer dogs. Infect. Immun. 2006, 74, 4778–4792. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Craven, M.; Egan, C.E.; Dowd, S.E.; McDonough, S.P.; Dogan, B.; Denkers, E.Y.; Bowman, D.; Scherl, E.; Simpson, K. Inflammation drives dysbiosis and bacterial invasion in murine models of ileal Crohn’s Disease. PLoS ONE 2012, 7, e0041594. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Kastl, A.J.; Terry, N.A.; Wu, G.D.; Albenberg, L.G. The Structure and Function of the Human Small Intestinal Microbiota: Current Understanding and Future Directions. CMGH 2020, 9, 33–45. [Google Scholar] [CrossRef] [Green Version]
- Zoetendal, E.G.; Raes, J.; Van Den Bogert, B.; Arumugam, M.; Booijink, C.C.; Troost, F.J.; Bork, P.; Wels, M.; De Vos, W.; Kleerebezem, M. The human small intestinal microbiota is driven by rapid uptake and conversion of simple carbohydrates. ISME J. 2012, 6, 1415–1426. [Google Scholar] [CrossRef]
- Elhenawy, W.; Tsai, C.N.; Coombes, B.K. Host-Specific Adaptive Diversification of Crohn’s Disease-Associated Adherent-Invasive Escherichia coli. Cell Host Microbe 2019, 25, 301–312. [Google Scholar] [CrossRef] [Green Version]
- Ormsby, M.J.; Logan, M.; Johnson, S.A.; McIntosh, A.; Fallata, G.; Papadopoulou, R.; Papachristou, E.; Hold, G.; Hansen, R.; Ijaz, U.; et al. Inflammation associated ethanolamine facilitates infection by Crohn’s disease-linked adherent-invasive Escherichia coli. EBioMedicine 2019, 43, 325–332. [Google Scholar] [CrossRef] [Green Version]
- Ormsby, M.J.; Johnson, S.A.; Carpena, N.; Meikle, L.M.; Goldstone, R.J.; McIntosh, A.; Wessel, H.; Hulme, H.; McConnachie, C.; Connolly, J.; et al. Propionic Acid Promotes the Virulent Phenotype of Crohn’s Disease-Associated Adherent-Invasive Escherichia coli. Cell Rep. 2020, 30, 2297–2305. [Google Scholar] [CrossRef] [Green Version]
- da Silva Santos, A.C.; Gomes Romeiro, F.; Yukie Sassaki, L.; Rodrigues, J. Escherichia coli from Crohn’s disease patient displays virulence features of enteroinvasive (EIEC), enterohemorragic (EHEC), and enteroaggregative (EAEC) pathotypes. Gut Pathog. 2015, 7, 2. [Google Scholar] [CrossRef] [Green Version]
- Baumgart, M.; Dogan, B.; Rishniw, M.; Weitzman, G.; Bosworth, B.; Yantiss, R.; Orsi, R.; Wiedmann, M.; McDonough, P.; Kim, S.; et al. Culture independent analysis of ileal mucosa reveals a selective increase in invasive Escherichia coli of novel phylogeny relative to depletion of Clostridiales in Crohn’s disease involving the ileum. ISME J. 2007, 1, 403–418. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Raisch, J.; Buc, E.; Bonnet, M.; Sauvanet, P.; Vazeille, E.; de Vallée, A.; Déchelotte, P.; Darcha, C.; Pezet, D.; Bonnet, R.; et al. Colon cancer-associated B2 Escherichia coli colonize gut mucosa and promote cell proliferation. World, J. Gastroenterol. 2014, 20, 6560–6572. [Google Scholar] [CrossRef] [PubMed]
- Long, A.G.; Lundsmith, E.T.; Hamilton, K.E. Inflammation and Colorectal Cancer. Curr. Colorectal Cancer Rep. 2017, 341–351. [Google Scholar] [CrossRef]
- Arthur, J.C.; Perez-Chanona, E.; Mühlbauer, M.; Tomkovich, S.; Uronis, J.M.; Fan, T.-J.; Campbell, B.; Abujamel, T.; Dogan, B.; Rogers, A.; et al. Intestinal inflammation targets cancer-inducing activity of the microbiota. Science 2012, 338, 120–123. [Google Scholar] [CrossRef] [Green Version]
- Dogan, B.; Scherl, E.; Bosworth, B.; Yantiss, R.; Altier, C.; McDonough, P.L.; Jiang, Z.; DuPont, H.; Garneau, P.; Harel, J.; et al. Multidrug resistance is common in Escherichia coli associated with ileal Crohn’s disease. Inflamm. Bowel Dis. 2013, 19, 141–150. [Google Scholar] [CrossRef] [PubMed]
- Abdelhalim, K.A.; Uzel, A.; Gülşen Ünal, N. Virulence determinants and genetic diversity of adherent-invasive Escherichia coli (AIEC) strains isolated from patients with Crohn’s disease. Microb. Pathog. 2020, 145, 104233. [Google Scholar] [CrossRef]
- Craven, M.; Dogan, B.; Schukken, A.; Volkman, M.; Chandler, A.; McDonough, P.L.; Simpson, K. Antimicrobial resistance impacts clinical outcome of granulomatous colitis in Boxer dogs. J. Vet. Intern. Med. 2010, 24, 819–824. [Google Scholar] [CrossRef]
- Manchester, A.C.; Hill, S.; Sabatino, B.; Armentano, R.; Carroll, M.; Kessler, B.; Miller, M.; Dogan, B.; McDonough, S.; Simpson, K. Association between Granulomatous Colitis in French Bulldogs and Invasive Escherichia coli and Response to Fluoroquinolone Antimicrobials. J. Vet. Intern. Med. 2013, 27, 56–61. [Google Scholar] [CrossRef]
- Darfeuille-Michaud, A.; Neut, C.; Barnich, N.; Lederman, E.; Di Martino, P.; Desreumaux, P.; Gambiez, L.; Joly, B.; Cortot, A.; Colombel, J. Presence of adherent Escherichia coli strains in ileal mucosa of patients with Crohn’s disease. Gastroenterology 1998, 115, 1405–1413. [Google Scholar] [CrossRef]
- Kothary, V.; Scherl, E.J.; Bosworth, B.; Jiang, Z.D.; DuPont, H.L.; Harel, J.; Simpson, K.; Dogan, B. Rifaximin Resistance in Escherichia coli Associated with Inflammatory Bowel Disease Correlates with Prior Rifaximin Use, Mutations in rpoB, and Activity of Phe-Arg-β-Naphthylamide-Inhibitable Efflux Pumps. Antimicrob. Agents Chemother. 2013, 57, 811–817. [Google Scholar] [CrossRef] [Green Version]
- Viladomiu, M.; Kivolowitz, C.; Abdulhamid, A.; Dogan, B.; Victorio, D.; Castellanos, J.G.; Woo, V.; Teng, F.; Tran, N.; Sczesnak, A.; et al. Maladie de CROHN et ARTHRITE: Et si le microbiome faisait le lien? IgA-coated E. coli enriched in Crohn’s disease spondyloarthritis promote TH17-dependent inflammation. Sci. Transl. Med. 2017, 9, eaaf9655. [Google Scholar] [CrossRef] [PubMed]
- Martin, H.M.; Campbell, B.J.; Hart, C.A.; Mpofu, C.; Nayar, M.; Singh, R.; Englyst, H.; Williams, H.; Rhodes, J. Enhanced Escherichia coli adherence and invasion in Crohn’s disease and colon cancer. Gastroenterology 2004, 127, 80–93. [Google Scholar] [CrossRef] [PubMed]
- Kim, S.C.; Tonkonogy, S.L.; Albright, C.A.; Tsang, J.; Balish, E.J.; Braun, J.; Huycke, M.; Sartor, R. Variable phenotypes of enterocolitis in interleukin 10-deficient mice monoassociated with two different commensal bacteria. Gastroenterology 2005, 128, 891–906. [Google Scholar] [CrossRef]
- Fallingborg, J.; Christensen La Ingeman-Nielsen, M.; Jacobsen Ba Abildgaard, K.; Rasmussen, H.H. pH-Profile and regional transit times of the normal gut measured by a radiotelemetry device. Aliment. Pharm. Ther. 2007, 3, 605–613. [Google Scholar] [CrossRef] [PubMed]
- Fallingborg, J.; Christensen, L.A.; Ingeman-Nielsen, M.; Jacobsen, B.A.; Abildgaard, K.; Rasmussen, H.H.; Rasmussen, S. Measurement of gastrointestinal pH and regional transit times in normal children. J. Pediatr. Gastroenterol. Nutr. 1990, 11, 211–214. [Google Scholar] [CrossRef] [PubMed]
- He, Y.; Xu, T.; Fossheim, L.E.; Zhang, X.H. FliC, a Flagellin Protein, Is Essential for the Growth and Virulence of Fish Pathogen Edwardsiella tarda. PLoS ONE 2012, 7, e0045070. [Google Scholar] [CrossRef] [Green Version]
- Sevrin, G.; Massier, S.; Chassaing, B.; Agus, A.; Delmas, J.; Denizot, J.; Billard, E.; Barnich, N. Adaptation of adherent-invasive E. coli to gut environment: Impact on flagellum expression and bacterial colonization ability. Gut Microbes 2018, 11, 364–380. [Google Scholar] [CrossRef]
- Hagberg, L.; Jodal, U.; Korhonen, T.K.; Lidin-Janson, G.; Lindberg, U.; Svanborg Edén, C. Adhesion, hemagglutination, and virulence of Escherichia coli causing urinary tract infections. Infect. Immun. 1981, 31, 564–570. [Google Scholar] [CrossRef] [Green Version]
- Céspedes, S.; Saitz, W.; Del Canto, F.; De la Fuente, M.; Quera, R.; Hermoso, M.; Muñoz, R.; Ginard, D.; Khorrami, S.; Girón, J.; et al. Genetic diversity and virulence determinants of Escherichia coli strains isolated from patients with Crohn’s disease in Spain and Chile. Front. Microbiol. 2017, 8, 639. [Google Scholar] [CrossRef]
- Stahlhut, S.G.; Tchesnokova, V.; Struve, C.; Weissman, S.J.; Chattopadhyay, S.; Yakovenko, O.; Aprikian, P.; Sokurenko, E.; Krogfelt, K. Comparative structure-function analysis of mannose-specific FimH adhesins from Klebsiella pneumoniae and Escherichia coli. J. Bacteriol. 2009, 191, 6592–6601. [Google Scholar] [CrossRef] [Green Version]
- Carvalho F a Barnich, N.; Sivignon, A.; Darcha, C.; Chan, C.H.F.; Stanners, C.P.; Darfeuille-Michaud, A. Crohn’s disease adherent-invasive Escherichia coli colonize and induce strong gut inflammation in transgenic mice expressing human CEACAM. J. Exp. Med. 2009, 206, 2179–2189. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Mossman, K.L.; Mian, M.F.; Lauzon, N.M.; Gyles, C.L.; Lichty, B.; Mackenzie, R.; Gill, N.; Ashkar, A. Cutting Edge: FimH Adhesin of Type 1 Fimbriae Is a Novel TLR4 Ligand. J. Immunol. 2008, 181, 6702–6706. [Google Scholar] [CrossRef] [PubMed]
- Dogan, B.; Suzuki, H.; Herlekar, D.; Sartor, R.B.; Campbell, B.J.; Roberts, C.L.; Stewart, K.; Scherl, E.; Araz, Y.; Bitar, P.; et al. Inflammation-associated adherent-invasive Escherichia coli are enriched in pathways for use of propanediol and iron and M-cell translocation. Inflamm. Bowel Dis. 2014, 20, 1919–1932. [Google Scholar] [CrossRef] [PubMed]
- Prorok-Hamon, M.; Friswell, M.K.; Alswied, A.; Roberts, C.L.; Song, F.; Flanagan, P.K.; Knight, P.; Codling, C.; Marchesi, J.; Winstanley, C.; et al. Colonic mucosa-associated diffusely adherent afaC+ Escherichia coli expressing lpfA and pks are increased in inflammatory bowel disease and colon cancer. Gut 2014, 61, 763–770. [Google Scholar] [CrossRef] [Green Version]
- Rolhion, N.; Carvalho, F.A.; Darfeuille-Michaud, A. OmpC and the σE regulatory pathway are involved in adhesion and invasion of the Crohn’s disease-associated Escherichia coli strain LF82. Mol. Microbiol. 2007, 63, 1684–1700. [Google Scholar] [CrossRef] [PubMed]
- Barnich, N.; Bringer, M.A.; Claret, L.; Daffeuille-Michaud, A. Involvement of Lipoprotein NlpI in the Virulence of Adherent Invasive Escherichia coli Strain LF82 Isolated from a Patient with Crohn’s Disease. Infect. Immun. 2004, 72, 2484–2493. [Google Scholar] [CrossRef] [Green Version]
- Rahman, M.M.; McFadden, G. Modulation of NF-kB signalling by microbial pathogens. Nat. Rev. Microbiol. 2011, 9, 291–306. [Google Scholar] [CrossRef]
- Zhang, S.; Fu, J.; Dogan, B.; Scherl, E.J.; Simpson, K.W. 5-Aminosalicylic acid downregulates the growth and virulence of Escherichia coli associated with IBD and colorectal cancer, and upregulates host anti-inflammatory activity. J. Antibiot. 2018, 71, 950–961. [Google Scholar] [CrossRef]
- Takaishi, H.; Matsuki, T.; Nakazawa, A.; Takada, T.; Kado, S.; Asahara, T.; Kamada, N.; Sakuraba, A.; Yajima, T.; Higuchi, H.; et al. Imbalance in intestinal microflora constitution could be involved in the pathogenesis of inflammatory bowel disease. Int. J. Med. Microbiol. 2008, 298, 463–472. [Google Scholar] [CrossRef]
- Martinez-Medina, M.; Aldeguer, X.; Lopez-Siles, M.; González-Huix, F.; López-Oliu, C.; Dahbi, G.; Bianco, J.; Bilanco, J.; Garcia-Gil, L.; Darfeuille-Michaud, A. Molecular diversity of Escherichia coli in the human gut: New ecological evidence supporting the role of adherent-invasive E. coli (AIEC) in Crohn’s disease. Inflamm. Bowel Dis. 2009, 15, 872–882. [Google Scholar] [CrossRef]
- Darfeuille-Michaud, A.; Boudeau, J.; Bulois, P.; Neut, C.; Glasser, A.L.; Barnich, N.; Bringer, M.; Swidsinski, A.; Beaugerie, L.; Colombel, J. High prevalence of adherent-invasive Escherichia coli associated with ileal mucosa in Crohn’s disease. Gastroenterology 2004, 127, 412–421. [Google Scholar] [CrossRef] [PubMed]
- Ormsby, M.; Johnson, S.; Meikle, L.; Goldstone, R.; McIntosh, A.; Wessel, H.; Hulme, H.; McConnachie, C.; Connolly, J.; Roe, A.; et al. Propionic acid promotes the virulent phenotype of Crohn’s disease-associated adherent-invasive Escherichia coli. bioRxiv 2018. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- McConnell, E.L.; Basit, A.W.; Murdan, S. Measurements of rat and mouse gastrointestinal pH, fluid and lymphoid tissue, and implications for in-vivo experiments. J. Pharm. Pharmacol. 2008, 60, 63–70. [Google Scholar] [CrossRef]
- Lawhon, S.D.; Maurer, R.; Suyemoto, M.; Altier, C. Intestinal short-chain fatty acids alter Salmonella typhimurium invasion gene expression and virulence through BarA/SirA. Mol. Microbiol. 2002, 46, 1451–1464. [Google Scholar] [CrossRef] [PubMed]
- Subramanian, S.; Rhodes, J.M.; Hart, C.A.; Tam, B.; Roberts, C.L.; Smith, S.L.; Corkill, J.; Winstanley, C.; Virji, M.; Campbell, B.; et al. Characterization of epithelial IL-8 response to inflammatory bowel disease mucosal E. coli and its inhibition by mesalamine. Inflamm. Bowel Dis. 2008, 14, 162–175. [Google Scholar] [CrossRef] [PubMed]
- Sethi, G.; Sung, B.; Aggarwal, B.B. TNF: A master switch for inflammation to cancer. Front. Biosci. 2008, 13, 5094–5107. [Google Scholar] [CrossRef] [Green Version]
- Hinton, M.; Linton, A.H. Control of salmonella infections in broiler chickens by the acid treatment of their feed. Vet. Rec. 1988, 123, 416–421. [Google Scholar] [CrossRef]
- Kwon, Y.M.; Ricke, S.C. Salmonella typhimurium poultry isolate growth response to propionic acid and sodium propionate under aerobic and anaerobic conditions. Int. Biodeterior. Biodegrad. 1999, 43, 161–165. [Google Scholar] [CrossRef]
- Anderson, C.J.; Satkovich, J.; Köseoğlu, V.K.; Agaisse, H.; Kendall, M.M. The ethanolamine permease EutH promotes vacuole adaptation of Salmonella enterica and Listeria monocytogenes during macrophage infection. Infect. Immun. 2018, 86, e00172-18. [Google Scholar] [CrossRef] [Green Version]
- Kendall, M.M.; Gruber, C.C.; Parker, C.T.; Sperandio, V. Ethanolamine controls expression of genes encoding components involved in interkingdom signaling and virulence in enterohemorrhagic escherichia coli O157:H7. MBio 2012, 3. [Google Scholar] [CrossRef] [Green Version]
- Vizcaino, M.I.; Crawford, J.M. The colibactin warhead crosslinks DNA. Nat. Chem. 2015, 7, 411–417. [Google Scholar] [CrossRef] [PubMed]
- Yusuf, F.; Adewiah, S.; Syam, A.F.; Fatchiyah, F. Altered profile of gut microbiota and the level short chain fatty acids in colorectal cancer patients. J. Phys. Conf. Ser. 2019, 1146, 012037. [Google Scholar] [CrossRef]
- Stumpff, F. A look at the smelly side of physiology: Transport of short chain fatty acids. Pflug. Arch. Eur. J. Physiol. 2018, 470, 571–598. [Google Scholar] [CrossRef] [PubMed]









{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
| Condition | Total SCFA | Molar Ratio (Acetate:Propionate:Butyrate) | Reference |
|---|---|---|---|
| Ileal, pH = 7.4 | 12 mM | 8:2.5:1.5 | [10] |
| Colonic, pH = 6.5 | 60 to 123 mM | 65:29:29 | [10] |
| Strain | Source | Phylogroup | AIEC | PKS | Reference |
|---|---|---|---|---|---|
| DH5α | ATCC | A | - | - | - |
| CUT75 | CD mucosa | A | - | - | [32] |
| LF82 | CD mucosa | B2 | + | - | [40] |
| CU24LW-1 | CD mucosa | A | + | - | [41] |
| CU524-2 | CD mucosa | B1 | + | - | [32] |
| CU541-1 | CD mucosa | B1 | + | - | [32] |
| CU541-15 | CD mucosa | B1 | + | - | [32] |
| CU578-1 | CD mucosa | D | + | - | [32] |
| CU576-1 | CD mucosa | D | + | - | [32] |
| 2A | CD-SpA mucosa | B2 | + | - | [42] |
| HM44 | CRC mucosa | B2 | - b | + | [43] |
| HM164 | CRC mucosa | B2 | - b | + | [43] |
| HM288 | CRC mucosa | B2 | - | - | [43] |
| HM334 | CRC mucosa | B2 | - | + | [43] |
| NC101 a | Healthy mouse feces | B2 | + | + | [35,44] |
| CUMT8 | mouse ileitis tissue | B1 | + | - | [25] |
| CUMSL1 | Agr2−/−mouse ileum | B2 | + | - | this study |
| CUMSL6 | Agr2−/−mouse ileum | B2 | + | - | this study |
| CUDC1 | GC dog colon | B1 | + | - | [38] |
| CUDLU1 | GC dog colon | B1 | + | - | [38] |
| CUKD1 | GC dog colon | B2 | + | - | [24] |
| CUKD2 | GC dog colon | D | + | - | [24] |
| Gene Function | Gene Name | Fold Change (2−ΔΔCt) | ||||||||||||
|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|
| Healthy Control | CD-associated E. coli (AIEC) | CRC-associated E. coli | ||||||||||||
| CUT75 b | LF82 | CU24LW-1 | CU524-2 | CU541-1 | CU541-15 | CU576-1 | CU578-1 | NC101 a | HM44 | HM164 | HM334 | HM288 | ||
| Motility | fliC | 0.34 | 0.31 * | 0. 73 * | 0.51 * | 0.41 * | 0.76 | 0.40 * | 0.17 * | 0.07 * | 5.73 * | 0.058 * | 0.50 * | 0.91 |
| Adhesion and invasion | fimH | 0.65 * | 0.63 * | 2.38 * | 0.37 * | 0.28 * | 0.79 * | 0.46 | 0.44 * | 0.63 | 1.25 | 0.43 * | 0.67 * | 1.04 |
| ompC | 1.48 * | 0.42 * | 1.99 * | 0.30 * | 0.31 * | 0.50 * | 0.60 | 0.49 * | 0.63 | 0.22 * | 0.74 * | 0.96 | 0.33 * | |
| yfgL | 1.1 | 0.77 * | 0.44 * | 0.62 * | 0.34 * | 0.70 | 0.37 * | 0.44 * | 0.32 * | 0.23 * | 0.45 * | 0.52 * | 0.60 * | |
| nlpL | 0.73 | 0.35 * | 2.46 * | 1.60 * | 0.52 * | 1.37 | 1.19 | 0.26 * | 0.54 * | 0.51 * | 2.05 * | 1.12 | 1.62 * | |
| lpfA141 | na | 0.39 * | na | na | na | na | na | na | na | na | na | na | na | |
| lpfA154 | na | 5.54 * | 0.90 | 0.24 * | 0.85 * | 0.54 | 0.48 * | na | na | na | - | - | ||
| Stress | htrA | 0.49 * | 0.91 * | 0.40 * | 0.58 * | 0.35 * | 0.45 * | 0.40 * | 0.32 * | 0.59 * | 0.24 * | 0.35 * | 0.36 * | 0.48 * |
| dsbA | 1.37 * | 1.05 | 1.75 * | 1.21 | 0.40 * | 1.01 | 0.44 * | 0.47 * | 0.40 * | 0.23 * | 0.71 * | 0.51 * | 0.71 * | |
| Iron acquisition | fyuA | na | 1.02 | na | na | na | 1.10 | 1.00 | 1.08 | 0.92 | 0.43 * | 1.200 | 0.44 * | 1.47 |
| chuA | na | 0.33 | na | na | na | na | 2.29 | 1.17 | 0.73 | 0.57 | 0.62 * | 0.55 * | 1.58 * | |
| Genotoxicity | pks | na | na | na | na | na | na | na | na | 0.54 | 0.058 * | 0.69 | 0.64 * | na |
| Gene | Protein | Function | Primer Sequences (5′→ 3′) |
|---|---|---|---|
| fliC * | Flagellin | Motility | F1: CAGCCTCTCGCTGATCACTC |
| R1: CCCGCTGCGTCATCCTTCGC | |||
| F2: CTGTCGCTGTTGACCCAGAA | |||
| R2: TGACCTGCTGCGTCATCTTT | |||
| fimH | Type 1 fimbrial subunit | Adhesion | F: CTTATGGCGGCGTGTTATCT |
| R: CGGCTTATCCGTTCTCGAATTA | |||
| ompC | Outer membrane protein C | Outer membrane protein | F: GGTGGTCTGAAATACGACGCTAAC |
| R: GTCGAACTGGTACTGAGCAACAGC | |||
| yfgL | Lipoprotein | Invasion | F: CCGGTGGTCAGCGACGGTCTGG |
| R: CGCCACGCAAAGAGAGCGAAGGC | |||
| nlpL | Lipoprotein | Invasion | F: GGCTCAAGGCGGACGCAACGG |
| R: GAACAGTGCCGTGGCGCTGTCC | |||
| lpfA141 | Long polar fimbrial protein A | M cell translocation | F: GCTGATGCAGGCGACGGTTCTG |
| R: CACAGACTTGTTCACCTGGCCC | |||
| lpfA154 | Long polar fimbrial protein A | M cell translocation | F: CAGGTGTAGGTAGTCTGGCGTC |
| R: GGTCGCCGTCGCCGCCAGGCGC | |||
| htrA | Periplasmic protease | Stress protein (Macrophage survival) | F: CGCAGATGGTGGAATACGGCCAGG |
| R: CCTGGCTTACGAAAGCACCGCGC | |||
| dsbA | Disulfide oxidoreductase | Oxidoreductase (Macrophage survival) | F: GGCGCAGTATGAAGATGGTAAAC |
| R: TTCAAACTGATAGCAGTGCGG | |||
| chuA | Outer membrane heme/hemoglobin receptor | Heme iron acquisition | F: CGGCGACAACTATGTCGTATAA |
| R: TAGGCCACATCAAGGCTAAAC | |||
| fyuA | Ferric Yersiniabactin uptake receptor | Iron acquisition | F: TCGTCGCCGAGAAATCCATCAACT |
| R: AAAGCTGCATGTCTTTGGTGTGGG | |||
| pks | Polyketide synthetase | Genotoxin production | F: ATCTTTCCGCCTAACCCGA |
| mdH | Malate dehydrogenase | Reference | F: CAACTGCCTTCAGGTTCA R: GCGTTCTGGATGCGTTTGGT |
| Name | Control | SCFA Treated |
|---|---|---|
| CU524-2 | 4 | 2 |
| CU576-1 | 4 | 2 |
| CU578-1 | 4 | 2 |
| CU541-1 | 3 | 2 |
| CU541-15 | 3 | 1 |
| LF82 | 3 | 2 |
| HM288 | 4 | 1 |
| NC101 | 3 | 2 |
© 2020 by the authors. Licensee MDPI, Basel, Switzerland. This article is an open access article distributed under the terms and conditions of the Creative Commons Attribution (CC BY) license (http://creativecommons.org/licenses/by/4.0/).
Share and Cite
Zhang, S.; Dogan, B.; Guo, C.; Herlekar, D.; Stewart, K.; Scherl, E.J.; Simpson, K.W. Short Chain Fatty Acids Modulate the Growth and Virulence of Pathosymbiont Escherichia coli and Host Response. Antibiotics 2020, 9, 462. https://doi.org/10.3390/antibiotics9080462
Zhang S, Dogan B, Guo C, Herlekar D, Stewart K, Scherl EJ, Simpson KW. Short Chain Fatty Acids Modulate the Growth and Virulence of Pathosymbiont Escherichia coli and Host Response. Antibiotics. 2020; 9(8):462. https://doi.org/10.3390/antibiotics9080462
Chicago/Turabian StyleZhang, Shiying, Belgin Dogan, Cindy Guo, Deepali Herlekar, Katrina Stewart, Ellen J. Scherl, and Kenneth W. Simpson. 2020. "Short Chain Fatty Acids Modulate the Growth and Virulence of Pathosymbiont Escherichia coli and Host Response" Antibiotics 9, no. 8: 462. https://doi.org/10.3390/antibiotics9080462
APA StyleZhang, S., Dogan, B., Guo, C., Herlekar, D., Stewart, K., Scherl, E. J., & Simpson, K. W. (2020). Short Chain Fatty Acids Modulate the Growth and Virulence of Pathosymbiont Escherichia coli and Host Response. Antibiotics, 9(8), 462. https://doi.org/10.3390/antibiotics9080462