1. Introduction
The plasma electrolytic oxidation (PEO) is an established process for generating oxidic conversion layers on metals, such as Al, Mg, Ti, and metal compounds of those. In contrast to conventional anodic oxidation, microarc discharges are created after initial substrate passivation by applying a sufficiently high anodic voltage and used for layer formation. In the center of the microarc discharge, there is a plasma temperature of about 7000 K [
1]. With increasing distance to the center, the temperature falls below the boiling point of the oxide. In this zone, the melted substrate material reacts with the anodically formed oxygen to form molten oxide [
2]. This is followed by the outer concentric zone in which the melting temperature of the oxide is no longer exceeded. In this area, thermally induced phase changes of the oxides take place. According to Rogov et al., the conversion proceeds in the direction of the Al
O
modification with the lowest Gibbs free energy [
3]:
More precisely, the Gibbs free energy of amorphous,
-, and
-alumina formation were quantified as −1625 kJ/mol, −1654 kJ/mol, and −1676 kJ/mol, respectively, by Naumann and Petzold [
4]. Furthermore, the repeated remelting and heat treatment leads to a compaction of the inner oxide area, especially on Al materials. The result is a three-layered structure consisting of a barrier layer of a few 100 nm thickness at the substrate/layer interface, an inner, compact layer, and an outer, porous layer. The barrier layer is repeatedly formed by electrochemical oxidation at the base of a former microarc discharge and mainly consists of amorphous aluminum oxide [
5,
6]. The phases
- and
-Al
O
dominate within the compact layer [
6,
7]. In the outer, porous layer, mixed oxides are increasingly found because of the incorporation of electrolyte constituents. They are characterized by a higher amorphous portion due to the lower thermal influence. In terms of maximum protection of the Al substrate against corrosion and/or abrasive wear, a high proportion of the compact layer of the total layer thickness is aimed for. Such PEO coatings are characterized by a low porosity and a high proportion of particularly hard and chemically resistant crystalline alumina phases. The chemical composition of the substrate alloy, the electrical regime, and the electrolyte composition were identified as the main influencing factors on the layer microstructure [
7,
8]. There is scientific consensus that the electrolyte composition influences the passivation behavior of the electrochemical system. With the help of potentiodynamic polarization tests, a distinction can be made between metal dissolution, metal passivation, or complex behavior, depending on the system [
9,
10]. Furthermore, electrolyte systems can be classified according to whether conversion of the substrate into an oxide, the incorporation of electrolyte anions into a mixed oxide, or even the deposition of a foreign oxide is sought. In the case of Al alloys, a pure conversion into the particularly wear- and corrosion-resistant, crystalline alumina phases [
11] is desirable. On the other hand, the inclusion of electrolyte anions such as aluminate in the layer-formation process enables the production of MgAl
O
spinel on Mg [
12] and the deposition of Al
O
layers on unalloyed steel [
13]. The scientific literature shows that the rate of oxide formation increases with increasing current density and that a higher thickness [
14] and oxide mass can be achieved due to increasing electrical charge [
15]. Besides the oxide formation, several electrochemical side reactions may occur during the PEO process. At the beginning of the process, electrochemical metal dissolution and gas evolution are competing with the anodic passive-layer formation [
8]. Anodic oxygen evolution can also be observed as a competition reaction of the oxide formation during the passive layer growth.
It has been observed that the rate of oxide formation decreases with increasing electrolyte temperature [
14,
16,
17], alkali concentration [
14,
18,
19], and process time [
5,
14,
20]. On the basis of this knowledge, the theory was developed that during PEO in strongly alkaline solutions, the chemical redissolution of the oxide occurs as a competitive reaction of the anodic oxide formation. Koroleveva et al. assigned this processes to reactions according to Equation (
2) [
21]:
This would be similar to the competition of oxide formation and oxide redissolution during conventional anodizing in acidic solutions. According to Nagayama and O’Sullivan [
22,
23], such reactions can be promoted by strong localization of electric field lines and are designated as field-assisted chemical redissolution. To describe the chemical redissolution of PEO coatings, Snizhko and Al Bosta also refer to the reaction according to Equation (
2) for the chemical dissolution of the natural passive layer [
18,
20]. Within the above-mentioned publications, the Al content of the electrolyte after PEO is often estimated in order to get an experimental access to conceivable redissolution reactions during the process. However, several additional reaction routes may lead to the elevated Al content after PEO. Al
ions can directly be emitted via field assisted anodic metal dissolution and emission of aluminum ions through the plasma channel [
15,
18]:
or by electrochemical etching of the metallic substrate in partial pulse periods without external polarization applied [
20]:
Therefore, it is not clear to what extent chemical redissolution of the plasma electrolytically formed oxide occurs during the PEO process and to what extent this affects the layer microstructure.
On the other hand, possible influences of the electrolyte temperature and the alkali concentration on the electrochemical passivation of the Al substrate were not considered in most of the studies on the chemical redissolution. The authors Moon and Pyun [
24] and Snizhko et al. [
25] showed that the steady state of the passive-layer growth is delayed due to increasing hydroxide concentration. This might promote side-reactions such as metal dissolution and oxygen evolution during the repassivation of the Al substrate at the base of a microarc discharge. Therefore, a decrease of the oxide growth rate and an increase of the Al ion concentration in the electrolyte might also be explained by the impairment of the substrate passivation and not necessarily point at the chemical redissolution reaction. Furthermore, it is questionable as to whether the chemical redissolution reaction described by Equation (
2), which originally refers to the chemical redissolution of amorphous alumina passive layers, can also be generalized for PEO layers consisting of a high proportion of crystalline alumina phases. Additionally, the redissolution behavior of mixed oxides, which arises through the incorporation of electrolyte anions, has not yet been taken into account. This is also indicated by the fact that there is a clear discrepancy in the scientific literature with regard to the significance of chemical redissolution in PEO processes. Several research groups observed an almost linear increase of the oxide thickness over a process time of up to 45 min [
26,
27,
28]. The authors Xue et al. found that the ratio of the outward growth and the total oxide thickness started to decrease after about 2 h and that the total oxide growth rate did not decrease until a process duration of about 5 h [
29]. In [
27,
28], compact and wear-resistant PEO layers are reported, which mainly consist of the crystalline phases
- and
-Al
O
. In contrast, the authors Al Bosta et al. observed a significant decrease of the layer growth rate after only about 20 min [
20]. After exceeding a process time of 50 min, the layer thickness even decreased. At this point, the PEO layers consisted of around 55%
-Al
O
, 18% Al
SiO
, 24% SiO
, and only 8%
-Al
O
[
20]. From the results of [
20,
26,
27,
28,
29] the conclusions might be drawn that the chemical redissolution of the oxide does not play a decisive role for PEO layers with a high proportion of crystalline alumina phases and technically relevant process times of less than one hour and that especially the presumably amorphous, Si-containing phases are characterized by a lower chemical resistance and a higher redissolution rate in alkaline solutions. This argumentation assumes that the rate of layer formation is constant over the entire process time, similar to conventional anodic oxidation, and that only the chemical redissolution rate determines the net growth rate and the maximum achievable oxide thickness. However, it is known that distribution and intensity of the microarc discharges change over the process duration due to the evolution of the oxide microstructure towards the formation of a more compact PEO layer [
8,
30]. The number of microarc discharges declines as the discharges focus on a decreasing number of defects in the oxide layer. The PEO process might even come to a complete standstill. In this case, redissolution would not be necessary to explain the declining rate of layer growth.
Within this work, it is intended to investigate the chemical redissolution of different oxide phases and the effect on the PEO coating’s microstructure and properties for the first time. The dissolution behavior of individual phases is assessed by comparing the phase composition and layer microstructure using diffraction methods and scanning electron microscopy before and after the exposition of the PEO coatings to an alkaline solution.
2. Materials and Methods
The PEO coatings were applied on the commercially available high-strength aluminum alloy EN AW-6082 T6, the chemical composition of which is listed in
Table 1. The sheets had geometrical dimension of 100 × 25 × 3 mm. The plasma electrolytic oxidation was carried out within a laboratory plant consisting of a rectifier
pe86CW-550-53-120/S (plating electronics, Sexau, Germany) and a capsuled treatment station (Scheigenpflug, Leipzig, Germany) with a basin for 12 L electrolyte, directly cooled by a heat exchanger. Two stainless steel sheets were used as counter electrodes. The electrolyte composition was 5 g/L KOH, 5 g/L Na
SiO
5H
O, and 1 g/L Na
HPO
. All substances were of analytical grade (Sigma-Aldrich, St. Louis, MI, USA). The electrical regime was defined by a symmetric, bipolar, rectangular, current controlled pulse of
=
= 30 A/dm
and
=
= 10 ms. The maximum anodic voltage was limited to 500 V, and the treatment time was 60 min.
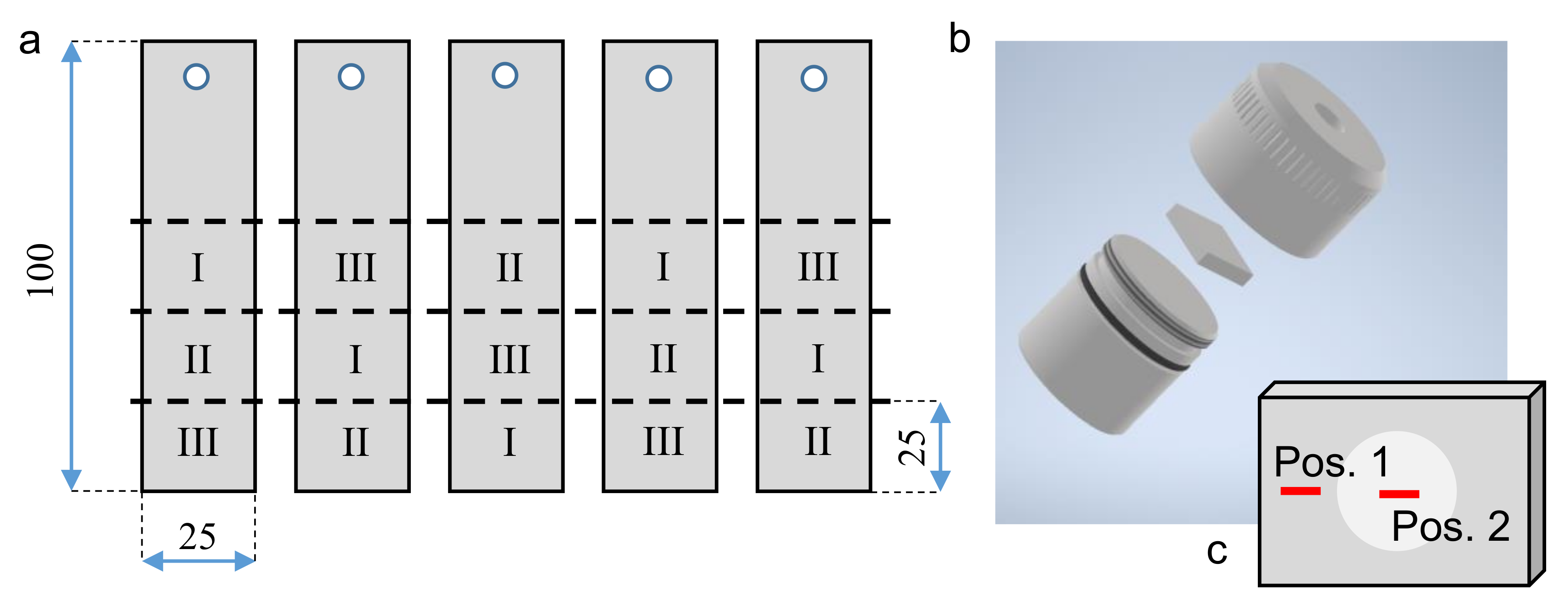
In order to provide the data resulting from the following investigations with a suitable statistical support, five aluminum sheets were treated under the same PEO conditions. Afterwards, three samples were cut from every sheet according to the schematic representation depicted in
Figure 1a.
Figure 1.
Schematic representation of the PEO-coated aluminum sheets, extraction positions of the individual samples and indication of the electrolytes (
Table 2) used for exposition experiments (
a) within a sample holder (
b) as well as the position of the further SEM and EBSD investigations (
c).
Figure 1.
Schematic representation of the PEO-coated aluminum sheets, extraction positions of the individual samples and indication of the electrolytes (
Table 2) used for exposition experiments (
a) within a sample holder (
b) as well as the position of the further SEM and EBSD investigations (
c).
The 15 samples prepared in this way were mounted in sample holders as exemplary shown in
Figure 1b and used for exposition experiments within three different electrolytes, indicated by the Roman numerals. The compositions of these are listed in
Table 2.
For the exposition experiments, the mounted samples were placed into glasses with 140 mL of the respective pre-heated electrolyte. Afterwards, the glasses were closed with a watch glass, positioned in a thermostat filled with water of 95 C, and were exposed to the electrolyte for 60 min. In order to determine the exact chemical aluminum concentrations of the testing solutions before and after the experiments, test volumes of 50 mL were extracted and investigated by inductively coupled plasma optical emission spectroscopy (ICP-OES) using a Optima 8300 (PerkinElmer, Waltham, MA, USA). In the course of these measurements, three undiluted partial volumes were shot into the flame in order to analyze the spectral information of the resulting light emission for 5 to 20 s. The characteristic Aluminium wave lengths of 394.40 nm and 396.15 nm were used to determine the Al content by use of a N9300184-standard of Al(NO) in 2% HNO with 1000 µg/mL Al (PerkinElmer, Waltham, MA, USA). The phase composition of every single coated sample was examined by X-ray diffraction (XRD) before and after the exposition experiment. The setup consists of a D8 Discover with an 1D LYNXEYE XE-T detector with 192 channels (Bruker, Germany) using Co-K radiation. A point focus with a collimator diameter of 2 mm was chosen to gain integral information without interfering signals from the samples edges. A step size of 0.02° and a step time of 3.9 s/step, which results in an effective measurement step time of 748.8 s/step due to the use of the 1D detector.
The morphology of the coating, as well as the spatial distributions of the single alumina phases within the exposed and non-exposed coating regions (Positions 1 and 2 in
Figure 1c), were investigated by using a scanning electron microscopy (SEM) and electron backscattering diffraction (EBSD). The chemical composition of the observed microstructure was determined by energy-dispersive X-ray spectroscopy (EDX) spot measurements and maps. For this purpose, one of the samples exposed to each of the electrolytes I, II, and III were prepared as listed in
Table 3.
The samples exposed to electrolyte I or II were prepared without embedding, while the sample immersed in electrolyte III, which showed layer delamination, was vacuum infiltrated with an epoxy resin (Epothin, Buehler, IL, USA). Slices of 2 mm thickness were detached by using a diamond cutting disk and mounted in a thermoplastic sample holder (Crystalbond, Buehler, IL, USA). Afterwards, the surfaces were grinded in five steps with increasing abrasive particle number. The following polishing steps were carried out on silk tie (MD-Dur, Struers, Willich, Germany), stretched on a steel disk. The resulting counterbody is relatively hard, and its use prevents roundings of the ceramic edge at the substrate/coating interface (which could be observed, for example, when using flow tie). In order to allow for Si-specific element maps during the subsequent investigations, diamond (instead of SiO) suspensions with decreasing particle size were used as abrasive media. The polishing and vibration polishing steps with 1 and 0.05 µm diamond particle size were carried out on neoprene tie (MD-Chem, Struers, Willich, Germany), the narrow mesh size of which allows to bind such small particles. After the sample surface was prepared for EBSD measurements in this way, it was dismounted from the sample holder and purified for investigations under vacuum conditions. Therefore, it was immersed in acetone and treated by ultrasound to dissolve the thermoplastic residuals. The procedure was repeated five times, each time using fresh acetone to gradually decrease the contamination of the sample surface. Since acetone itself showed a certain tendency to adsorb at the surfaces of the sample, they were afterwards purified in isopropyl following the same procedure. Since the typical micro- and nano sized defect structure of PEO coatings results in an intrinsic capillary hygroscopy, the samples were dried for 24 h under vacuum at 60 C. Afterwards, an electrical conductive layer was sputtered onto the sample surface. The Kikuchi lines showed an unsatisfying band contrast by use of carbon layers, which can be attributed to their excessive thickness and uneven thickness distribution. Therefore, thin layers of gold, invisible to the naked eye, were applied.
The micro-structure analysis was carried out by using a field-emission SEM (NEON 40EsB, Zeiss, Jena, Germany) equipped with an EDX (GENESIS, EDAX, Mahwah, NJ, USA) and EBSD system (OIM 5.31, EDAX TSL, Mahwah, NJ, USA). The SEM micrographs were taken in backscatter (BS) and secondary (SE) electron mode using an acceleration voltage of 15 kV and a working distance of 10 mm. For the EBSD measurements, the acceleration voltage was 15 kV. The samples were tilted by 70°, and the aperture was opened to 120 µm in high current mode. The sampling step size was set to 150 nm, while the sampling time was limited to some seconds in order to prevent a deterioration of the EBSD data as a result of local charging (leading to electron-beam drift) and contamination. During this measurement, the parameters of which were optimized for electron backscattering diffraction, EDX data were recorded as well. They allow for a qualitative representation of the spatial distribution of the chemical elements detected. However, the short spot measurement prohibits a quantitative interpretation.
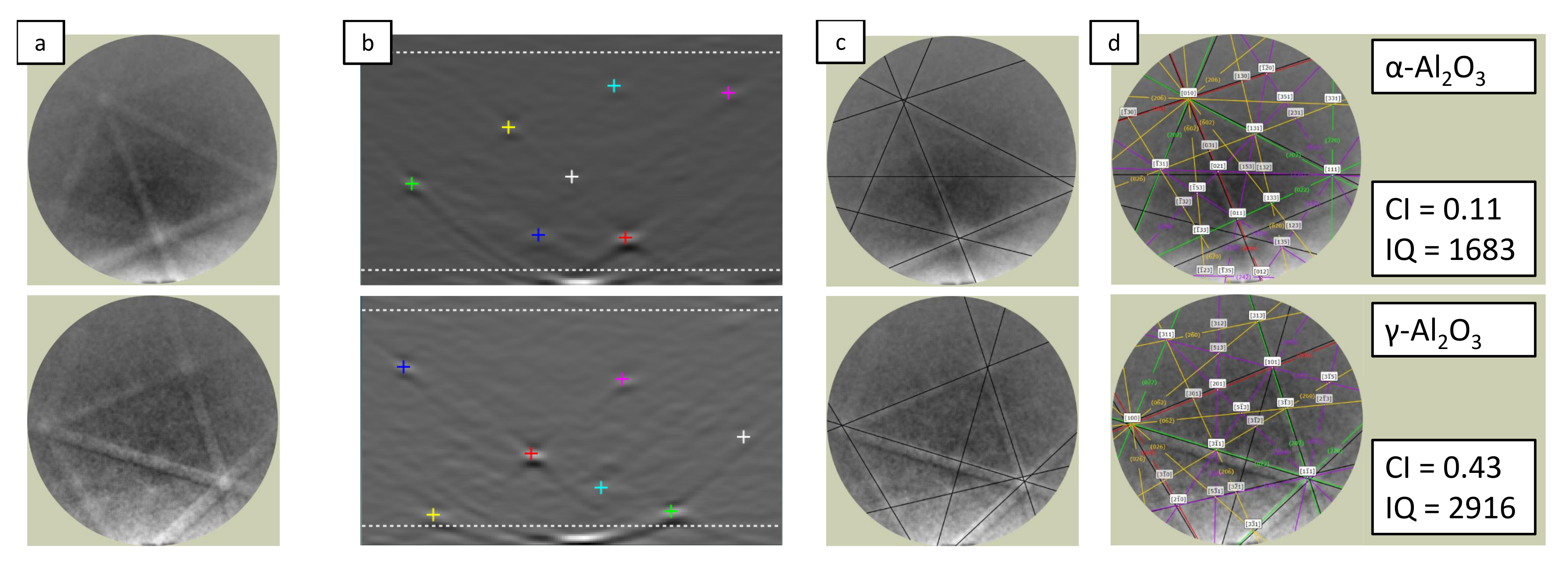
Figure 2 summarizes the procedure used for processing and indexing the data obtained by the EBSD measurement. The raw signal intensity map with diffraction bands visible for the naked eye,
Figure 2a, was transferred by using Hough transform into an dual space shown in
Figure 2b. Here, seven local maxima, indicated by colored markers, were used to identify the main characteristic diffraction lines of the spot under investigation, depicted in
Figure 2c. Afterwards, the OIM software was used to assign those incomplete Kikuchi patterns to one of the two crystalline aluminum oxide modifications
- and
-Al
O
previously identified by XRD. The corresponding complete Kikuchi patterns are presented in
Figure 2d. This combined use of diffraction-based material-analytic methods allows for an EBSD phase map, despite the confidence index (CI) being relatively low (between 0.1 and 0.5) in this study. The CI value quantifies, via a complex algorithm, how well the detected diffraction pattern matches the indexed diffraction pattern [
31,
32]. As a side effect of this procedure, some grains of the aluminum-substrate alloy were indexed as alumina phases as well, which has to be considered negligible artifacts of this methodology.
Figure 2.
Graphical representation of the procedure used for indexing EBSD data, showing the raw signal intensity map with diffraction bands (a), the dual space resulting from the Hough transform with colored markers (b), indicating the characteristic diffraction lines of the spot under investigation (c), which were used to assign the data to the characteristic Kikuchi pattern of - (top) or -AlO (button) (d).
Figure 2.
Graphical representation of the procedure used for indexing EBSD data, showing the raw signal intensity map with diffraction bands (a), the dual space resulting from the Hough transform with colored markers (b), indicating the characteristic diffraction lines of the spot under investigation (c), which were used to assign the data to the characteristic Kikuchi pattern of - (top) or -AlO (button) (d).
During the EBSD measurement, regions were identified in which only a few to no diffraction bands were present, which showed only a low band contrast. This leads to a reduction of the peak number and strength in the Hough space and is quantified by the so-called image quality (IQ) [
31]. In addition to numerous influencing factors on the part of preparation (topography, artefacts, edges) and the experimental setup (acceleration voltage, inclination angle, spot measurement time), the IQ is influenced by various structural features (residual stresses, distortions, dopants) along the area under investigation.
Furthermore, a low IQ can indicate amorphous areas or zones with a low crystalline order. Therefore, areas that did not allow for clear assignment to the crystalline aluminum-oxide modifications determined by XRD have been categorized into three subclasses in this work.
- i
IQ = 0–500: no band contrast, no pattern detectable, areas with practical no signal response, cavities, edges, amorphous zones
- ii
IQ ≥ 500–750: very low band contrast, weakly delectable diffraction patterns, no indexing possible
- iii
IQ ≥ 750–1000: low band contrast, detectable diffraction patterns, no phase assignable
4. Discussion
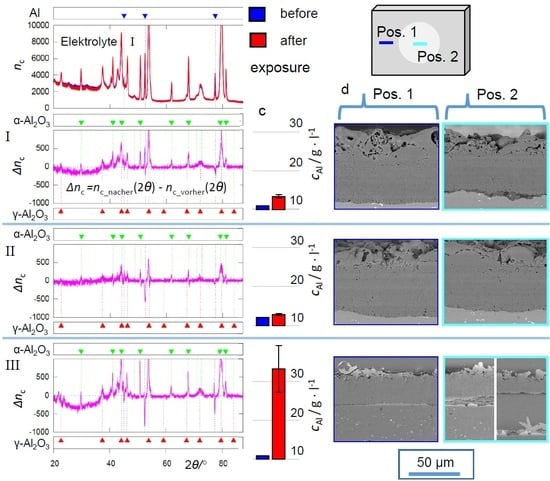
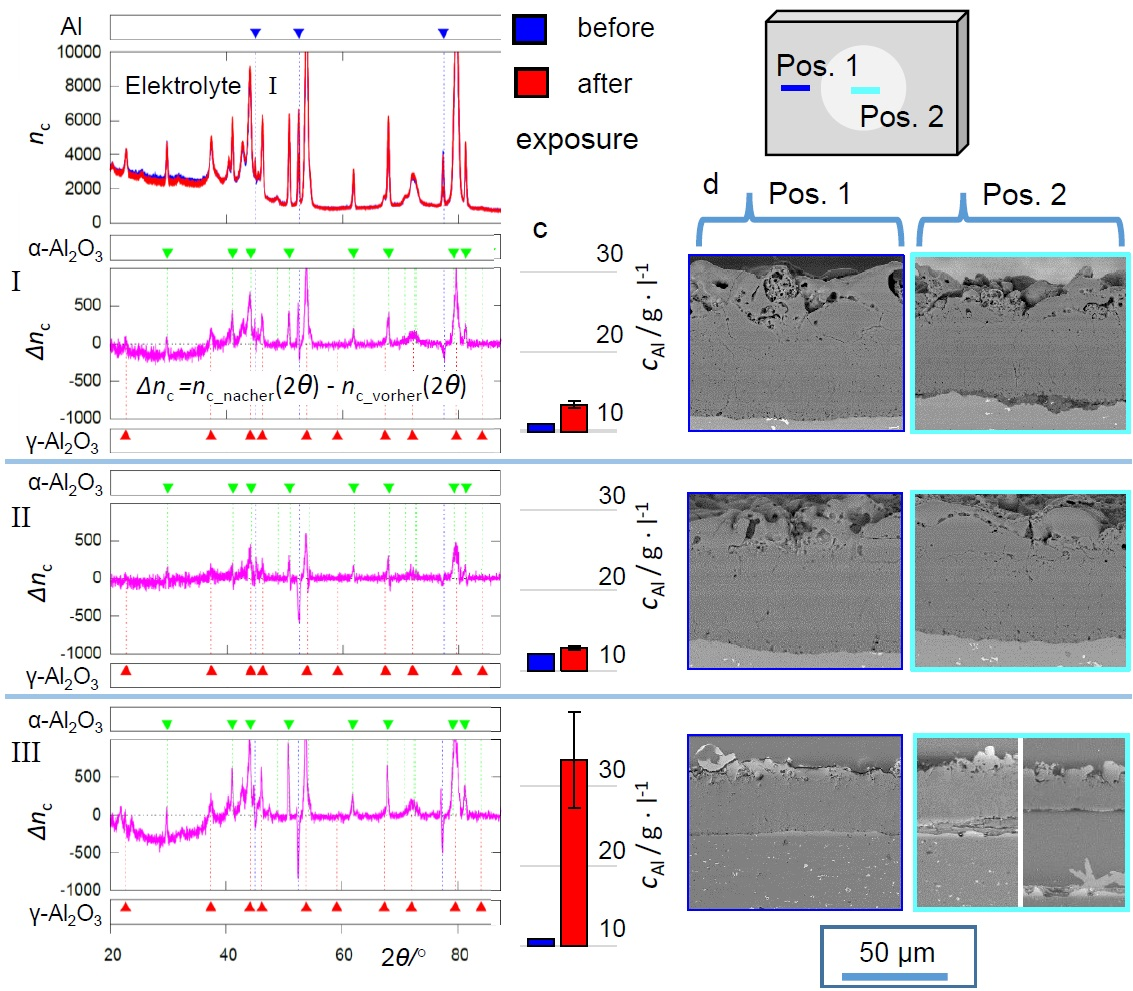
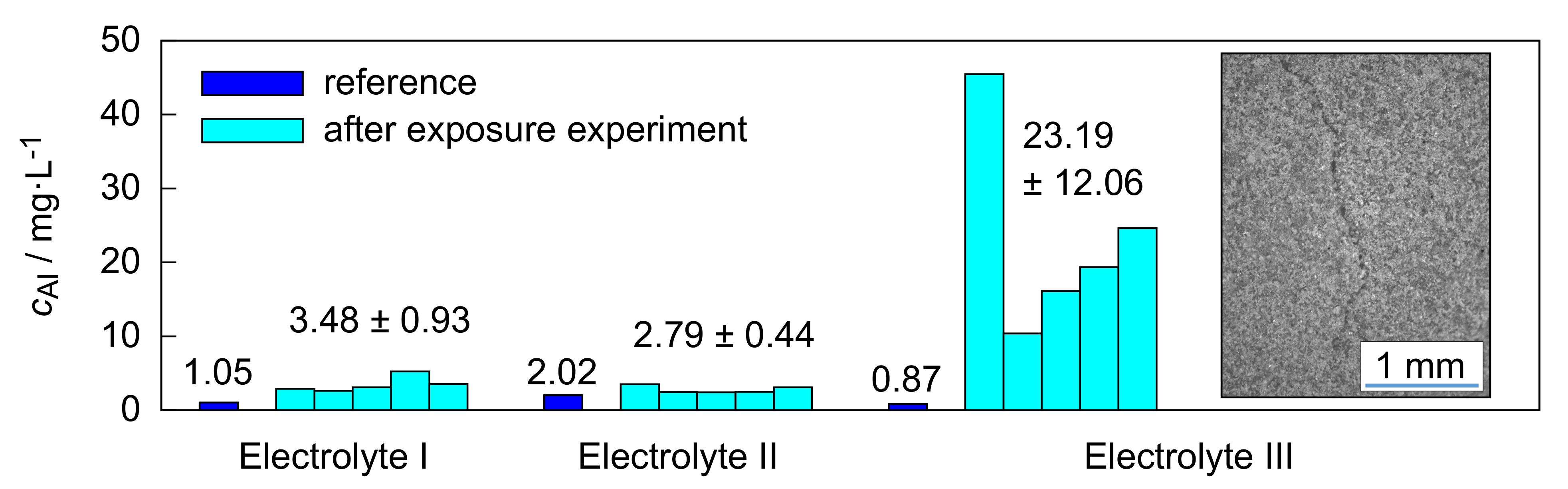
The ICP measurements, represented in
Figure 3, as well as the micrographs summarized in
Figure 5,
Figure 8 and
Figure 11 show that the exposition to electrolyte II barely affected the PEO coating, while electrolyte I and III lead to Al release by dissolution of the substrate/coating interface and the substrate itself. The dissolution of the substrate/coating interface can be attributed to reactions according to Equation (
2). The observed substrate attack can be interpreted as reactions according to Equation (
4). Since these processes require hydroxide ions, the effects were more pronounced in electrolyte III with a pH of 14 than in electrolyte I with a pH of 13. The silicate content of electrolyte II (pH ≈ 13,
= 5 g/L) suppressed the above-mentioned reactions by passivation. Furthermore, the spatially highly localized occurrence of the dissolution processes allows for the following conclusions. On the one hand, it is proven that the present intrinsic cavity network of the fully formed PEO coating still allows the electrolyte access to the layer/substrate interface. This supports the assumption that there is an electrochemically active zone in this area over the entire process time, the characteristics of which influence the PEO to a decisive extent [
34,
35,
36]. On the other hand, it becomes clear that an oxide modification must be present at the substrate/layer interface in and above the passive film, which is more prone to chemical dissolution than the oxide of the unaffected bulk coating.
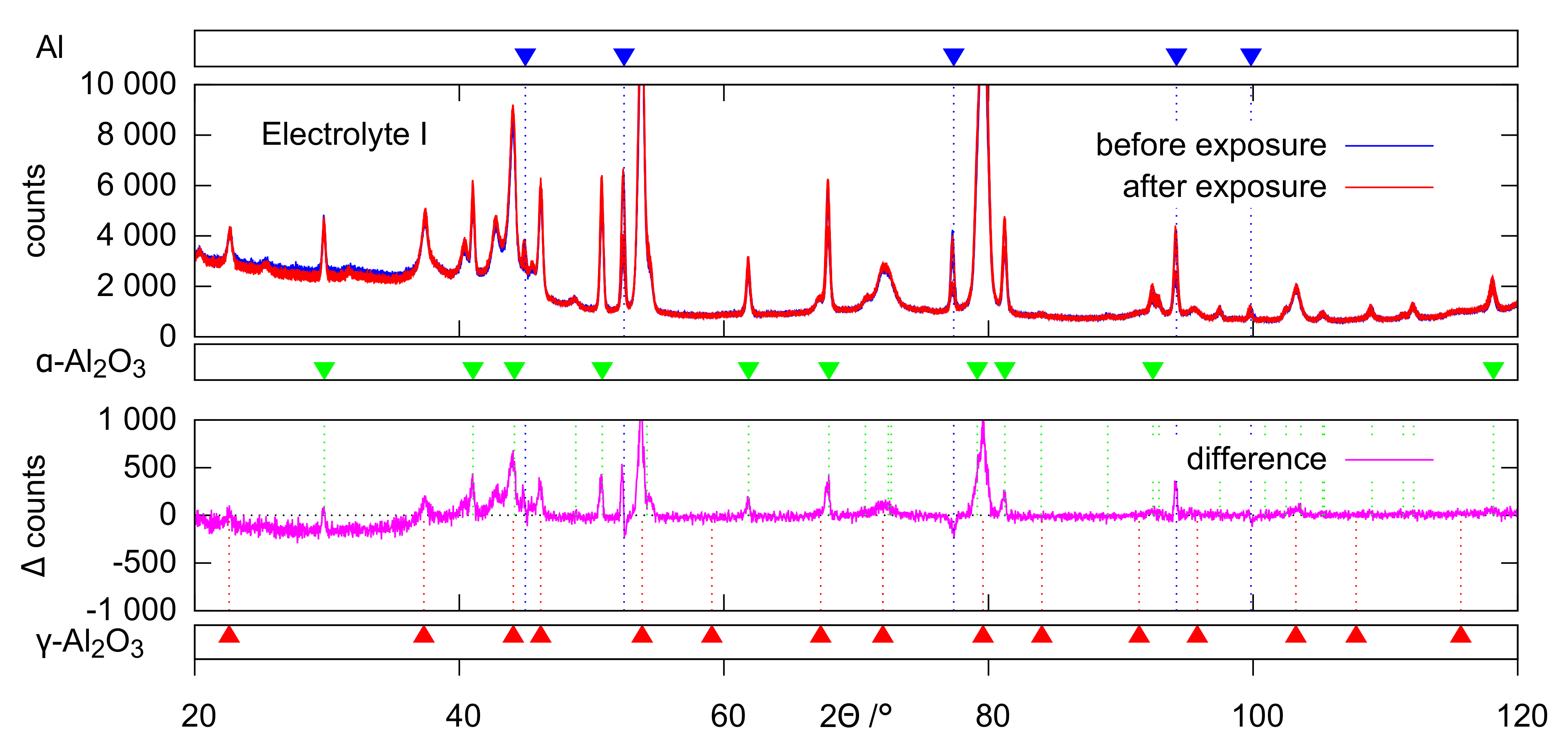
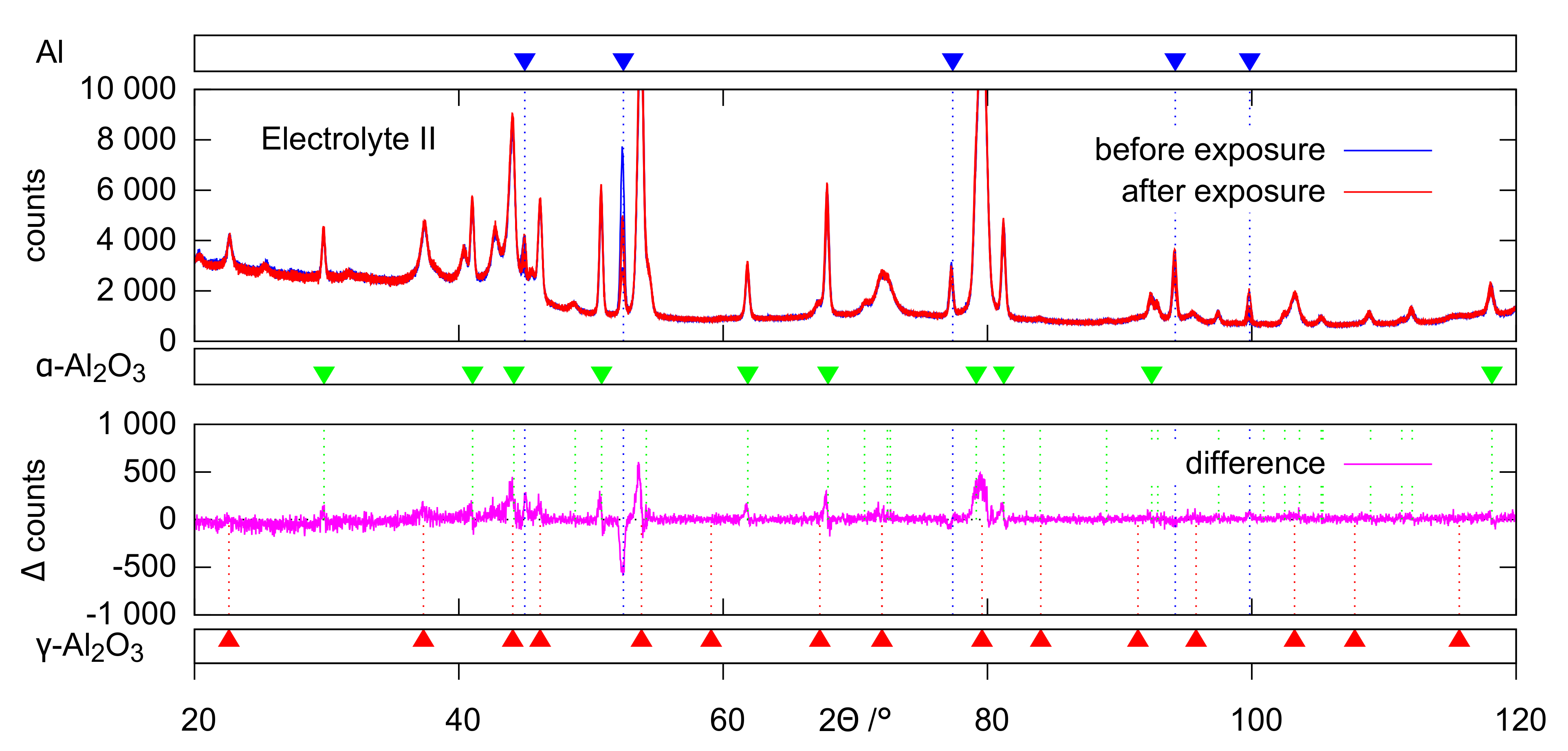
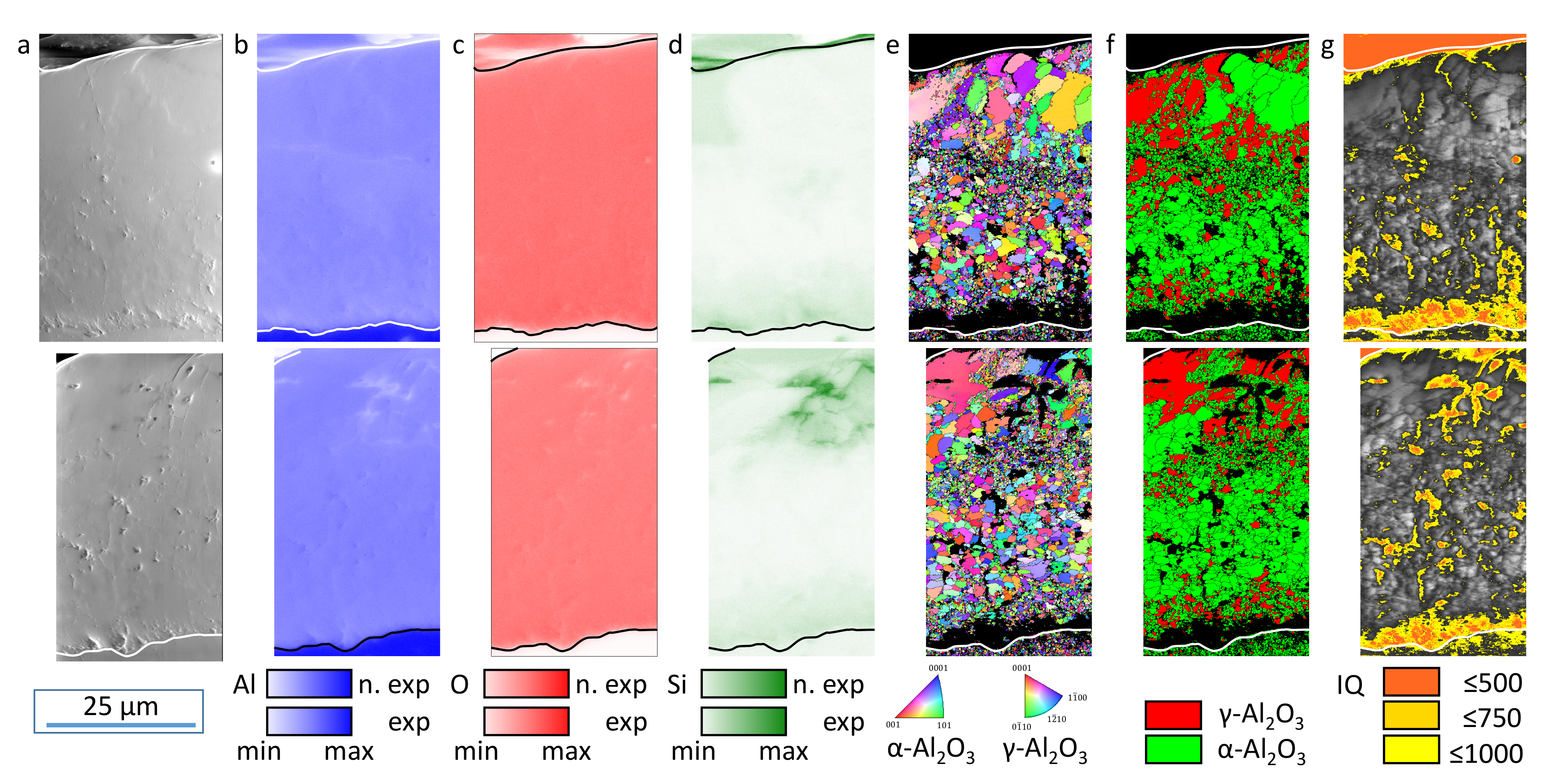
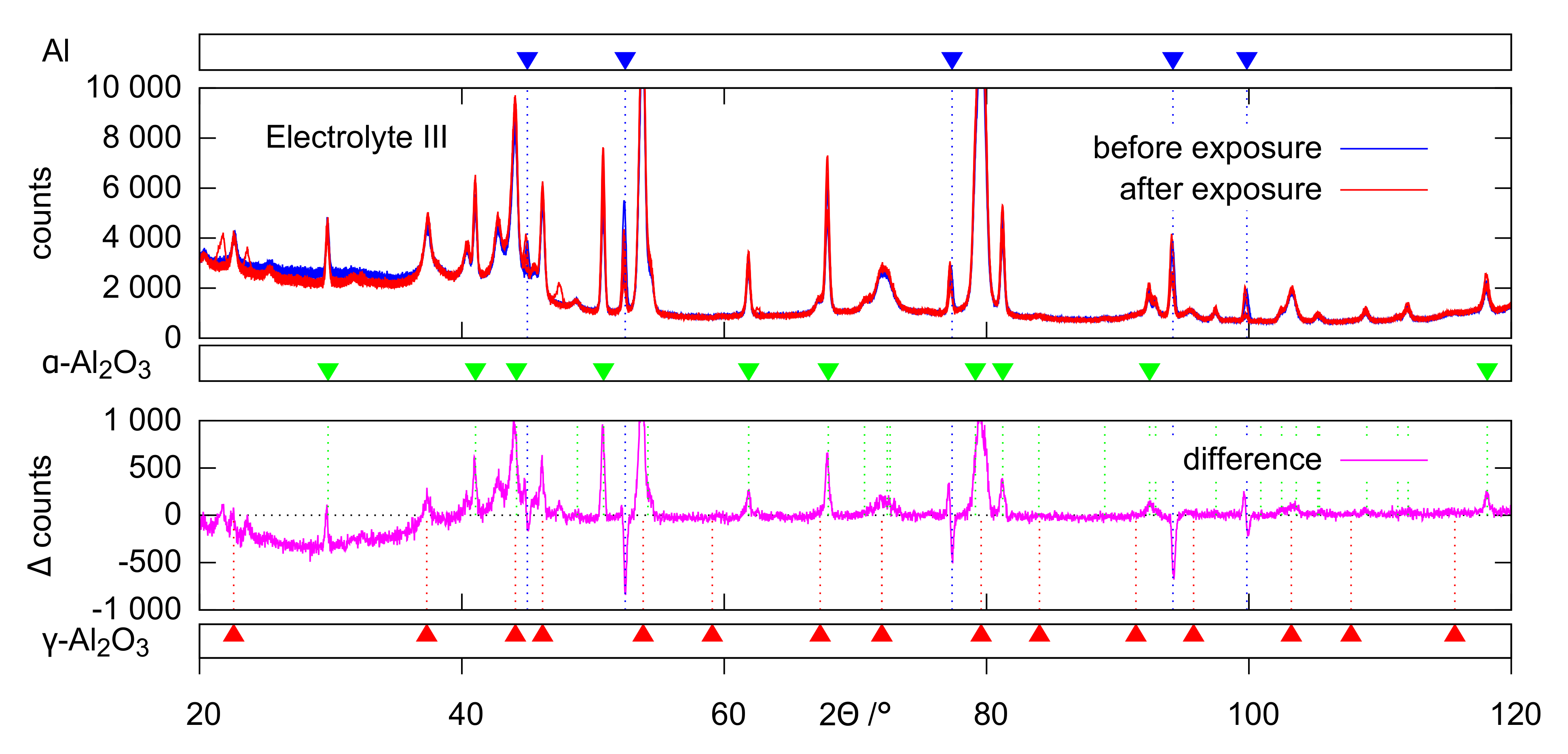
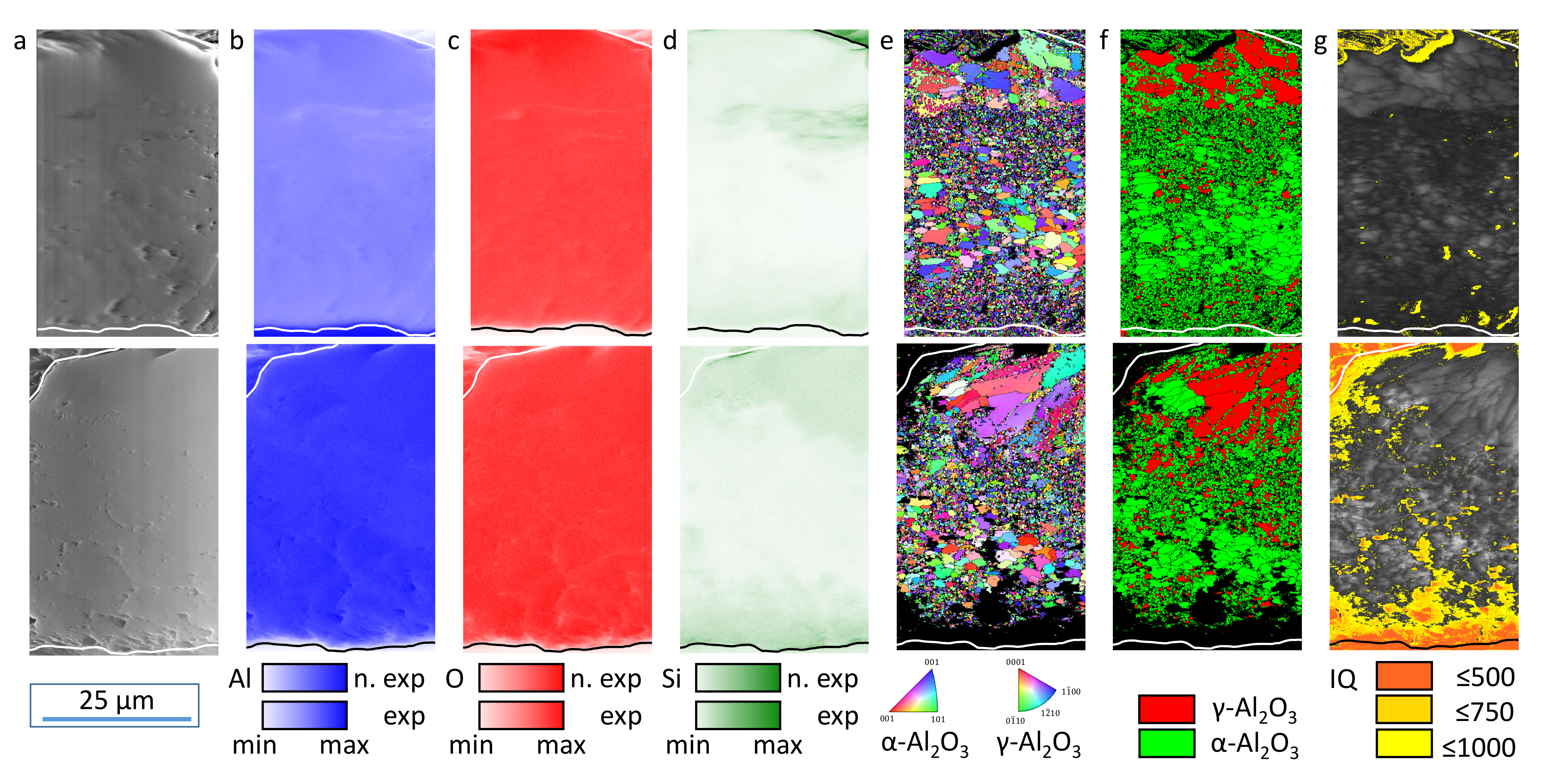
The evaluation of the XRD (
Figure 4,
Figure 7 and
Figure 10) and EBSD (
Figure 6,
Figure 9 and
Figure 12) data provides a conclusive explanation for this. The occurrence of the characteristic deflections in the purple intensity difference curves of the diffractograms is to be interpreted as the disappearance of amorphous parts after the exposition experiments. Accordingly, the corresponding non-crystalline oxide phases are affected preferentially by dissolution. The extent of this effect follows the same pattern that has already become clear from the ICP measurements and SEM images: hardly any oxide dissolution in electrolyte II, significant dissolution in electrolyte I, which is even stronger in electrolyte III. These observations can likewise be explained by increased dissolution of (amorphous) alumina at increased pH and inhibition of this reaction in the presence of passivating silicate constituents. The leaching of the amorphous coating parts leads to a slight relative increase of the generally much more stable crystalline oxide modifications. This directs to positive deflections in the 2
range of the crystalline peaks in the intensity difference curves. Since this peak’s superelevations are more pronounced for
-Al
O
than for
-Al
O
, at least the XRD data indicate that
-Al
O
is slightly affected by dissolution as well.
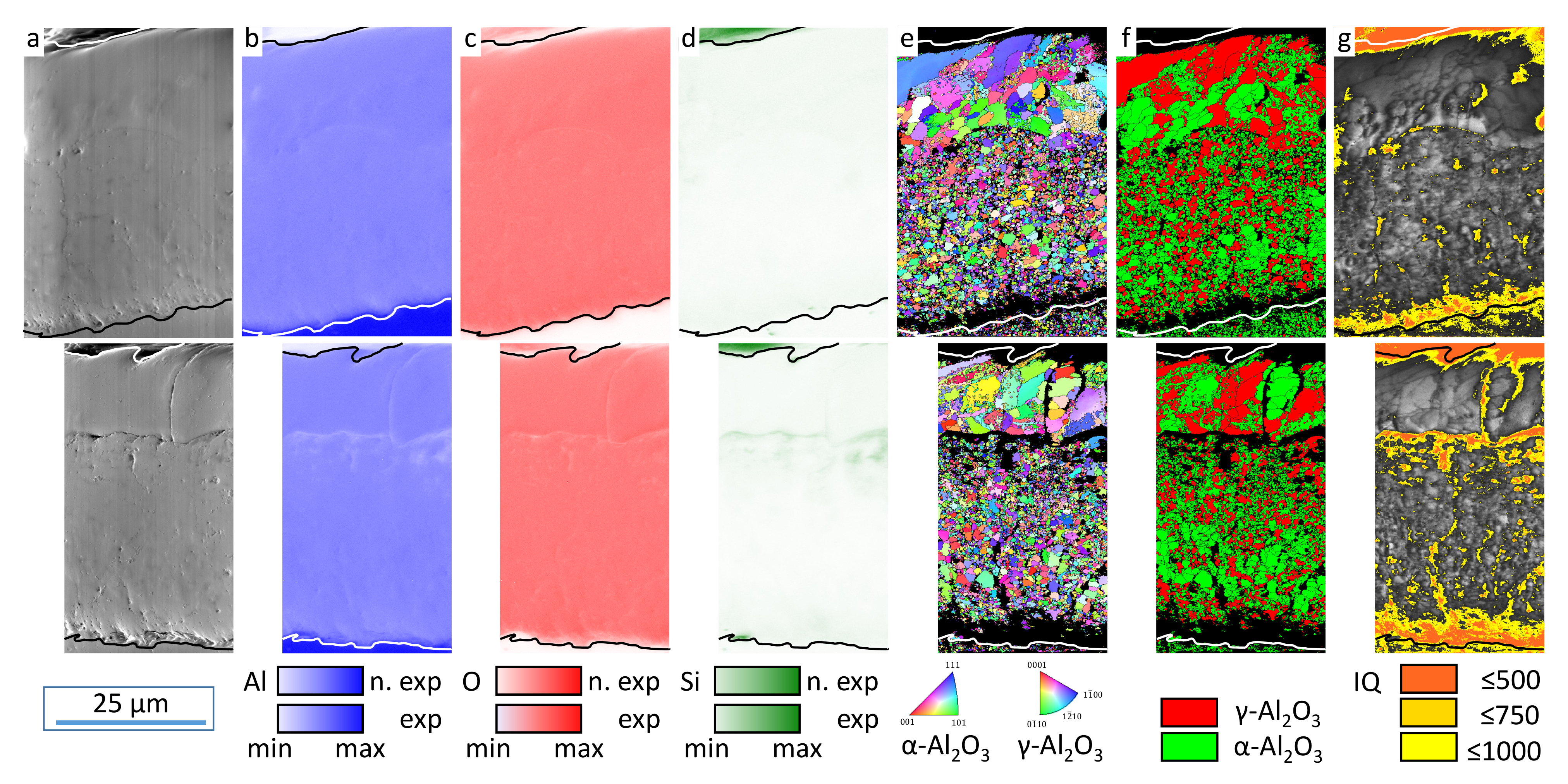
However, this observation cannot be supported based on the EBSD data. The phase and IQ maps give a good overview regarding the spatial distribution of the oxide modifications. However, their statistical reliability is too low for allowing a statement as to whether the volume ratio of the crystalline oxides has changed as a result of the exposition experiments. Nevertheless, the element, EBSD, and IQ maps in
Figure 6 and
Figure 9 clearly show a zone of non (IQ < 500) and weakly (IQ 500–1000) crystalline oxide above the substrate/coating interface with an extension of some micrometers before the exposition experiments. This amorphous alumina can be attributed to be the freshest electrochemical formed oxide before the plasma-chemical phase-transformation processes according to Equation (
1). The actual bonding character within the present Al
O
modifications, as well as a conceivable gradation between the layer/substrate interface and the crystalline layer regions, could be elucidated in course of further investigations by X-ray photoelectron spectroscopy (XPS). While the extension of the non- and weakly crystalline zones remains unchanged after exposition to electrolyte II (
Figure 6g), the area of low IQ is increased and oblong zones are added after exposition to electrolyte I (
Figure 9g). For electrolyte III, this effect is more pronounced (
Figure 12g). Therefore, it can be concluded that the amorphous oxide components, whose disappearance is proven by the deflection of the purple intensity difference curve in the X-ray diffractogram, were indeed localized directly above the layer/substrate interface. Additional areas of low IQ were created by leaching these amorphous or weakly crystalline oxides and etching the surrounding crystalline oxides. This is also in accordance with the corresponding SEM images.
The elevated Si content in porous coating zones and the vicinity of the layer/substrate interface can be explained by electrolyte residues in layer cavities and the contribution of the silicate components to the passivation reactions [
13]. It is an interesting observation that within the crystalline oxide modifications, Si is preferentially bound in
-Al
O
but rarely in
-Al
O
. However, no significant influence on the chemical dissolution of these oxides could be observed within this study. The effect is very small, if any, since the crystalline modifications have proven to be chemically stable in general. The exposition experiments used in this study do not fully represent the conditions during PEO in that the high electric fields actually present were not applied. However, since the electric permittivity of crystalline alumina is about 10 times that of aqueous solutions, electric field lines would have been localized along the cavity network around the crystalline regions and in the layer/substrate interface. Therefore, effects of a field-assisted chemical dissolution according to [
22,
23] would have increased the selective dissolution of amorphous alumina, if at all.
The observations regarding substrate-related XRD peaks within the intensity difference curves summarized in
Table 4 could not be clearly explained within this study and require further investigations. A changed state of internal stress as a result of the continuous or partial layer delamination, which would cause a peak shift, would be conceivable. However, the deviations in the peak positions are comparably small. The changed height of the substrate material as a result of the formation of etch pits could also have influenced the angular position of the peaks. No clear interpretation is possible at this point. Further investigations are neccessary in order to clarify the observation.

















{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}