
Chemical Vapor Deposition of Zirconium Compounds: A Review

{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
Abstract
:1. Introduction
1.1. Chemical Vapor Deposition
1.2. Chemistry of Zirconium Compounds
2. Metallic Zirconium
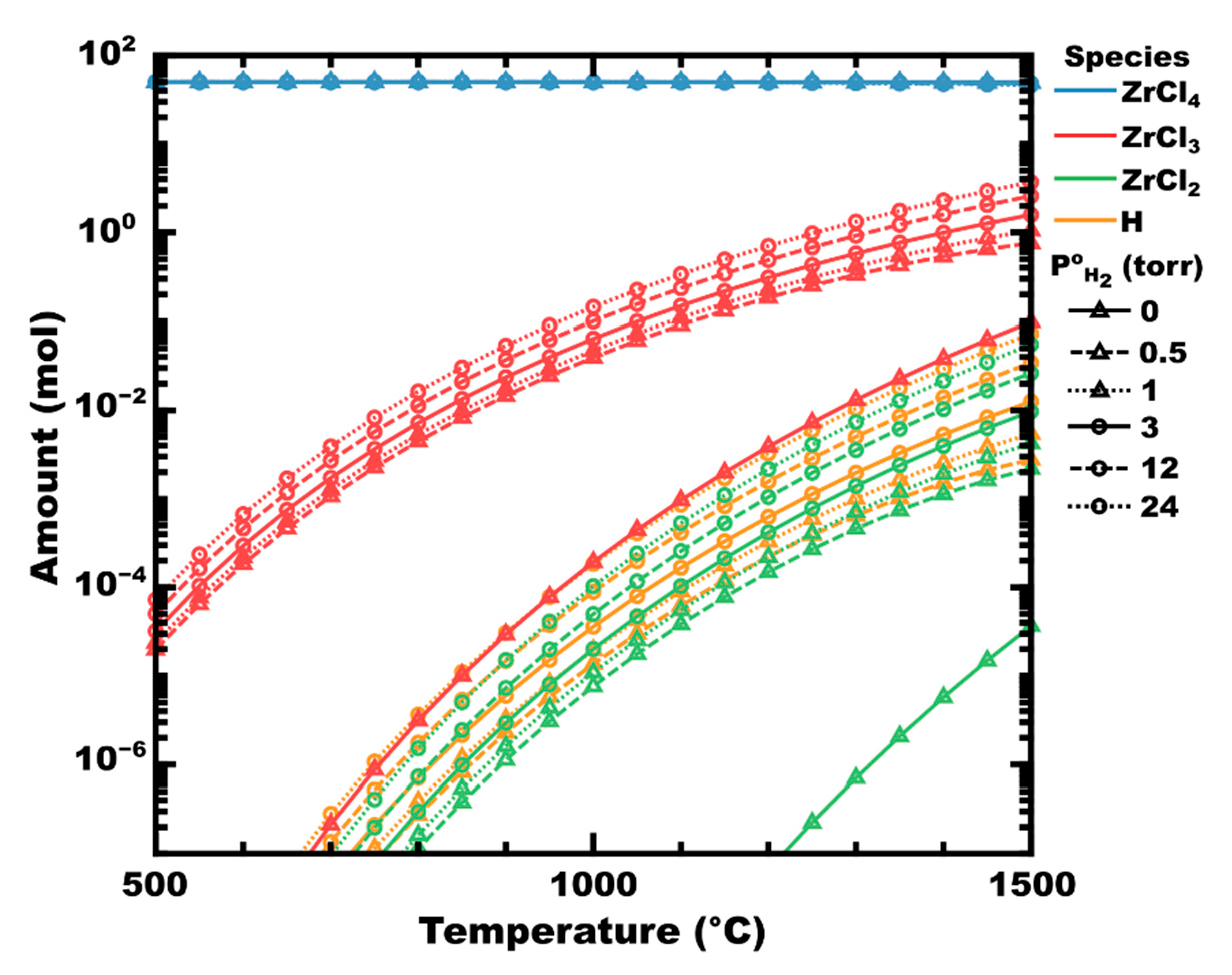
Deposition from Zirconium Halides
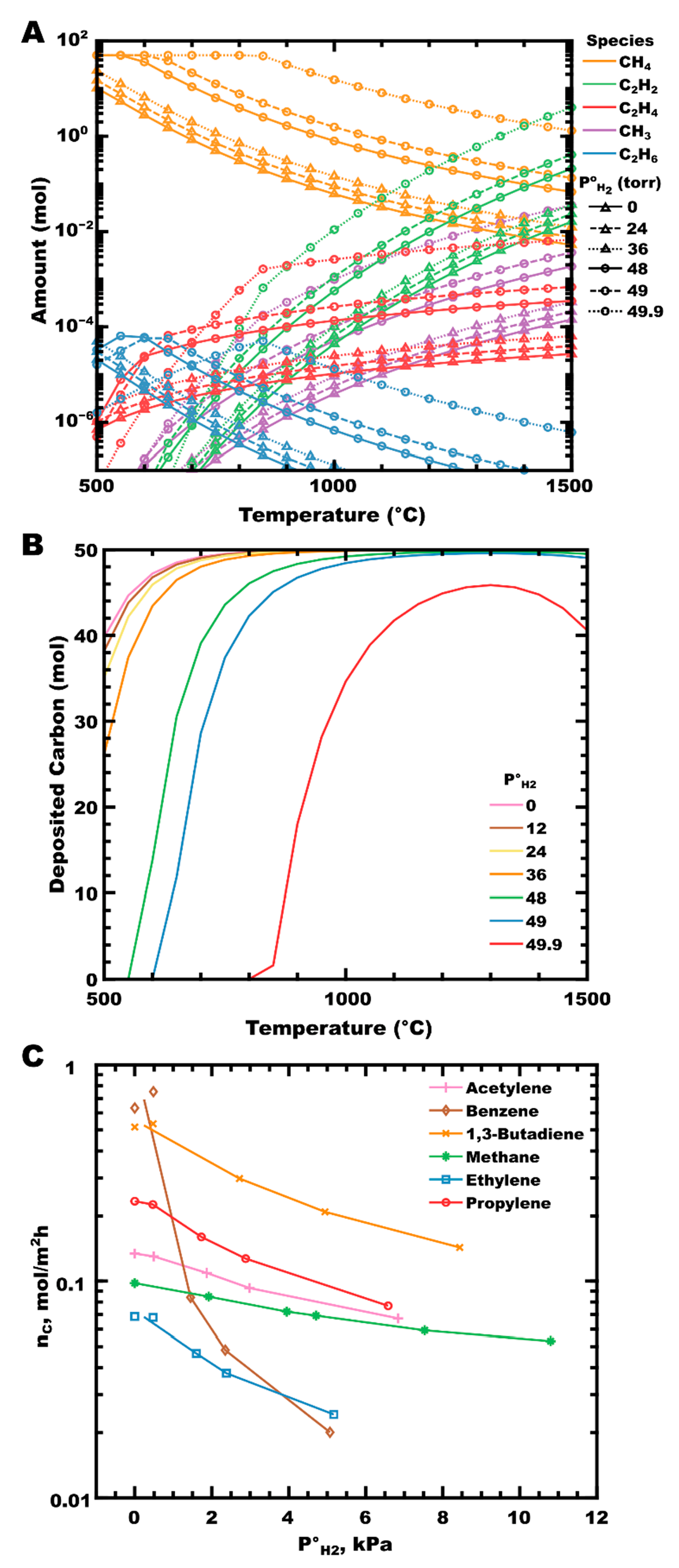
3. Zirconium Carbide
3.1. Diffusion Mechanism
3.2. Droplet Mechanism
3.3. The Role of Hydrogen
4. Zirconium Nitride
4.1. Inorganic CVD
4.2. Organometallic CVD
5. Zirconium Dioxide
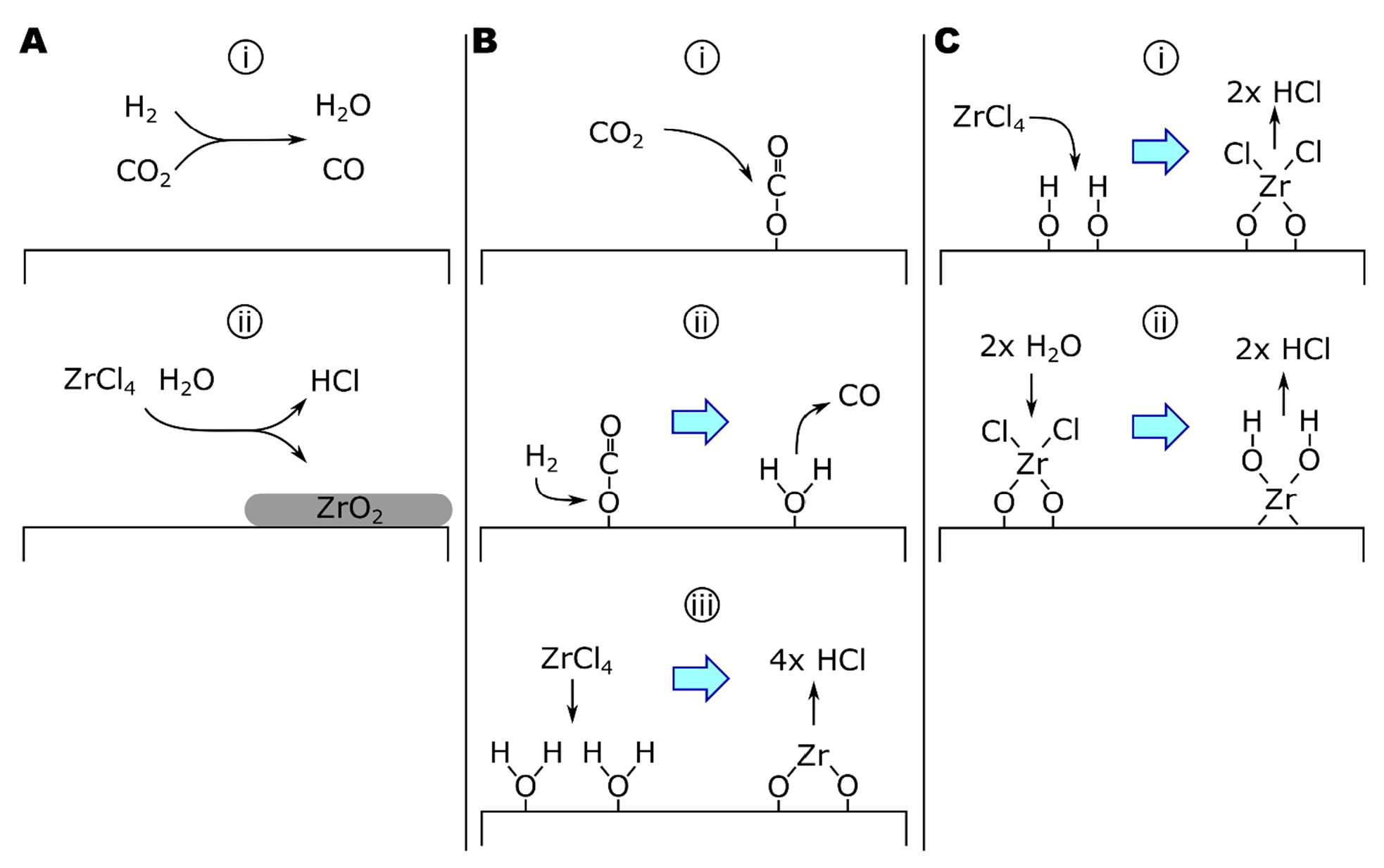
5.1. ZrCl4-CO2-H2
5.2. ZrCl4-H2O
6. Zirconium Diboride
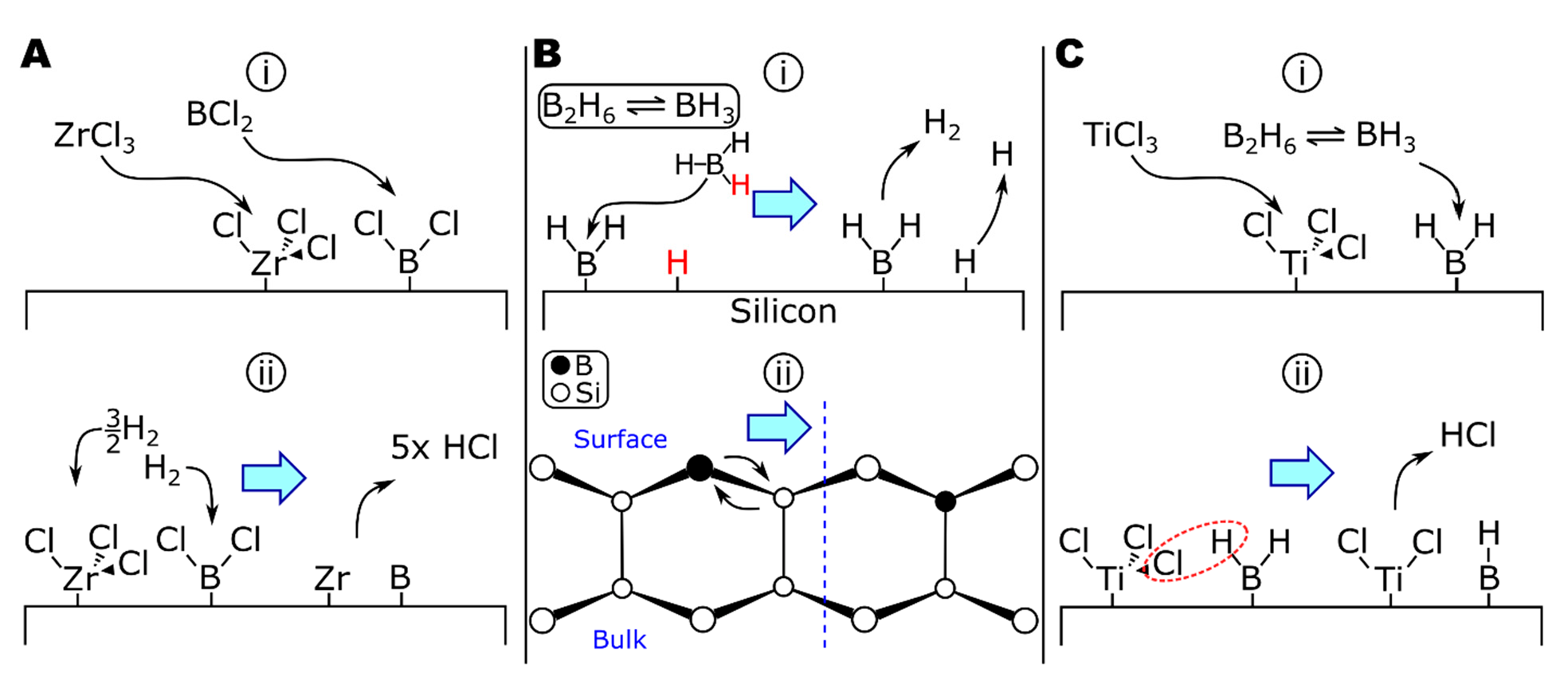
6.1. ZrB2 from Independent Precursors
6.2. ZrB2 from Single-Source Precursor
7. Zirconium Silicides
8. Conclusions
Author Contributions
Funding
Institutional Review Board Statement
Informed Consent Statement
Data Availability Statement
Acknowledgments
Conflicts of Interest
References
- Campbell, I.E.; Powell, C.F.; Nowicki, D.H.; Gonser, B.W. The Vapor-Phase Deposition of Refractory Materials. J. Electrochem. Soc. 1949, 96, 318. [Google Scholar] [CrossRef]
- Lemaignan, C. Zirconium Alloys: Properties and Characteristics. In Material Properties/Oxide Fuels for Light Water Reactors and Fast Neutron Reactors, 2nd ed.; Allen, T.R., Stoller, R.E., Yamanaka, S., Konings, R.J.M., Eds.; Comprehensive Nuclear Materials; Elsevier Ltd.: Amsterdam, The Netherlands, 2012; pp. 217–232. [Google Scholar]
- Ogata, T. Metal Fuel. In Advanced Fuels/Fuel Cladding/Nuclear Fuel Performance Modeling and Simulation, 2nd ed.; Allen, T.R., Stoller, R.E., Yamanaka, S., Konings, R.J.M., Eds.; Comprehensive Nuclear Materials; Elsevier Ltd.: Amsterdam, The Netherlands, 2012; pp. 1–40. [Google Scholar]
- Wooding, S.J.; Bacon, D.J. A molecular dynamics study of displacement cascades in α-zirconium. Philos. Mag. A 1997, 76, 1033–1051. [Google Scholar] [CrossRef]
- Bacon, D.J.; Gao, F.; Osetsky, Y.N. The primary damage state in fcc, bcc and hcp metals as seen in molecular dynamics simulations. J. Nucl. Mater. 2000, 276, 1–12. [Google Scholar] [CrossRef]
- Voskoboinikov, R.E.; Osetsky, Y.N.; Bacon, D.J. Statistics of primary damage creation in high-energy displacement cascades in copper and zirconium. Nucl. Instrum. Methods Phys. Res. Sect. B Beam Interact. Mater. At. 2006, 242, 68–70. [Google Scholar] [CrossRef]
- Stoller, R.E. Primary Radiation Damage Formation. In Basic Aspects of Radiation Effects in Solids/Basic Aspects of Multi-Scale Modeling, 2nd ed.; Allen, T.R., Stoller, R.E., Yamanaka, S., Konings, R.J.M., Eds.; Comprehensive Nuclear Materials; Elsevier Ltd.: Amsterdam, The Netherlands, 2012; pp. 293–332. [Google Scholar]
- Yamanaka, S.; Yamada, K.; Kurosaki, K.; Uno, M.; Takeda, K.; Anada, H.; Matsuda, T.; Kobayashi, S. Characteristics of zirconium hydride and deuteride. J. Alloy. Compd. 2002, 330–332, 99–104. [Google Scholar] [CrossRef]
- Yamanaka, S.; Yamada, K.; Kurosaki, K.; Uno, M.; Takeda, K.; Anada, H.; Matsuda, T.; Kobayashi, S. Thermal properties of zirconium hydride. J. Nucl. Mater. 2001, 294, 94–98. [Google Scholar] [CrossRef]
- Yamanaka, S.; Yoshioka, K.; Uno, M.; Katsura, M.; Anada, H.; Matsuda, T.; Kobayashi, S. Isotope effects on the physicochemical properties of zirconium hydride. J. Alloy. Compd. 1999, 293–295, 908–914. [Google Scholar] [CrossRef]
- Olander, D.R.; Konashi, K.; Yamawaki, M. Uranium-Zirconium Hydride Fuel. In Advanced Fuels/Fuel Cladding/Nuclear Fuel Performance Modeling and Simulation, 2nd ed.; Allen, T.R., Stoller, R.E., Yamanaka, S., Konings, R.J.M., Eds.; Comprehensive Nuclear Materials; Elsevier Ltd: Amsterdam, The Netherlands, 2012; pp. 313–357. [Google Scholar]
- Minato, K.; Ogawa, T. Advanced Concepts in TRISO Fuel. In Advanced Fuels/Fuel Cladding/Nuclear Fuel Performance Modeling and Simulation, 2nd ed.; Allen, T.R., Stoller, R.E., Yamanaka, S., Konings, R.J.M., Eds.; Comprehensive Nuclear Materials; Elsevier Ltd.: Amsterdam, The Netherlands, 2012; pp. 216–236. [Google Scholar]
- Wagner, P. High-Temperature Fuel Technology for Nuclear Process Heat: ZrC-Containing Coated Particle Fuels and High-Density Graphite Fuel Matrices; LA-6984; Los Alamos Scientific Laboratory: Los Alamos, NM, USA, 1977.
- Arai, Y. Nitride Fuel. In Advanced Fuels/Fuel Cladding/Nuclear Fuel Performance Modeling and Simulation, 2nd ed.; Allen, T.R., Stoller, R.E., Yamanaka, S., Konings, R.J.M., Eds.; Comprehensive Nuclear Materials; Elsevier Ltd.: Amsterdam, The Netherlands, 2012; pp. 41–54. [Google Scholar]
- Arai, Y.; Minato, K. Fabrication and electrochemical behavior of nitride fuel for future applications. J. Nucl. Mater. 2005, 344, 180–185. [Google Scholar] [CrossRef]
- Canel, J.; Zaman, J.; Bettembourg, J.; Flem, M.; Poissonnet, S. Composite Zirconium Silicides Through an In Situ Process. Int. J. Appl. Ceram. Technol. 2006, 3, 23–31. [Google Scholar] [CrossRef]
- Cheol Lee, G.; Noh, H.; Yeom, H.; Jo, H.; Kyun Kim, T.; Kim, M.; Sridharan, K.; Sun Park, H. Zirconium-silicide coating on zircaloy-4 substrate for accident tolerance: Effects on oxidation resistance and boiling. Ann. Nucl. Energy 2019, 126, 350–358. [Google Scholar] [CrossRef]
- Yeom, H.; Lockhart, C.; Mariani, R.; Xu, P.; Corradini, M.; Sridharan, K. Evaluation of steam corrosion and water quenching behavior of zirconium-silicide coated LWR fuel claddings. J. Nucl. Mater. 2018, 499, 256–267. [Google Scholar] [CrossRef]
- Raison, P.E.; Haire, R.G. Structural investigation of the pseudo-ternary system AmO2–Cm2O3–ZrO2 as potential materials for transmutation. J. Nucl. Mater. 2003, 320, 31–35. [Google Scholar] [CrossRef]
- Poeml, P.; Konings, R.J.M.; Somers, J.; Wiss, T.; de Haas, G.J.L.M.; Klaassen, F.C. Inter Matrix Fuel. In Advanced Fuels/Fuel #ladding/Nuclear Fuel Performance Modeling and Simulation, 2nd ed.; Allen, T.R., Stoller, R.E., Yamanaka, S., Konings, R.J.M., Eds.; Comprehensive Nuclear Materials; Elsevier Ltd.: Amsterdam, The Netherlands, 2012; pp. 237–256. [Google Scholar]
- Eggers, G.H. Method of Making ZrH Fuel Element. US407 1587A, 1978.
- Murata, Y. Cutting Tool Tips and Ceramics Containing HfN and ZrB2. US348 7594A, 1970.
- Hintermann, H.E. Tribological and protective coatings by chemical vapour deposition. Thin Solid Film. 1981, 84, 215–243. [Google Scholar] [CrossRef]
- Hirose, M.; Yasui, T.; Ochi, Y.; Nakagawa, M. Method of Forming a Decorative Metallic Nitride Coating. US442 0498A, 1982.
- Johnson, P.C.; Randhawa, H. Zirconium nitride films prepared by cathodic arc plasma deposition process. Surf. Coat. Technol. 1987, 33, 53–62. [Google Scholar] [CrossRef]
- Johansson, B.O.; Sundgren, J.E.; Helmersson, U.; Hibbs, M.K. Structure of reactively magnetron sputtered Hf-N films. Appl. Phys. Lett. 1984, 44, 670–672. [Google Scholar] [CrossRef]
- Karlsson, B.; Shimshock, R.P.; Seraphin, B.O.; Haygarth, J.C. Optical properties of CVD-coated TiN, ZrN and HfN. Sol. Energy Mater. 1983, 7, 401–411. [Google Scholar] [CrossRef]
- Schlegel, A.; Wachter, P.; Nickl, J.J.; Lingg, H. Optical properties of TiN and ZrN. J. Phys. C Solid State Phys. 1977, 10, 4889–4896. [Google Scholar] [CrossRef]
- Namavar, F.; Wang, G.; Cheung, C.L.; Sabirianov, R.F.; Zeng, X.C.; Mei, W.N.; Bai, J.; Brewer, J.R.; Haider, H.; Garvin, K.L. Thermal stability of nanostructurally stabilized zirconium oxide. Nanotechnology 2007, 18, 415702. [Google Scholar] [CrossRef]
- Lepselter, M.P.; Andrews, J.M. Ohmic Contacts to Silicon. In Ohmic Contacts to Semiconductors, Schwartz, B., Ed.; The Electrochemical Society: New York, NY, USA, 1969; pp. 159–186. [Google Scholar]
- Roy, S.; Paul, A. Growth of hafnium and zirconium silicides by reactive diffusion. Mater. Chem. Phys. 2014, 143, 1309–1314. [Google Scholar] [CrossRef]
- Sung, J.; Goedde, D.M.; Girolami, G.S.; Abelson, J.R. Remote-plasma chemical vapor deposition of conformal ZrB2 films at low temperature: A promising diffusion barrier for ultralarge scale integrated electronics. J. Appl. Phys. 2002, 91, 3904–3911. [Google Scholar] [CrossRef] [Green Version]
- Sung, J.; Goedde, D.M.; Girolami, G.S.; Abelson, J.R. Diffusion Barrier Characteristics of Zirconium Diboride Films Grown by Remote Plasma CVD. MRS Proc. 1999, 563, 39–44. [Google Scholar] [CrossRef]
- Suni, I.; Mäenpää, M.; Nicolet, M.A.; Luomajärvi, M. Thermal Stability of Hafnium and Titanium Nitride Diffusion Barriers in Multilayer Contacts to Silicon. J. Electrochem. Soc. 2019, 130, 1215–1218. [Google Scholar] [CrossRef]
- Wittmer, M. Properties and microelectronic applications of thin films of refractory metal nitrides. J. Vac. Sci. Technol. A Vac. Surf. Film. 1985, 3, 1797–1803. [Google Scholar] [CrossRef]
- Mäenpää, M.; Suni, I.; Sigurd, D.; Finetti, M.; Nicolet, M.A. Stable Metallization Systems for Solar Cells. Phys. Status Solidi A 1982, 72, 763–769. [Google Scholar] [CrossRef]
- Tauber, R.N.; Dumbri, A.C.; Caffrey, R.E. Preparation and Properties of Pyrolytic Zirconium Dioxide Films. J. Electrochem. Soc. 1971, 118, 747. [Google Scholar] [CrossRef]
- Wilk, G.D.; Wallace, R.M.; Anthony, J.M. High-κ gate dielectrics: Current status and materials properties considerations. J. Appl. Phys. 2001, 89, 5243–5275. [Google Scholar] [CrossRef]
- Espinoza-Pérez, L.J.; López-Honorato, E.; González, L.A. Development of ZrO2 and YSZ coatings deposited by PE-CVD below 800 °C for the protection of Ni alloys. Ceram. Int. 2020, 46, 15621–15630. [Google Scholar] [CrossRef]
- Ohlhorst, C.W.; Glass, D.E.; Bruce, W.E., III; Lindell, M.C.; Vaugn, W.L.; Dirling, R.B., Jr.; Hogenson, P.A.; Nichols, J.M.; Risner, N.W.; Thompson, D.R. Development of X-43A Mach 10 Leading Edges. In Proceedings of the 56th International Astronautical Congress, Fukuoka, Japan, 17–21 October 2005. [Google Scholar]
- Fahrenholtz, W.G.; Hilmas, G.E. Oxidation of ultra-high temperature transition metal diboride ceramics. Int. Mater. Rev. 2012, 57, 61–72. [Google Scholar] [CrossRef]
- Fahrenholtz, W.G.; Hilmas, G.E. Ultra-high temperature ceramics: Materials for extreme environments. Scr. Mater. 2017, 129, 94–99. [Google Scholar] [CrossRef] [Green Version]
- Fahrenholtz, W.G.; Hilmas, G.E.; Talmy, I.G.; Zaykoski, J.A. Refractory Diborides of Zirconium and Hafnium. J. Am. Ceram. Soc. 2007, 90, 1347–1364. [Google Scholar] [CrossRef]
- Blumenthal, W.B. The Element, Zirconium. In The Chemical Behavior of Zirconium; D. van Nostrand Co. Inc.: Princeton, NJ, USA, 1958; pp. 1–45. [Google Scholar]
- Stojilovic, N.; Bender, E.T.; Ramsier, R.D. Surface chemistry of zirconium. Prog. Surf. Sci. 2005, 78, 101–184. [Google Scholar] [CrossRef]
- Xu, L.; Xiao, Y.; van Sandwijk, A.; Xu, Q.; Yang, Y. Production of nuclear grade zirconium: A review. J. Nucl. Mater. 2015, 466, 21–28. [Google Scholar] [CrossRef]
- Lustman, B.; Kerze, F.J. (Eds.) The Metallurgy of Zirconium, 1st ed.; McGraw-Hill: New York, NY, USA, 1955; Volume 4. [Google Scholar]
- Blumenthal, W.B. The Chemical Behavior of Zirconium; D. Van Nostrand Co. Inc.: Princeton, NJ, USA, 1958. [Google Scholar]
- Pierson, H.O. Introduction and General Considerations. In Handbook of Chemical Vapor Deposition: Principles, Technology, and Applications, 2nd ed.; Materials Science and Process Technology Series; Noyes Publications: Saddle River, NJ, USA, 1999; pp. 25–35. [Google Scholar]
- Jairath, R.; Jain, A.; Tolles, R.D.; Hampden-Smith, M.J.; Kodas, T.T. Introduction. In The Chemistry of Metal CVD; Kodas, T.T., Hampden-Smith, M.J., Eds.; VCH Publishers Inc.: Weinheim, Germany, 1994; pp. 1–44. [Google Scholar]
- Pierson, H.O. Handbook of Chemical Vapor Deposition: Principles, Technology, and Applications, 2nd ed.; Noyes Publications: Norwich, NY, USA, 1999; p. 506. [Google Scholar]
- Xu, Y.; Yan, X.-T. Chemical Vapour Deposition; Springer: London, UK, 2010; p. 352. [Google Scholar]
- Kodas, T.T.; Hampden-Smith, M.J. (Eds.) The Chemistry of Metal CVD.; Wiley: Weinheim, Germany, 1994. [Google Scholar]
- Pierson, H.O. CVD Processes and Equipment. In Handbook of Chemical Vapor Deposition: Principles, Technology, and Applications, 2nd ed.; Materials Science and Process Technology Series; Noyes Publications: Saddle River, NJ, USA, 1999; pp. 108–146. [Google Scholar]
- Xu, Y.; Yan, X.-T. Chemical Vapor Deposition Systems Design. In Chemical Vapour Deposition; Engineering Materials and Processes; Springer: London, UK, 2010; pp. 73–128. [Google Scholar]
- Pierson, H.O. Fundamentals of Chemical Vapor Deposition. In Handbook of Chemical Vapor Deposition: Principles, Technology, and Applications, 2nd ed.; Materials Science and Process Technology Series; Noyes Publications: Saddle River, NJ, USA, 1999; pp. 36–67. [Google Scholar]
- Thornton, J.A. High Rate Thick Film Growth. Annu. Rev. Mater. Sci. 1977, 7, 239–260. [Google Scholar] [CrossRef]
- Thornton, J.A. Influence of apparatus geometry and deposition conditions on the structure and topography of thick sputtered coatings. J. Vac. Sci. Technol. 1974, 11, 666–670. [Google Scholar] [CrossRef]
- Xu, Y.; Yan, X.-T. Microstructure Evolution and Process Control. In Chemical Vapour Deposition; Engineering Materials and Processes; Springer: London, UK, 2010; pp. 215–270. [Google Scholar]
- Martinu, L.; Zabeida, O.; Klemberg-Sapieha, J.E. Plasma-Enhanced Chemical Vapor Deposition of Functional Coatings. In Handbook of Deposition Technologies for Films and Coatings; Martin, P.M., Ed.; Science, Applications and Technology; Elsevier: Amsterdam, The Netherlands, 2010; pp. 392–465. [Google Scholar]
- Walton, S.G.; Greene, J.E. Plasmas in Deposition Processes. In Handbook of Deposition Technologies for Films and Coatings; Martin, P.M., Ed.; Science, Applications, and Technology; Elsevier: Amsterdam, The Netherlands, 2010; pp. 32–92. [Google Scholar]
- Pierson, H.O. Metallo-Organic CVD (MOCVD). In Handbook of Chemical Vapor Deposition: Principles, Technology, and Applications, 2nd ed.; Materials Science and Process Technology Series; Noyes Publications: Norwich, NY, USA, 1999; pp. 84–107. [Google Scholar]
- Blumenthal, W.B. Interstitial Solutions and Intermetallic Compounds. In The Chemical Behavior of Zirconium; D. Van Nostrand Co, Inc: Princeton, NJ, USA, 1958; pp. 46–101. [Google Scholar]
- Fix, R.M.; Gordon, R.G.; Hoffman, D.M. Solution-phase reactivity as a guide to the low-temperature chemical vapor deposition of early-transition-metal nitride thin films. J. Am. Chem. Soc. 1990, 112, 7833–7835. [Google Scholar] [CrossRef]
- Wagner, P. Research, Development, and Production of Substoichiometric Zirconium Carbide for High-Temperature Insulation; LA-5224; Los Alamos National Laboratory: Los Alamos, NM, USA, 1973. [Google Scholar]
- Gasparrini, C.; Rana, D.S.; Le Brun, N.; Horlait, D.; Markides, C.N.; Farnan, I.; Lee, W.E. On the stoichiometry of zirconium carbide. Sci. Rep. 2020, 10, 6347. [Google Scholar] [CrossRef] [Green Version]
- Zhou, Y.; Heitmann, T.W.; Fahrenholtz, W.G.; Hilmas, G.E. Synthesis of ZrCx with controlled carbon stoichiometry by low temperature solid state reaction. J. Eur. Ceram. Soc. 2019, 39, 2594–2600. [Google Scholar] [CrossRef]
- Miller, J.H.; Hunn, J.D.; Jolly, B.C.; Menchhofer, P.A. Development of ZrC Coating in a Fluidized Bed Chemical Vapor Deposition Furnace; ORNL/TM-2009/214; Oak Ridge National Laboratory: Oak Ridge, TN, USA, 2009. [Google Scholar]
- Aylsworth, J.W. Art of Manufacturing Electrical Incandescing Conductors. U.S. Patent US553296A, 21 January 1896. [Google Scholar]
- Weintraub, E. Reduction of Chemical Compounds. U.S. Patent US1019394A, 5 March 1912. [Google Scholar]
- Langmuir, I. Chemical Reactions at Low Pressures. J. Am. Chem. Soc. 1915, 37, 1139–1167. [Google Scholar] [CrossRef]
- Van Arkel, A.E.; de Boer, J.H. Darstellung von reinem Titanium-, Zirkonium-, Hafnium- und Thoriummetall. Z. Fuer Anorg. Und Allg. Chem. 1925, 148, 345–350. [Google Scholar] [CrossRef]
- Van Arkel, A.E.; de Boer, J.H. Process of Precipitating Metals on an Incandescing Body. U.S. Patent US1671213A, 29 May 1928. [Google Scholar]
- Shapiro, Z.M. Iodide-decomposition process for production of zirconium. In The Metallurgy of Zirconium; Lustman, B., Kerze, F.J., Eds.; National Nuclear Energy Series; McGraw-Hill: New York, NY, USA, 1955; Volume 4, pp. 135–215. [Google Scholar]
- Campbell, I.E.; Sherwood, E.M.; Powell, C.F.; Jones, R.P. Protection of Uranium: Vapor-Deposited Coatings; Battelle Memorial Institute: Columbus, OH, USA, 1953. [Google Scholar]
- Kroll, W.J. How commercial titanium and zirconium were born. J. Frankl. Inst. 1955, 260, 169–192. [Google Scholar] [CrossRef]
- Sale, F.R. The transport and deposition reactions involved in the production of zirconium coatings from mixed iodide vapours. J. Less Common Met. 1969, 19, 53–62. [Google Scholar] [CrossRef]
- Bhatti, M.A.; Copley, D.B.; Shelton, R.A.J. A reinvestigation of the disproportionation of zirconium triiodide. J. Less Common Met. 1977, 55, 293–296. [Google Scholar] [CrossRef]
- Copley, D.B.; Shelton, R.A.J. The disproportionation and non-stoichiometry of zirconium trichloride. J. Less Common Met. 1970, 20, 359–366. [Google Scholar] [CrossRef]
- Normanton, A.S.; Shelton, R.A.J. The disproportionation of zirconium tribromide. J. Less Common Met. 1973, 32, 111–116. [Google Scholar] [CrossRef]
- Becker, A.; Hüttinger, K.J. Chemistry and kinetics of chemical vapor deposition of pyrocarbon—IV pyrocarbon deposition from methane in the low temperature regime. Carbon 1998, 36, 213–224. [Google Scholar] [CrossRef]
- Miller, J.A.; Melius, C.F. Kinetic and thermodynamic issues in the formation of aromatic compounds in flames of aliphatic fuels. Combust. Flame 1992, 91, 21–39. [Google Scholar] [CrossRef]
- Hu, C.; Shen, H.; Zhang, S.; Li, H. Methane pyrolysis in preparation of pyrolytic carbon: Thermodynamic and kinetic analysis by density functional theory. Chin. J. Aeronaut. 2020, 33, 1064–1073. [Google Scholar] [CrossRef]
- Pierson, J.F.; Czerwiec, T.; Belmonte, T.; Michel, H. Diagnostic of Ar–BCl3 microwave discharges by optical emission spectroscopy. Surf. Coat. Technol. 1997, 97, 749–754. [Google Scholar] [CrossRef]
- Pierson, J.F.; Belmonte, T.; Michel, H. Low temperature growth mechanism of zirconium diboride films synthesised in flowing microwave Ar–BCl3 post-discharges. Surf. Coat. Technol. 1999, 116–119, 1049–1054. [Google Scholar] [CrossRef]
- Ohshita, Y.; Ishitani, A.; Takada, T. Surface reaction mechanism of SiCl2 with carrier gas H2 in silicon vapor phase epitaxial growth. J. Cryst. Growth 1991, 108, 499–507. [Google Scholar] [CrossRef]
- Ohshita, Y.; Ishitani, A.; Takada, T. Theoretical studies of Si vapor-phase epitaxial growth by ab initio molecular-orbital calculations. Phys. Rev. B Condens. Matter 1990, 41, 12720–12727. [Google Scholar] [CrossRef]
- Wagner, P.; Wahman, L.A.; White, R.W.; Hollabaugh, C.M.; Reiswig, R.D. Factors influencing the chemical vapor deposition of ZrC. J. Nucl. Mater. 1976, 62, 221–228. [Google Scholar] [CrossRef]
- Hollabaugh, C.M.; Wahman, L.A.; Reiswig, R.D.; White, R.W.; Wagner, P. Chemical Vapor Deposition of ZrC Made by Reactions of ZrCl4 with CH4 and with C3H6. Nucl. Technol. 1977, 35, 527–535. [Google Scholar] [CrossRef]
- Ikawa, K. Vapor deposition of zirconium carbide-carbon composites by the chloride process. J. Less Common Met. 1972, 29, 233–239. [Google Scholar] [CrossRef]
- Ogawa, T.; Ikawa, K.; Iwamoto, K. Chemical vapor deposition of ZrC within a spouted bed by bromide process. J. Nucl. Mater. 1981, 97, 104–112. [Google Scholar] [CrossRef]
- Charollais, F.; Fonquernie, S.; Perrais, C.; Perez, M.; Dugne, O.; Cellier, F.; Harbonnier, G.; Vitali, M.-P. CEA and AREVA R&D on HTR fuel fabrication and presentation of the CAPRI experimental manufacturing line. Nucl. Eng. Des. 2006, 236, 534–542. [Google Scholar] [CrossRef]
- Reynolds, G.H. Chemical vapor deposition of ZrC on pyrocarbon-coated fuel particles. J. Nucl. Mater. 1974, 50, 215–216. [Google Scholar] [CrossRef]
- Wallace, T.C. Chemical Vapor Deposition of ZrC in Small Bore Carbon-Composite Tubes; LA-UR-73-692; LANL: Los Alamos, NM, USA, 1973.
- Caputo, A.J. Vapor Deposition of Metal Carbides; Y-1852 (pt 4); Oak Ridge Y-12 Plant: Oak Ridge, TN, USA, 1973. [Google Scholar]
- Don, J.; Wright, M.A. Investigations of Oxidation Protection Systems for Carbon-Carbon Composites Formed by Chemical Vapor Deposition and Plasma-Assisted Chemical Vapor Deposition Techniques; ADA232800; Bolling AFB: Washington, DC, USA, 1991. [Google Scholar]
- Samoilenko, V.G.; Pereselentseva, L.N. Deposition of zirconium carbide coatings acting as diffusion barriers in composites consisting of a metallic matrix and refractory metal fibers. Sov. Powder Metall. Met. Ceram. 1975, 14, 725–728. [Google Scholar] [CrossRef]
- Park, J.H.; Jung, C.H.; Kim, D.J.; Park, J.Y. Effect of H2 dilution gas on the growth of ZrC during low pressure chemical vapor deposition in the ZrCl4–CH4–Ar system. Surf. Coat. Technol. 2008, 203, 87–90. [Google Scholar] [CrossRef]
- Park, J.H.; Jung, C.H.; Kim, D.J.; Park, J.Y. Temperature dependency of the LPCVD growth of ZrC with the ZrCl4–CH4–H2 system. Surf. Coat. Technol. 2008, 203, 324–328. [Google Scholar] [CrossRef]
- Liu, Q.; Zhang, L.; Cheng, L.; Wang, Y. Morphologies and growth mechanisms of zirconium carbide films by chemical vapor deposition. J. Coat. Technol. Res. 2009, 6, 269–273. [Google Scholar] [CrossRef]
- Kim, J.G.; Park, S.J.; Park, J.Y.; Choi, D.J. The effect of temperature on the growth and properties of chemical vapor deposited ZrC films on SiC-coated graphite substrates. Ceram. Int. 2015, 41, 211–216. [Google Scholar] [CrossRef]
- Hollabaugh, C.M.; Reiswig, R.D.; Wagner, P.; Wahman, L.A.; White, R.W. A new method for coating microspheres with zirconium carbide and zirconium carbide-carbon graded coats. J. Nucl. Mater. 1975, 57, 325–332. [Google Scholar] [CrossRef] [Green Version]
- Wang, Y.; Liu, Q.; Liu, J.; Zhang, L.; Cheng, L. Deposition Mechanism for Chemical Vapor Deposition of Zirconium Carbide Coatings. J. Am. Ceram. Soc. 2008, 91, 1249–1252. [Google Scholar] [CrossRef]
- Lamm, B.W.; McMurray, J.W.; Cakmak, E.; Lance, M.J.; Mitchell, D.J. Leveraging computational thermodynamics to guide SiC-ZrC chemical vapor deposition process development. Surf. Coat. Tech. 2022, 444, 128672. [Google Scholar] [CrossRef]
- Ikawa, K. Vapor deposition of zirconium carbide-carbon composites by the iodide process. J. Less Common Met. 1972, 27, 325–332. [Google Scholar] [CrossRef]
- Ikawa, K.; Iwamoto, K. Coating Microspheres with Zirconium Carbide-Carbon Alloy by Iodide Process. J. Nucl. Sci. Technol. 1974, 11, 263–267. [Google Scholar] [CrossRef]
- Ogawa, T.; Ikawa, K.; Iwamoto, K. Effect of gas composition on the deposition of ZrC-C mixtures: The bromide process. J. Mater. Sci. 1979, 14, 125–132. [Google Scholar] [CrossRef]
- Ikawa, K. Co-deposition of zirconium with carbon by the bromide process. J. Less Common Met. 1976, 44, 207–213. [Google Scholar] [CrossRef]
- Benzinger, W.; Becker, A.; Hüttinger, K.J. Chemistry and kinetics of chemical vapour deposition of pyrocarbon: I. Fundamentals of kinetics and chemical reaction engineering. Carbon 1996, 34, 957–966. [Google Scholar] [CrossRef]
- Becker, A.; Hüttinger, K.J. Chemistry and kinetics of chemical vapor deposition of pyrocarbon—III pyrocarbon deposition from propylene and benzene in the low temperature regime. Carbon 1998, 36, 201–211. [Google Scholar] [CrossRef]
- Jackson, H.F.; Lee, W.E. Properties and Characteristics of ZrC. In Material Properties/Oxide Fuels of Light Water Reactors and Fast Neutron Reactors, 2nd ed.; Allen, T.R., Stoller, R.E., Yamanaka, S., Konings, R.J.M., Eds.; Comprehensive Nuclear Materials; Elsevier: Amsterdam, The Netherlands, 2012; pp. 339–372. [Google Scholar]
- Liu, Q.M.; Zhang, L.T.; Meng, Z.X.; Cheng, L.F. Chemical Vapor Deposition (CVD) of ZrC Coatings from ZrCl4-C3H6-H2. Adv. Mater. Res. 2011, 189–193, 648–652. [Google Scholar] [CrossRef]
- Liu, Q.; Liu, J.; Luan, X. Preparation of ZrC-SiC composite coatings by chemical vapor deposition and study of co-deposition mechanism. J. Mater. Sci. Technol. 2019, 35, 2942–2949. [Google Scholar] [CrossRef]
- Liu, Q.; Zhang, L.; Cheng, L.; Wang, Y. Chemical vapour deposition of zirconium carbide and silicon carbide hybrid whiskers. Mater. Lett. 2010, 64, 552–554. [Google Scholar] [CrossRef]
- Liu, Q.M.; Zhang, L.T.; Liu, J.; Wang, Y.G. Thermodynamic study on codeposition of ZrC-SiC from MTS-ZrCl4-CH4-H2. Inorg. Mater. 2010, 46, 1090–1095. [Google Scholar] [CrossRef]
- Grisdale, R.O.; Pfister, A.C.; Van Roosbroeck, W. Pyrolytic Film Resistors: Carbon and Borocarbon. Bell Syst. Tech. J. 1951, 30, 271–314. [Google Scholar] [CrossRef]
- Grisdale, R.O. The Formation of Black Carbon. J. Appl. Phys. 1953, 24, 1082–1091. [Google Scholar] [CrossRef]
- Haynes, W.M.; Lide, D.R.; Bruno, T.J. (Eds.) Thermochemistry, Electrochemistry, and Solution Chemistry. In CRC Handbook of Chemistry and Physics, 97th ed.; CRC Press: Boca Raton, FL, USA, 2016. [Google Scholar]
- Baker, F.B.; Storms, E.K.; Holley, C.E. Enthalpy of formation of zirconium carbide. J. Chem. Eng. Data 1969, 14, 244–246. [Google Scholar] [CrossRef]
- Becker, A.; Hüttinger, K.J. Chemistry and kinetics of chemical vapor deposition of pyrocarbon—II pyrocarbon deposition from ethylene, acetylene and 1,3-butadiene in the low temperature regime. Carbon 1998, 36, 177–199. [Google Scholar] [CrossRef]
- Tamari, N.; Kato, A. Catalytic effect of nickel on the growth of zirconium carbide whiskers by chemical vapor deposition. J. Less Common Met. 1978, 58, 147–160. [Google Scholar] [CrossRef]
- Loumagne, F.; Langlais, F.; Naslain, R. Reactional mechanisms of the chemical vapour deposition of SiC-based ceramics from CH3SiCl3-H2 gas precursor. J. Cryst. Growth 1995, 155, 205–213. [Google Scholar] [CrossRef]
- Fix, R.; Gordon, R.G.; Hoffman, D.M. Chemical vapor deposition of titanium, zirconium, and hafnium nitride thin films. Chem. Mater. 1991, 3, 1138–1148. [Google Scholar] [CrossRef]
- Motojima, S.; Kani, E.; Takahashi, Y.; Sugiyama, K. Impurity activated whisker growth of zirconium nitride by chemical vapour deposition. J. Mater. Sci. 1979, 14, 1495–1499. [Google Scholar] [CrossRef]
- Yajima, A.; Segawa, Y.; Matsuzaki, R.; Saeki, Y. Reaction Process of Zirconium Tetrachloride with Ammonia in the Vapor Phase and Properties of the Zirconium Nitride Formed. Bull. Chem. Soc. Jpn. 1983, 56, 2638–2642. [Google Scholar] [CrossRef] [Green Version]
- Sugiyama, K.; Pac, S.; Takahashi, Y.; Motojima, S. Low Temperature Deposition of Metal Nitrides by Thermal Decomposition of Organometallic Compounds. J. Electrochem. Soc. 1975, 122, 1545–1549. [Google Scholar] [CrossRef]
- Chiu, H.-T.; Huang, C.-C. Low-pressure chemical vapor deposition of titanium and zirconium carbonitride thin films from M(NEt2)4 (M = Ti and Zr). Mater. Lett. 1993, 16, 194–199. [Google Scholar] [CrossRef]
- Berndt, H.; Zeng, A.Q.; Stock, H.R.; Mayr, P. Zirconium carbonitride films produced by plasma-assisted metal organic chemical vapour deposition. Surf. Coat. Technol. 1995, 74–75, 369–374. [Google Scholar] [CrossRef]
- Arrieta, M. Low Temperature Chemical Vapor Deposition of Zirconium Nitride in a Fluidized Bed. Doctoral dissertation, Texas A&M University, College Station, TX, USA, 2012. [Google Scholar]
- Arrieta, M.Y.; Keiser, D.D.; Perez-Nunez, D.; McDeavitt, S.M. Fluidized Bed Chemical Vapor Deposition of Zirconium Nitride Films. Nucl. Technol. 2017, 199, 219–226. [Google Scholar] [CrossRef]
- Sudderth, L.; Perez-Nunez, D.; Keiser, D.; McDeavitt, S. Fabrication of ZrN Barrier Coatings for U-Mo Microspheres Via Fluidized Bed Chemical Vapor Deposition Using a Metalorganic Precursor. Nucl. Technol. 2018, 202, 81–93. [Google Scholar] [CrossRef]
- Mittemeijer, E.J. Fundamentals of Nitriding and Nitrocarburizing. In Steel Heat Treating Fundamentals and Processes; Dossett, J.L., Totten, G.E., Eds.; ASM International: Almere, The Netherlands, 2013; Volume 4A. [Google Scholar]
- Mittemeijer, E.J.; Slycke, J.T. Chemical Potentials and Activities of Nitrogen and Carbon Imposed by Gaseous Nitriding and Carburising Atmospheres. Surf. Eng. 1996, 12, 152–162. [Google Scholar] [CrossRef]
- Mittemeijer, E.J.; Somers, M.A.J. Thermodynamics, kinetics, and process control of nitriding. Surf. Eng. 1997, 13, 483–497. [Google Scholar] [CrossRef]
- Grabke, H.J. Reaktionen von Ammoniak, Stickstoff und Wasserstoff an der Oberfläche von Eisen I. Zur Kinetik der Nitrierung von Eisen mit NH3-H2-Gemischen und der Denitrierung. Ber. Bunsenges. Phys. Chem. 1968, 74, 533–541. [Google Scholar] [CrossRef]
- Agte, C.; Moers, K. Methoden zur Reindarstellung hochschmelzender Carbide, Nitride und Boride und Beschreibung einiger ihrer Eigenschaften. Z. Fuer Anorg. Allg. Chem. 1931, 198, 233–275. [Google Scholar] [CrossRef]
- Haynes, W.E.; Lide, D.R.; Bruno, T.J. Properties of the Elements and Inorganic Compounds. In CRC Handbook of Chemistry and Physics, 97th ed.; Haynes, W.M., Ed.; CRC Press: Boca Raton, FL, USA, 2014; pp. 4–71. [Google Scholar]
- Futamoto, M.; Yuito, I.; Kawabe, U. Hafnium carbide and nitride whisker growth by chemical vapor deposition. J. Cryst. Growth 1983, 61, 69–74. [Google Scholar] [CrossRef]
- Wagner, R.S.; Ellis, W.C. Vapor-Liquid-Solid Mechanism of Single Crystal Growth. Appl. Phys. Lett. 1964, 4, 89–90. [Google Scholar] [CrossRef]
- Laidler, K.J. Elementary Reactions in Solution. In Chemical Kinetics, 3rd ed.; Harper & Row: New York, NY, USA, 1987; pp. 183–228. [Google Scholar]
- Rabinowitch, E.; Wood, W.C. The collison mechanism and the primary photochemical process in solutions. Trans. Faraday Soc. 1936, 32, 1381–1387. [Google Scholar] [CrossRef]
- Franck, J.; Rabinowitsch, E. Some remarks about free radicals and the photochemistry of solutions. Trans. Faraday Soc. 1934, 30, 120–130. [Google Scholar] [CrossRef]
- Wedekind, E. Studien über das elementare Zirkonium II. Justus Liebigs Ann. Der Chem. 1913, 395, 149–194. [Google Scholar] [CrossRef] [Green Version]
- Alexander, P.P. Production of Zirconium Nitride. U.S. Patent US2461019A, 8 February 1949. [Google Scholar]
- Tibbetts, G.G. Role of nitrogen atoms in "ion-nitriding". J. Appl. Phys. 1974, 45, 5072–5073. [Google Scholar] [CrossRef]
- Roliński, E.; Konieczny, A.; Sharp, G. Influence of nitriding mechanisms on surface roughness of plasma and gas nitrided/nitrocarburized gray cast iron. Heat Treat. Prog. 2007, 7, 39–46. [Google Scholar]
- Hudis, M. Study of ion-nitriding. J. Appl. Phys. 1973, 44, 1489–1496. [Google Scholar] [CrossRef]
- Michel, H.; Czerwiec, T.; Gantois, M.; Ablitzer, D.; Ricard, A. Progress in the analysis of the mechanisms of ion nitriding. Surf. Coat. Technol. 1995, 72, 103–111. [Google Scholar] [CrossRef]
- Dietrich, H.; Geng, P.; Jacobi, K.; Ertl, G. Sticking coefficient for dissociative adsorption of N2 on Ru single-crystal surfaces. J. Chem. Phys. 1996, 104, 375–381. [Google Scholar] [CrossRef]
- Smith, T. Effect of Surface Coverage and Temperature on the Sticking Coefficient. J. Chem. Phys. 1964, 40, 1805–1812. [Google Scholar] [CrossRef]
- Rie, K.T.; Wöhle, J.; Gebauer, A. Synthesis of thin coatings by plasma-assisted chemical vapour deposition using metallo-organic compounds as precursors. Surf. Coat. Technol. 1993, 59, 202–206. [Google Scholar] [CrossRef]
- Becker, J.S.; Kim, E.; Gordon, R.G. Atomic Layer Deposition of Insulating Hafnium and Zirconium Nitrides. Chem. Mater. 2004, 16, 3497–3501. [Google Scholar] [CrossRef]
- Bhattacharya, S.; Jamison, L.; Seidman, D.N.; Mohamed, W.; Bei, Y.; Pellin, M.J.; Yacout, A.M. Nanocrystalline ZrN thin film development via atomic layer deposition for U-Mo powder. J. Nucl. Mater. 2019, 526, 151770. [Google Scholar] [CrossRef]
- Bradley, D.C.; Thomas, I.M. Metallo-organic compounds containing metal–nitrogen bonds. Part I. Some dialkylamino-derivatives of titanium and zirconium. J. Chem. Soc. 1960, 0, 3857–3861. [Google Scholar] [CrossRef]
- Bradley, D.C.; Torrible, E.G. Metallo-Organic Compounds Containing Metal–Nitrogen Bonds: Part IV. Some Bis-(Primary Amino)-Titanium Compounds. Can. J. Chem. 1963, 41, 134–138. [Google Scholar] [CrossRef]
- Bartlett, R.K. The reaction of primary amines with tetrakis(diethylamino)zirconium. J. Inorg. Nucl. Chem. 1966, 28, 2448–2449. [Google Scholar] [CrossRef]
- Elam, J.W.; Schuisky, M.; Ferguson, J.D.; George, S.M. Surface chemistry and film growth during TiN atomic layer deposition using TDMAT and NH3. Thin Solid Film. 2003, 436, 145–156. [Google Scholar] [CrossRef]
- George, S.M. Atomic layer deposition: An overview. Chem. Rev. 2010, 110, 111–131. [Google Scholar] [CrossRef]
- Amato-Wierda, C.; Wierda, D.A. Chemical vapor deposition of titanium nitride thin films from tetrakis(dimethylamido)titanium and hydrazine as a coreactant. J. Mater. Res. 2000, 15, 2414–2424. [Google Scholar] [CrossRef]
- Truong, C.M.; Chen, P.J.; Corneille, J.S.; Oh, W.S.; Goodman, D.W. Low-Pressure Deposition of TiN Thin Films from a Tetrakis(dimethylamido)titanium Precursor. J. Phys. Chem. 1995, 99, 8831–8842. [Google Scholar] [CrossRef]
- Becker, K.; Ebert, F. Die Kristallstruktur einiger binärer Carbide und Nitride. Z. Phys. 1925, 31, 268–272. [Google Scholar] [CrossRef]
- Wahl, G.; Schlosser, S.; Schmaderer, F. Kinetics of the chlorination of Y and Zr and the deposition of Y- and Zr-oxides by reaction of the chlorides with oxygen. In Proceedings of the The Seventh International Conference on Chemical Vapor Deposition, Los Angeles, CA, USA, 14–19 October 1979; pp. 536–543. [Google Scholar]
- Brennfleck, K.; Fitzer, E.; Mack, G. Basic study of CVD of ZrO2-layers on metal substrates. In Proceedings of the 8th International Conference on Chemical Vapor Deposition, Gouvieux, France, 15–18 September 1981; pp. 672–684. [Google Scholar]
- Sipp, E.; Langlais, F.; Naslain, R. Kinetics of deposition of zirconia-based ceramics from ZrCl4-H2-CO2-Ar gas mixtures. J. Alloy. Compd. 1992, 186, 65–76. [Google Scholar] [CrossRef]
- Minet, J.; Langlais, F.; Naslain, R. Chemical vapor infiltration of zirconia within the pore network of fibrous ceramic materials from ZrCl4H2CO2 gas mixtures. Compos. Sci. Technol. 1990, 37, 79–107. [Google Scholar] [CrossRef]
- Minet, J.; Langlais, F.; Naslain, R.; Bernard, C. On the chemical vapour deposition of zirconia from ZrCl4-H2-CO2-Ar gas mixtures: I. A thermodynamic approach. J. Less Common Met. 1986, 119, 219–235. [Google Scholar] [CrossRef]
- Minet, J.; Langlais, F.; Naslain, R. On the chemical vapour deposition of zirconia from ZrCl4-H2-CO2-Ar gas mixture: II. An experimental approach. J. Less Common Met. 1987, 132, 273–287. [Google Scholar] [CrossRef]
- Tingey, G.L. Kinetics of the Water—Gas Equilibrium Reaction. I. The Reaction of Carbon Dioxide with Hydrogen. J. Phys. Chem. 1966, 70, 1406–1412. [Google Scholar] [CrossRef]
- Bradford, B.W. The water-gas reaction in low-pressure explosions. J. Chem. Soc. (Resumed) 1933, 1557–1563. [Google Scholar] [CrossRef]
- Bustamante, F.; Enick, R.M.; Cugini, A.V.; Killmeyer, R.P.; Howard, B.H.; Rothenberger, K.S.; Ciocco, M.V.; Morreale, B.D.; Chattopadhyay, S.; Shi, S. High-temperature kinetics of the homogeneous reverse water-gas shift reaction. AIChE J. 2004, 50, 1028–1041. [Google Scholar] [CrossRef]
- Graven, W.M.; Long, F.J. Kinetics and Mechanisms of the Two Opposing Reactions of the Equilibrium CO + H2O = CO2 + H2. J. Am. Chem. Soc. 1954, 76, 2602–2607. [Google Scholar] [CrossRef]
- Holgate, H.R.; Tester, J.W. Oxidation of hydrogen and carbon monoxide in sub- and supercritical water: Reaction kinetics, pathways, and water-density effects. 2. Elementary reaction modeling. J. Phys. Chem. 1994, 98, 810–822. [Google Scholar] [CrossRef]
- Fujita, S. Mechanism of the reverse water gas shift reaction over Cu/ZnO catalyst. J. Catal. 1992, 134, 220–225. [Google Scholar] [CrossRef]
- Chen, C.S.; Cheng, W.H.; Lin, S.S. Mechanism of CO formation in reverse water-gas shift reaction over Cu/Al2O3 catalyst. Catal. Lett. 2000, 68, 45–48. [Google Scholar] [CrossRef]
- Rahtu, A.; Ritala, M. Reaction mechanism studies on the zirconium chloride–water atomic layer deposition process. J. Mater. Chem. 2002, 12, 1484–1489. [Google Scholar] [CrossRef]
- Kukli, K.; Forsgren, K.; Aarik, J.; Uustare, T.; Aidla, A.; Niskanen, A.; Ritala, M.; Leskelä, M.; Hårsta, A. Atomic layer deposition of zirconium oxide from zirconium tetraiodide, water and hydrogen peroxide. J. Cryst. Growth 2001, 231, 262–272. [Google Scholar] [CrossRef]
- Kukli, K.; Forsgren, K.; Ritala, M.; Leskela, M.; Aarik, J.; Harsta, A. Dielectric Properties of Zirconium Oxide Grown by Atomic Layer Deposition from Iodide Precursor. J. Electrochem. Soc. 2001, 148, F227. [Google Scholar] [CrossRef]
- Aarik, J.; Aidla, A.; Mändar, H.; Uustare, T.; Sammelselg, V. Growth kinetics and structure formation of ZrO2 thin films in chloride-based atomic layer deposition process. Thin Solid Film. 2002, 408, 97–103. [Google Scholar] [CrossRef]
- Ritala, M.; Leskelä, M. Zirconium dioxide thin films deposited by ALE using zirconium tetrachloride as precursor. Appl. Surf. Sci. 1994, 75, 333–340. [Google Scholar] [CrossRef]
- Houssa, M.; Tuominen, M.; Naili, M.; Afanas’ev, V.; Stesmans, A.; Haukka, S.; Heyns, M.M. Trap-assisted tunneling in high permittivity gate dielectric stacks. J. Appl. Phys. 2000, 87, 8615–8620. [Google Scholar] [CrossRef]
- Li, B.; Griffiths, K.; Zhang, C.S.; Norton, P.R. The autocatalytic decomposition of water on Zr(0001). Surf. Sci. 1997, 384, 70–80. [Google Scholar] [CrossRef]
- Wuchina, E.; Opila, E.; Opeka, M.; Fahrenholtz, W.G.; Talmy, I.G. UHTCs: Ultra-High Temperature Ceramic Materials for Extreme Environment Applications. Interface 2007, 16, 30–36. [Google Scholar] [CrossRef]
- Pierson, J.F.; Belmonte, T.; Michel, H. Thermodynamic and experimental study of low temperature ZrB2 chemical vapor deposition. Le J. De Phys. IV 2001, 11, Pr3-85–Pr83-91. [Google Scholar] [CrossRef]
- Pierson, H.O.; Mullendore, A.W. Thick boride coatings by chemical vapor deposition. Thin Solid Film. 1982, 95, 99–104. [Google Scholar] [CrossRef]
- Mukaida, M.; Goto, T.; Hirai, T. Preferred orientation of TiB2 plates prepared by CVD of the TiCl4 + B2H6 system. J. Mater. Sci. 1991, 26, 6613–6617. [Google Scholar] [CrossRef]
- Wang, A.; Malé, G. Experimental investigation of the ZrBClH CVD system. J. Eur. Ceram. Soc. 1993, 11, 241–251. [Google Scholar] [CrossRef]
- Jensen, J.A.; Gozum, J.E.; Pollina, D.M.; Girolami, G.S. Titanium, zirconium, and hafnium tetrahydroborates as "tailored" CVD precursors for metal diboride thin films. J. Am. Chem. Soc. 1988, 110, 1643–1644. [Google Scholar] [CrossRef]
- Rice, G.W.; Woodin, R.L. Zirconium Borohydride as a Zirconium Boride Precursor. J. Am. Ceram. Soc. 1988, 71, C-181–C-183. [Google Scholar] [CrossRef]
- Wayda, A.L.; Schneemeyer, L.F.; Opila, R.L. Low-temperature deposition of zirconium and hafnium boride films by thermal decomposition of the metal borohydrides (M[BH4]4). Appl. Phys. Lett. 1988, 53, 361–363. [Google Scholar] [CrossRef]
- Kaufman, L.; Clougherty, E.V. Investigation of Boride Compounds for Very High Temperature Applications Part 2; RTD-TDR-63-4096; ManLabs Inc.: Cambridge, MA, USA, 1965. [Google Scholar]
- Clougherty, E.V.; Hill, R.J.; Rhodes, W.H.; Peters, E.T. Processing and Characterization; ManLabs Inc.: Cambridge, MA, USA; Avco Corp.: Greenwich, CT, USA, 1970. [Google Scholar]
- Pierson, J.F.; Belmonte, T.; Czerwiec, T.; Hertz, D.; Michel, H. Low temperature ZrB2 remote plasma enhanced chemical vapor deposition. Thin Solid Film. 2000, 359, 68–76. [Google Scholar] [CrossRef]
- Zhou, C.; Wang, P.; Wei, C.; Han, W.; Zhang, X.; Xu, B. ZrB 2 grains synthesized on graphite by chemical vapor deposition. J. Alloy. Compd. 2017, 698, 27–32. [Google Scholar] [CrossRef]
- Caputo, A.J.; Lackey, W.J.; Wright, I.G.; Angelini, P. Chemical Vapor Deposition of Erosion-Resistant TiB2 Coatings. J. Electrochem. Soc. 1985, 132, 2274–2280. [Google Scholar] [CrossRef]
- Choy, K.L.; Derby, B. The CVD of TiB2 Protective Coating on SiC Monofilament Fibres. Le J. De Phys. IV 1991, 2, C2-697–C2-703. [Google Scholar] [CrossRef]
- Jayaraman, S.; Yang, Y.; Kim, D.Y.; Girolami, G.S.; Abelson, J.R. Hafnium diboride thin films by chemical vapor deposition from a single source precursor. J. Vac. Sci. Technol. A: Vac. Surf. Film. 2005, 23, 1619–1625. [Google Scholar] [CrossRef] [Green Version]
- Sulyaeva, V.S.; Shestakov, V.A.; Rumyantsev, Y.M.; Kosinova, M.L. Synthesis of Zirconium Diboride Films and ZrB2/BCxNy Heterostructures. Inorg. Mater. 2018, 54, 133–139. [Google Scholar] [CrossRef]
- Wang, A.; Male, G. Thermodynamics of the heterogeneous system ZrCl4—BCl3—H2. Calphad 1992, 16, 243–254. [Google Scholar] [CrossRef]
- Naslain, R.; Thebault, J.; Hagenmuller, P.; Bernard, C. The thermodynamic approach to boron chemical vapour deposition based on a computer minimization of the total gibbs free energy. J. Less Common Met. 1979, 67, 85–100. [Google Scholar] [CrossRef]
- Lengyel, I.; Jensen, K.F. A chemical mechanism for in situ boron doping during silicon chemical vapor deposition. Thin Solid Film. 2000, 365, 231–241. [Google Scholar] [CrossRef]
- Lamborn, D.R.; Snyder, D.W.; Xi, X.X.; Redwing, J.M. Modeling studies of the chemical vapor deposition of boron films from B2H6. J. Cryst. Growth 2007, 299, 358–364. [Google Scholar] [CrossRef]
- Reynolds, G.J.; Cooper III, C.B.; Gaczi, P.J. Selective titanium disilicide by low-pressure chemical vapor deposition. J. Appl. Phys. 1989, 65, 3212–3218. [Google Scholar] [CrossRef]
- Mohammadi, V.; de Boer, W.B.; Nanver, L.K. Temperature dependence of chemical-vapor deposition of pure boron layers from diborane. Appl. Phys. Lett. 2012, 101, 111906. [Google Scholar] [CrossRef] [Green Version]
- Mohammadi, V.; de Boer, W.B.; Nanver, L.K. An analytical kinetic model for chemical-vapor deposition of pureB layers from diborane. J. Appl. Phys. 2012, 112, 113501. [Google Scholar] [CrossRef]
- Komatsu, S.; Moriyoshi, Y. Influence of atomic hydrogen on the growth reactions of amorphous boron films in a low-pressure B2H6+He+H2plasma. J. Appl. Phys. 1988, 64, 1878–1884. [Google Scholar] [CrossRef]
- Wang, Y.; Hamers, R.J. Boron-induced reconstructions of Si(001) investigated by scanning tunneling microscopy. J. Vac. Sci. Technol. A Vac. Surf. Film. 1995, 13, 1431–1437. [Google Scholar] [CrossRef]
- Wang, Y.; Hamers, R.J.; Kaxiras, E. Atomic structure and bonding of boron-induced reconstructions on Si(001). Phys Rev Lett 1995, 74, 403–406. [Google Scholar] [CrossRef]
- Hamers, R.J.; Wang, Y. Atomically-Resolved Studies of the Chemistry and Bonding at Silicon Surfaces. Chem. Rev. 1996, 96, 1261–1290. [Google Scholar] [CrossRef] [PubMed]
- Wang, Y.; Shan, J.; Hamers, R.J. Combined scanning tunneling microscopy and infrared spectroscopy study of the interaction of diborane with Si(001). J. Vac. Sci. Technol. B Microelectron. Nanometer Struct. 1996, 14, 1038–1042. [Google Scholar] [CrossRef]
- Headrick, R.L.; Weir, B.E.; Bevk, J.; Freer, B.S.; Eaglesham, D.J.; Feldman, L.C. Influence of surface reconstruction on the orientation of homoepitaxial silicon films. Phys. Rev. Lett. 1990, 65, 1128–1131. [Google Scholar] [CrossRef] [PubMed]
- Headrick, R.L.; Levi, A.F.; Luftman, H.S.; Kovalchick, J.; Feldman, L.C. Electrical conduction in the Si(111):B-(sqrt 3 x sqrt 3)R30 degrees/a-Si interface reconstruction. Phys. Rev. B Condens. Matter 1991, 43, 14711–14714. [Google Scholar] [CrossRef] [PubMed]
- Fernández, H.; Grotewold, J.; Previtali, C.M. Thermal decomposition of diborane. Part I. The decomposition mechanism at low conversion and temperature and the inhibiting effect of accumulated hydrogen. J. Chem. Soc. Dalton Trans. 1973, 20, 2090–2095. [Google Scholar] [CrossRef]
- Grimes, R.N. Boron. In Advanced Inorganic Chemistry, 6th ed.; Cotton, F.A., Wilkinson, G., Murillo, C.A., Bochmann, M., Eds.; John Wiley & Sons, Inc.: New York, NY, USA, 1999; pp. 131–174. [Google Scholar]
- Yu, M.L.; Vitkavage, D.J.; Meyerson, B.S. Doping reaction of PH3 and B2H6 with Si(100). J. Appl. Phys. 1986, 59, 4032–4037. [Google Scholar] [CrossRef]
- Pierson, H.O.; Mullendore, A.W. The chemical vapor deposition of TiB2 from diborane. Thin Solid Film. 1980, 72, 511–516. [Google Scholar] [CrossRef]
- James, B.D.; Nanda, R.K.; Walbridge, M.G.H. Reactions of Lewis bases with tetrahydroborate derivatives of the Group IVa elements. Preparation of new zirconium hydride species. Inorg. Chem. 1967, 6, 1979–1983. [Google Scholar] [CrossRef]
- Fryzuk, M.D.; Rettig, S.J.; Westerhaus, A.; Williams, H.D. Synthesis, stability, and fluxional behavior of binuclear mixed-hydride-tetrahydroborate complexes of hafnium(IV): X-ray crystal structure of [[(Me2PCH2SiMe2)2N]Hf(BH4)2](.mu.-H)3[Hf(BH4)[N(SiMe2CH2PMe2)2. Inorg. Chem. 1985, 24, 4316–4325. [Google Scholar] [CrossRef]
- Bird, P.H.; Churchill, M.R. The crystal structure of zirconium(IV) borohydride (at –160°). Chem. Commun. 1967, 403. [Google Scholar] [CrossRef]
- Hedberg, K.; Plato, V. Electron-diffraction investigation of zirconium tetraborohydride, Zr(BH4)4. Inorg. Chem. 1971, 10, 590–594. [Google Scholar] [CrossRef]
- Marks, T.J.; Kolb, J.R. Covalent transition metal, lanthanide, and actinide tetrahydroborate complexes. Chem. Rev. 1977, 77, 263–293. [Google Scholar] [CrossRef]
- Marks, T.J.; Kolb, J.R. Dynamic interligand hydrogen transfer in some η5-cyclopentadienylzirconium and -hafnium tetrahydroborates. J. Am. Chem. Soc. 1975, 97, 3397–3401. [Google Scholar] [CrossRef]
- Temperton, R.H.; Gibson, A.; O’Shea, J.N. In situ XPS analysis of the atomic layer deposition of aluminium oxide on titanium dioxide. Phys. Chem. Chem. Phys. 2019, 21, 1393–1398. [Google Scholar] [CrossRef]
- Kokkonen, E.; Kaipio, M.; Nieminen, H.E.; Rehman, F.; Miikkulainen, V.; Putkonen, M.; Ritala, M.; Huotari, S.; Schnadt, J.; Urpelainen, S. Ambient pressure x-ray photoelectron spectroscopy setup for synchrotron-based in situ and operando atomic layer deposition research. Rev. Sci. Instrum. 2022, 93, 013905. [Google Scholar] [CrossRef]
- Kircher, C.J.; Mayer, J.W.; Tu, K.N.; Ziegler, J.F. Analysis of formation of hafnium silicide on silicon. Appl. Phys. Lett. 1973, 22, 81–83. [Google Scholar] [CrossRef]
- Ziegler, J.F.; Mayer, J.W.; Kircher, C.J.; Tu, K.N. Kinetics of the formation of hafnium silicides on silicon. J. Appl. Phys. 1973, 44, 3851–3857. [Google Scholar] [CrossRef]
- Murarka, S.P. Thermodynamic Considerations. In Silicides for VLSI Applications; Academic Press: New York, NY, USA, 1983; pp. 71–98. [Google Scholar]
- Kematick, R.J.; Myers, C.E. Thermodynamics of the Phase Formation of the Titanium Silicides. Chem. Mater. 1996, 8, 287–291. [Google Scholar] [CrossRef]







Disclaimer/Publisher’s Note: The statements, opinions and data contained in all publications are solely those of the individual author(s) and contributor(s) and not of MDPI and/or the editor(s). MDPI and/or the editor(s) disclaim responsibility for any injury to people or property resulting from any ideas, methods, instructions or products referred to in the content. |
© 2023 by the authors. Licensee MDPI, Basel, Switzerland. This article is an open access article distributed under the terms and conditions of the Creative Commons Attribution (CC BY) license (https://creativecommons.org/licenses/by/4.0/).
Share and Cite
Lamm, B.W.; Mitchell, D.J. Chemical Vapor Deposition of Zirconium Compounds: A Review. Coatings 2023, 13, 266. https://doi.org/10.3390/coatings13020266
Lamm BW, Mitchell DJ. Chemical Vapor Deposition of Zirconium Compounds: A Review. Coatings. 2023; 13(2):266. https://doi.org/10.3390/coatings13020266
Chicago/Turabian StyleLamm, Benjamin Weitkamp, and David Joseph Mitchell. 2023. "Chemical Vapor Deposition of Zirconium Compounds: A Review" Coatings 13, no. 2: 266. https://doi.org/10.3390/coatings13020266
APA StyleLamm, B. W., & Mitchell, D. J. (2023). Chemical Vapor Deposition of Zirconium Compounds: A Review. Coatings, 13(2), 266. https://doi.org/10.3390/coatings13020266