
Proteolytic Cleavages in the VEGF Family: Generating Diversity among Angiogenic VEGFs, Essential for the Activation of Lymphangiogenic VEGFs



Abstract
:Simple Summary
Abstract
1. Introduction
- Tissue drainage for fluid balance and waste disposal
- Immune surveillance, including hosting and trafficking of immune cells
- Uptake of dietary long-chain fatty acids and other highly lipophilic compounds in the intestine
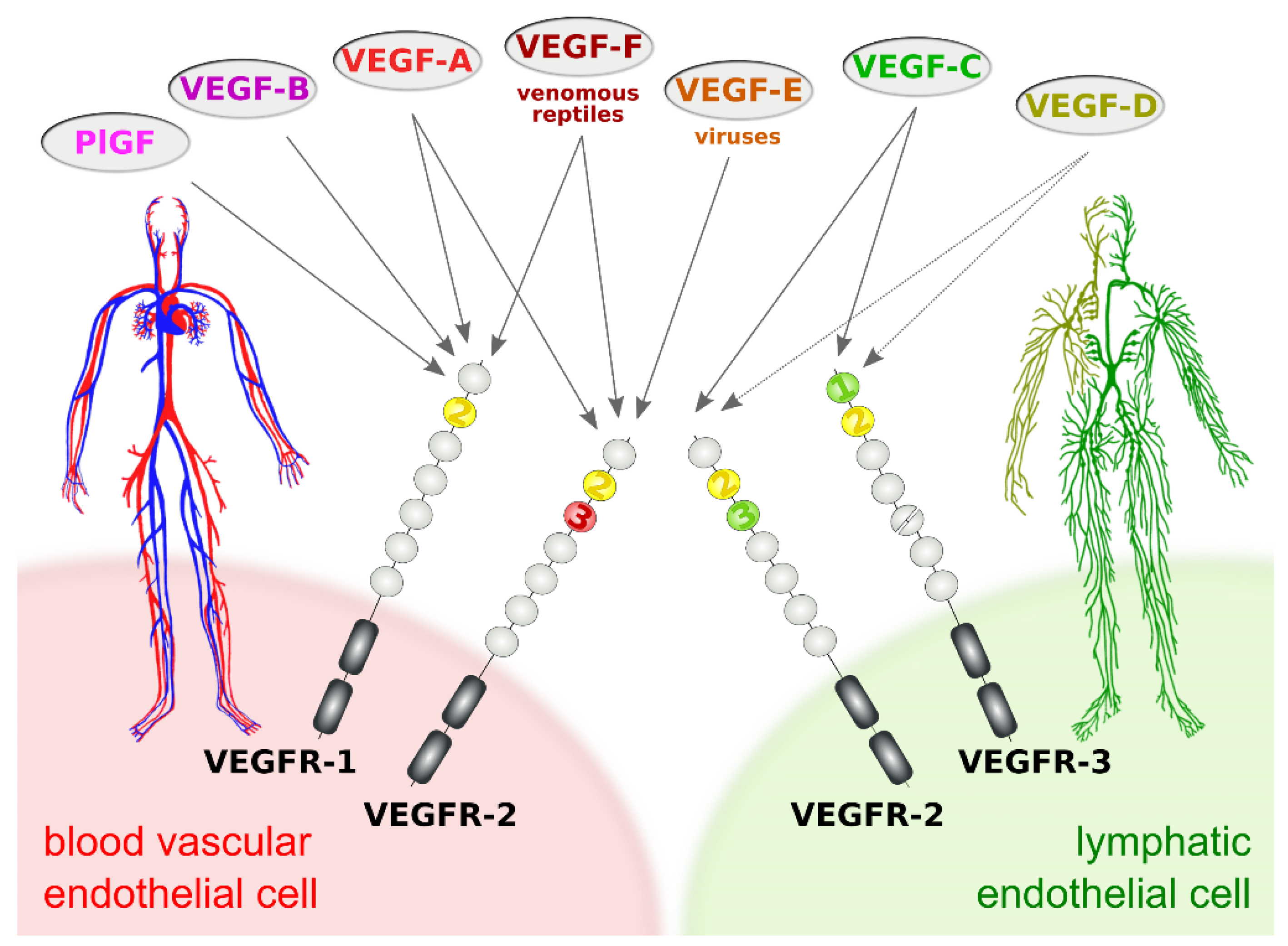
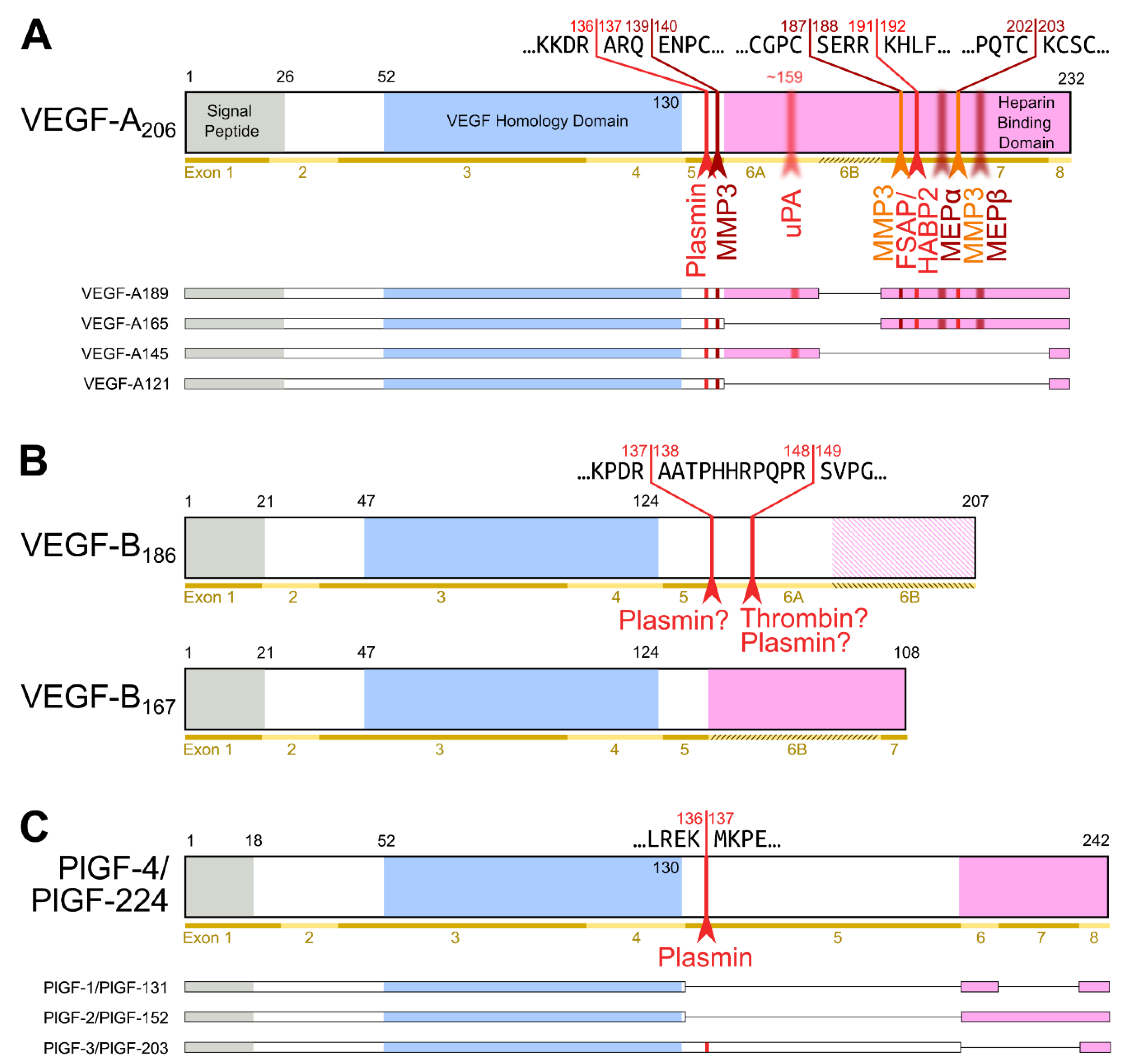
2. Proteolytic Processing of the Hemangiogenic VEGFs
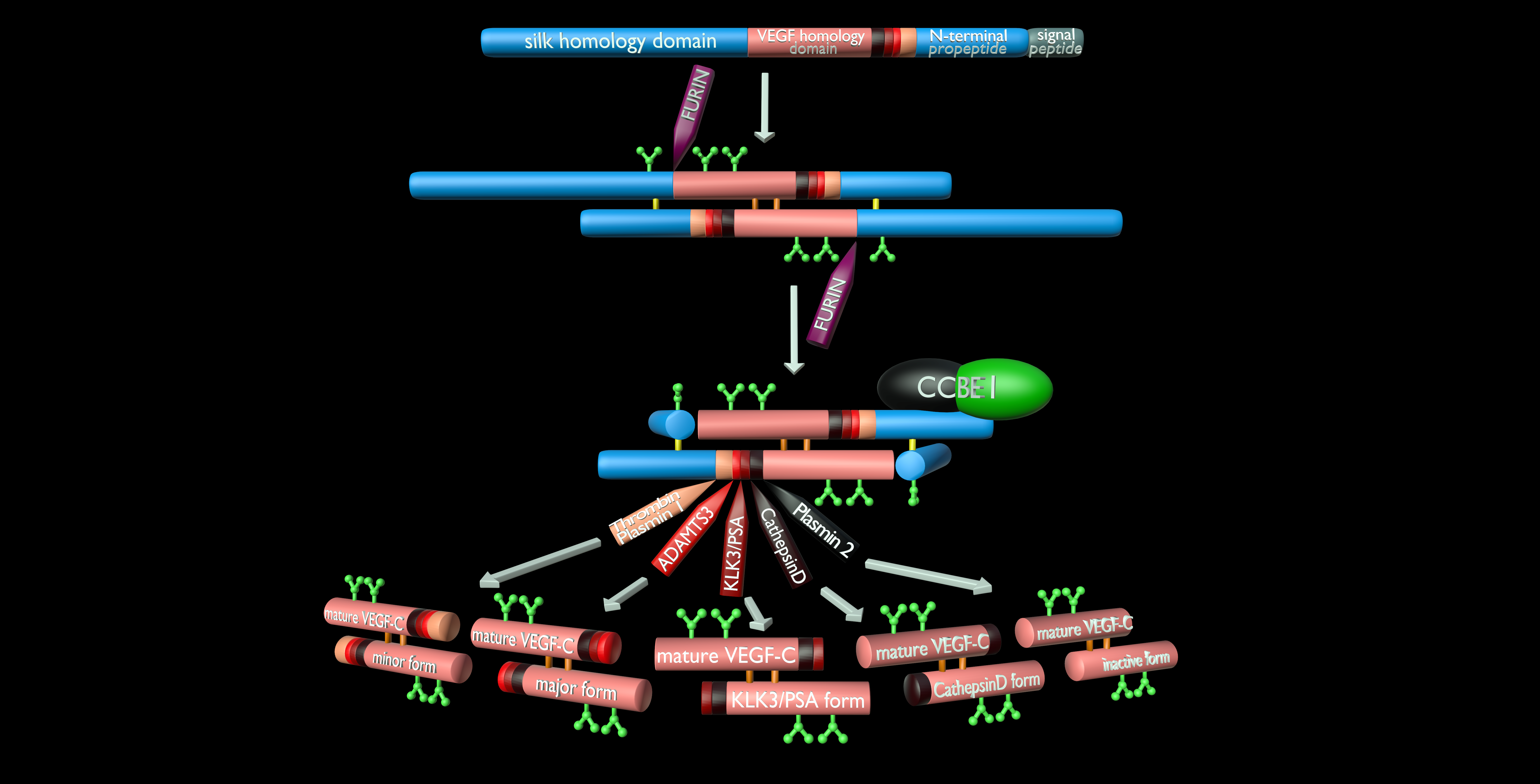
3. The Lymphangiogenic Growth Factors VEGF-C and VEGF-D
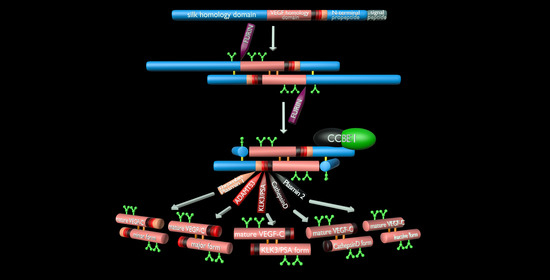
- Protein convertases constitutively cleave VEGF-C before secretion. This intracellular cleavage occurs between the central VEGF homology domain (VHD) and the C-terminal propeptide. However, it does not remove the C-terminal propeptide because it remains covalently attached to the rest of the molecule by disulfide bonds [38,39,40].
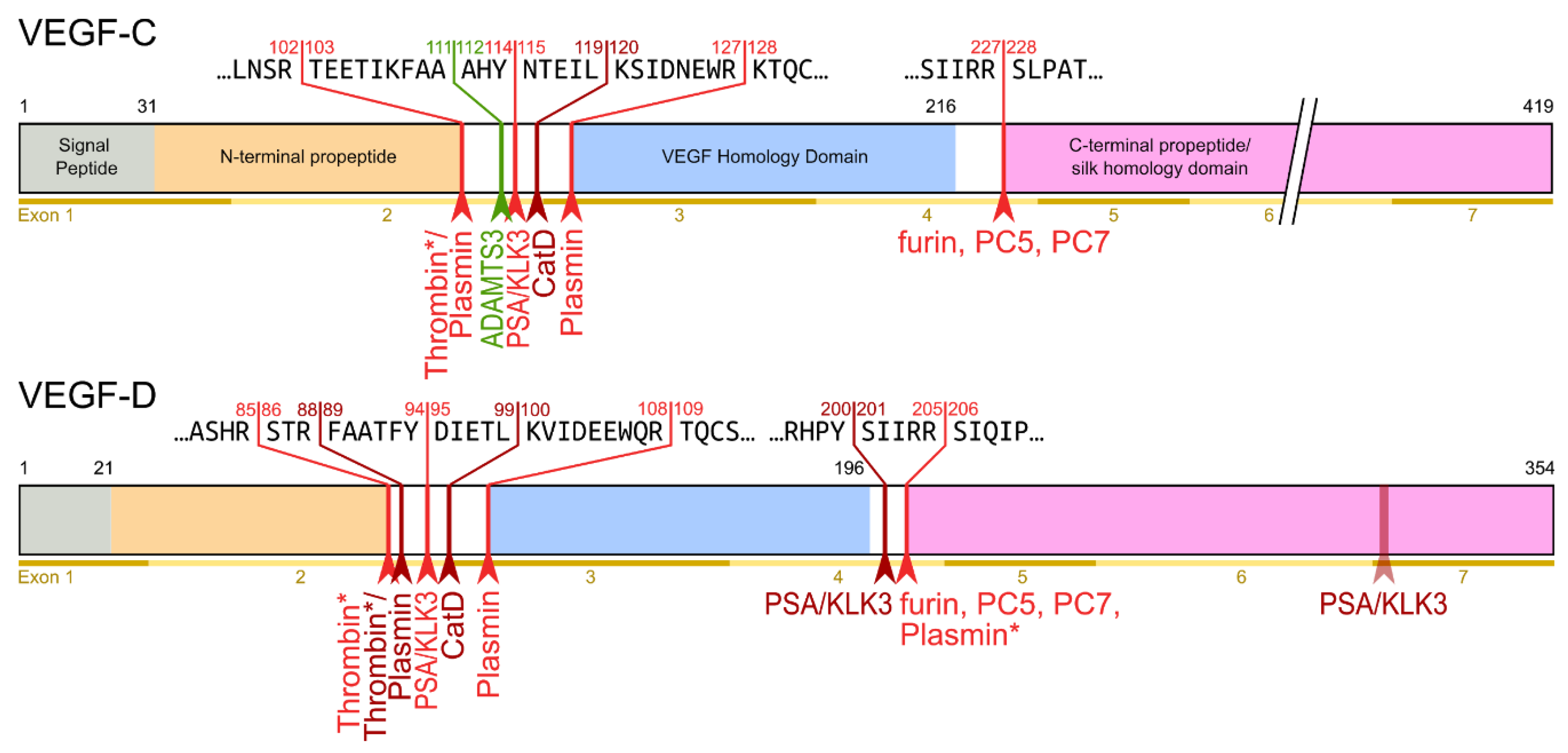
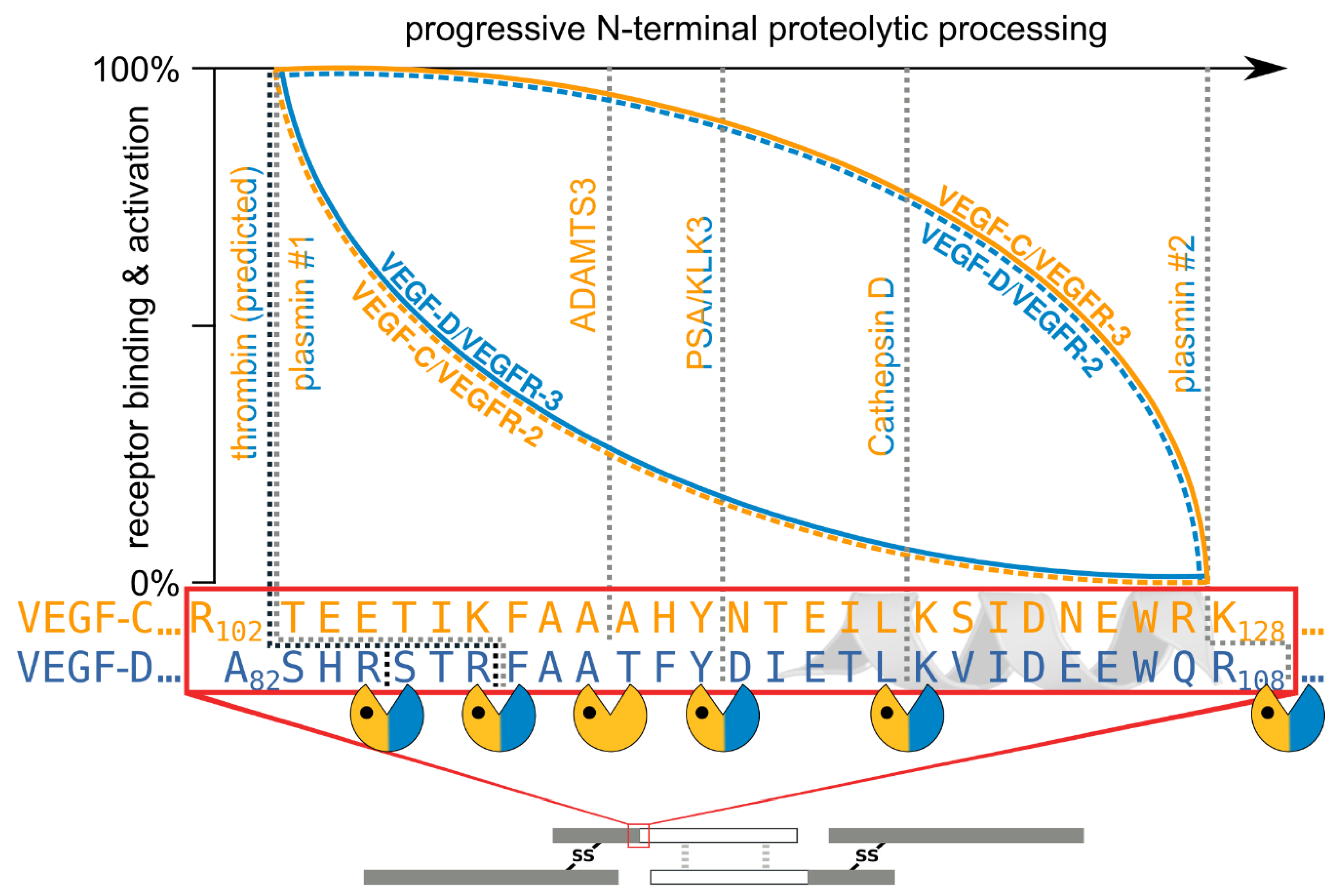
- The second, extracellular cleavage activates the protein. This cleavage occurs between the N-terminal propeptide and the VHD [38] and can be mediated by different proteases. ADAMTS3 mediates VEGF-C activation in the embryonic development of the mammalian lymphatic system [41,42,43]. ADAMTS3 is specific for VEGF-C and does not activate VEGF-D. All other activating proteases target both VEGF-C and VEGF-D: plasmin [43,44], prostate-specific antigen (KLK3/PSA), cathepsin D (CatD) [45], and thrombin [46]. The resulting forms of VEGF-C and VEGF-D are referred to as active, mature, or short forms. However, they differ from each other at their N-termini because different proteases cleave at different positions within the linker between the N-terminal propeptide and the VHD (see Figure 4).
4. Plasmin and Thrombin
5. ADAMTS3 and the Cofactor CCBE1
6. Species-Specific Differences
7. Which Cell Types Provide ADAMTS3 and CCBE1?
8. Enigmatic Propeptides
- The presence of the C-terminal propeptide is required for efficient cleavage of the N-terminal propeptide by ADAMTS3 [63].
9. Changing Receptor Preferences with KLK3 and Cathepsin D
- The shorter the N-terminus of the resulting mature growth factor, the lower its receptor binding affinity and receptor activation potential.
- N-terminal shortening affects VEGF-C and VEGF-D very differently. While VEGF-C rapidly loses its potential to activate VEGFR-2 (through activation by ADAMTS3 or PSA), VEGF-D maintains much of its VEGFR-2 binding and activation potential. Vice versa, VEGF-D rapidly loses its VEGFR-3 binding and activation potential, whereas VEGF-C maintains much of it when processed to a similar degree.
- Both VEGF-C and VEGF-D are completely inactivated with respect to their receptor tyrosine kinase activity by complete removal of their N-terminal helices, which, e.g., can be achieved by prolonged exposure to plasmin.
10. Secondary Processing and Inactivation
11. Other Cleavages
12. Possible Involvement in Reproduction and Wound Healing
13. Activating VEGF-C and VEGF-D in Cell Culture
14. Truncated cDNAs Are Used to Recombinantly Express Pre-Activated VEGF-C and VEGF-D
15. Modulation of Proteolytic Processing
16. Proteolytic Activation of VEGF-C and VEGF-D in Cancer
17. Blocking VEGF-C and VEGF-D Activation
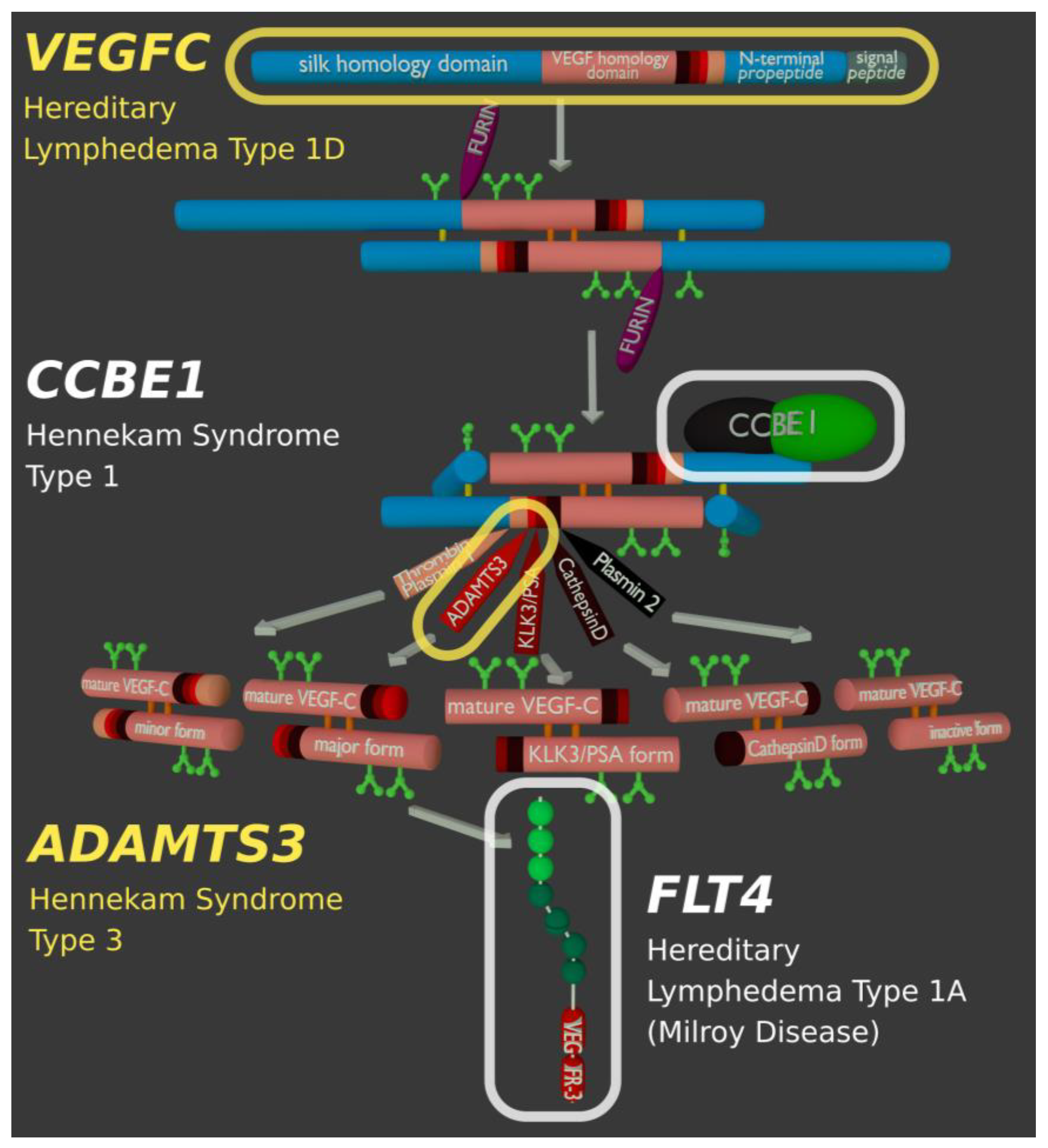
18. Lymphedema and Genetic Lesions Affecting the Activation of VEGF-C
19. Outlook: Molecular Nudging
Author Contributions
Funding
Institutional Review Board Statement
Informed Consent Statement
Data Availability Statement
Conflicts of Interest
References
- de Groof, A.; Guelen, L.; Deijs, M.; van der Wal, Y.; Miyata, M.; Ng, K.S.; van Grinsven, L.; Simmelink, B.; Biermann, Y.; Grisez, L.; et al. A Novel Virus Causes Scale Drop Disease in Lates Calcarifer. PLOS Pathog. 2015, 11, e1005074. [Google Scholar] [CrossRef] [Green Version]
- Lyttle, D.J.; Fraser, K.M.; Fleming, S.B.; Mercer, A.A.; Robinson, A.J. Homologs of Vascular Endothelial Growth Factor Are Encoded by the Poxvirus Orf Virus. J. Virol. 1994, 68, 84–92. [Google Scholar] [CrossRef] [Green Version]
- Meyer, M.; Clauss, M.; Lepple-Wienhues, A.; Waltenberger, J.; Augustin, H.G.; Ziche, M.; Lanz, C.; Büttner, M.; Rziha, H.-J.; Dehio, C. A Novel Vascular Endothelial Growth Factor Encoded by Orf Virus, VEGF-E, Mediates Angiogenesis via Signalling through VEGFR-2 (KDR) but Not VEGFR-1 (Flt-1) Receptor Tyrosine Kinases. EMBO J. 1999, 18, 363–374. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Ogawa, S.; Oku, A.; Sawano, A.; Yamaguchi, S.; Yazaki, Y.; Shibuya, M. A Novel Type of Vascular Endothelial Growth Factor, VEGF-E (NZ-7 VEGF), Preferentially Utilizes KDR/Flk-1 Receptor and Carries a Potent Mitotic Activity without Heparin-Binding Domain. J. Biol. Chem. 1998, 273, 31273–31282. [Google Scholar] [CrossRef] [Green Version]
- Yamazaki, Y.; Matsunaga, Y.; Tokunaga, Y.; Obayashi, S.; Saito, M.; Morita, T. Snake Venom Vascular Endothelial Growth Factors (VEGF-Fs) Exclusively Vary Their Structures and Functions among Species. J. Biol. Chem. 2009, 284, 9885–9891. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Wiesmann, C.; Fuh, G.; Christinger, H.W.; Eigenbrot, C.; Wells, J.A.; de Vos, A.M. Crystal Structure at 1.7 Å Resolution of VEGF in Complex with Domain 2 of the Flt-1 Receptor. Cell 1997, 91, 695–704. [Google Scholar] [CrossRef] [Green Version]
- Fuh, G.; Li, B.; Crowley, C.; Cunningham, B.; Wells, J.A. Requirements for Binding and Signaling of the Kinase Domain Receptor for Vascular Endothelial Growth Factor. J. Biol. Chem. 1998, 273, 11197–11204. [Google Scholar] [CrossRef] [Green Version]
- Leppänen, V.-M.; Prota, A.E.; Jeltsch, M.; Anisimov, A.; Kalkkinen, N.; Strandin, T.; Lankinen, H.; Goldman, A.; Ballmer-Hofer, K.; Alitalo, K. Structural Determinants of Growth Factor Binding and Specificity by VEGF Receptor 2. Proc. Natl. Acad. Sci. USA 2010, 107, 2425–2430. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Leppänen, V.-M.; Tvorogov, D.; Kisko, K.; Prota, A.E.; Jeltsch, M.; Anisimov, A.; Markovic-Mueller, S.; Stuttfeld, E.; Goldie, K.N.; Ballmer-Hofer, K.; et al. Structural and Mechanistic Insights into VEGF Receptor 3 Ligand Binding and Activation. Proc. Natl. Acad. Sci. USA 2013, 110, 12960–12965. [Google Scholar] [CrossRef] [Green Version]
- Apte, R.S.; Chen, D.S.; Ferrara, N. VEGF in Signaling and Disease: Beyond Discovery and Development. Cell 2019, 176, 1248–1264. [Google Scholar] [CrossRef] [Green Version]
- Simons, M.; Gordon, E.; Claesson-Welsh, L. Mechanisms and Regulation of Endothelial VEGF Receptor Signalling. Nat. Rev. Mol. Cell Biol. 2016, 17, 611–625. [Google Scholar] [CrossRef] [PubMed]
- Carmeliet, P.; Ferreira, V.; Breier, G.; Pollefeyt, S.; Kieckens, L.; Gertsenstein, M.; Fahrig, M.; Vandenhoeck, A.; Harpal, K.; Eberhardt, C.; et al. Abnormal Blood Vessel Development and Lethality in Embryos Lacking a Single VEGF Allele. Nature 1996, 380, 435–439. [Google Scholar] [CrossRef] [PubMed]
- Ferrara, N.; Carver-Moore, K.; Chen, H.; Dowd, M.; Lu, L.; O’Shea, K.S.; Powell-Braxton, L.; Hillan, K.J.; Moore, M.W. Heterozygous Embryonic Lethality Induced by Targeted Inactivation of the VEGF Gene. Nature 1996, 380, 439–442. [Google Scholar] [CrossRef] [Green Version]
- Karkkainen, M.J.; Haiko, P.; Sainio, K.; Partanen, J.; Taipale, J.; Petrova, T.V.; Jeltsch, M.; Jackson, D.G.; Talikka, M.; Rauvala, H.; et al. Vascular Endothelial Growth Factor C Is Required for Sprouting of the First Lymphatic Vessels from Embryonic Veins. Nat. Immunol. 2003, 5, 74–80. [Google Scholar] [CrossRef]
- Rodamilans, B.; Shan, H.; Pasin, F.; García, J.A. Plant Viral Proteases: Beyond the Role of Peptide Cutters. Front. Plant Sci. 2018, 9. [Google Scholar] [CrossRef] [Green Version]
- van der Hoorn, R.A.L.; Rivas, S. Unravelling the Mode of Action of Plant Proteases. New Phytol. 2018, 218, 879–881. [Google Scholar] [CrossRef] [Green Version]
- Künnapuu, J.; Björkgren, I.; Shimmi, O. The Drosophila DPP Signal Is Produced by Cleavage of Its Proprotein at Evolutionary Diversified Furin-Recognition Sites. Proc. Natl. Acad. Sci. USA 2009, 106, 8501–8506. [Google Scholar] [CrossRef] [Green Version]
- Turk, B.; Turk, D.; Turk, V. Protease Signalling: The Cutting Edge. EMBO J. 2012, 31, 1630–1643. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Vempati, P.; Popel, A.S.; Mac Gabhann, F. Extracellular Regulation of VEGF: Isoforms, Proteolysis, and Vascular Patterning. Cytokine Growth Factor Rev. 2014, 25, 1–19. [Google Scholar] [CrossRef] [Green Version]
- Bridgett, S.; Dellett, M.; Simpson, D.A. RNA-Sequencing Data Supports the Existence of Novel VEGFA Splicing Events but Not of VEGFA Xxx b Isoforms. Sci. Rep. 2017, 7, 58. [Google Scholar] [CrossRef] [Green Version]
- Keyt, B.A.; Berleau, L.T.; Nguyen, H.V.; Chen, H.; Heinsohn, H.; Vandlen, R.; Ferrara, N. The Carboxyl-Terminal Domain(111165) of Vascular Endothelial Growth Factor Is Critical for Its Mitogenic Potency. J. Biol. Chem. 1996, 271, 7788–7795. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Park, J.E.; Keller, G.A.; Ferrara, N. The Vascular Endothelial Growth Factor (VEGF) Isoforms: Differential Deposition into the Subepithelial Extracellular Matrix and Bioactivity of Extracellular Matrix-Bound VEGF. Mol. Biol. Cell 1993, 4, 1317–1326. [Google Scholar] [CrossRef] [Green Version]
- Carmeliet, P.; Ng, Y.-S.; Nuyens, D.; Theilmeier, G.; Brusselmans, K.; Cornelissen, I.; Ehler, E.; Kakkar, V.V.; Stalmans, I.; Mattot, V.; et al. Impaired Myocardial Angiogenesis and Ischemic Cardiomyopathy in Mice Lacking the Vascular Endothelial Growth Factor Isoforms VEGF 164 and VEGF 188. Nat. Med. 1999, 5, 495–502. [Google Scholar] [CrossRef]
- Ruhrberg, C.; Gerhardt, H.; Golding, M.; Watson, R.; Ioannidou, S.; Fujisawa, H.; Betsholtz, C.; Shima, D.T. Spatially Restricted Patterning Cues Provided by Heparin-Binding VEGF-A Control Blood Vessel Branching Morphogenesis. Genes Dev. 2002, 16, 2684–2698. [Google Scholar] [CrossRef] [Green Version]
- Gerhardt, H.; Golding, M.; Fruttiger, M.; Ruhrberg, C.; Lundkvist, A.; Abramsson, A.; Jeltsch, M.; Mitchell, C.; Alitalo, K.; Shima, D.; et al. VEGF Guides Angiogenic Sprouting Utilizing Endothelial Tip Cell Filopodia. J. Cell Biol. 2003, 161, 1163–1177. [Google Scholar] [CrossRef] [PubMed]
- Plouët, J.; Moro, F.; Bertagnolli, S.; Coldeboeuf, N.; Mazarguil, H.; Clamens, S.; Bayard, F. Extracellular Cleavage of the Vascular Endothelial Growth Factor 189-Amino Acid Form by Urokinase Is Required for Its Mitogenic Effect. J. Biol. Chem. 1997, 272, 13390–13396. [Google Scholar] [CrossRef] [Green Version]
- Houck, K.A.; Leung, D.W.; Rowland, A.M.; Winer, J.; Ferrara, N. Dual Regulation of Vascular Endothelial Growth Factor Bioavailability by Genetic and Proteolytic Mechanisms. J. Biol. Chem. 1992, 267, 26031–26037. [Google Scholar] [CrossRef]
- Ferrara, N. Binding to the Extracellular Matrix and Proteolytic Processing: Two Key Mechanisms Regulating Vascular Endothelial Growth Factor Action. Mol. Biol. Cell 2010, 21, 687–690. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Uslu, Ö.; Herold, J.; Kanse, S.M. VEGF-A-Cleavage by FSAP and Inhibition of Neo-Vascularization. Cells 2019, 8, 1396. [Google Scholar] [CrossRef] [Green Version]
- Lee, S.; Jilani, S.M.; Nikolova, G.V.; Carpizo, D.; Iruela-Arispe, M.L. Processing of VEGF-A by Matrix Metalloproteinases Regulates Bioavailability and Vascular Patterning in Tumors. J. Cell Biol. 2005, 169, 681–691. [Google Scholar] [CrossRef] [Green Version]
- Eming, S.A.; Krieg, T. Molecular Mechanisms of VEGF-A Action during Tissue Repair. J. Investig. Dermatol. Symp. Proc. 2006, 11, 79–86. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Hoffmann, D.C.; Willenborg, S.; Koch, M.; Zwolanek, D.; Müller, S.; Becker, A.-K.A.; Metzger, S.; Ehrbar, M.; Kurschat, P.; Hellmich, M.; et al. Proteolytic Processing Regulates Placental Growth Factor Activities. J. Biol. Chem. 2013, 288, 17976–17989. [Google Scholar] [CrossRef] [Green Version]
- Makinen, T.; Olofsson, B.; Karpanen, T.; Hellman, U.; Soker, S.; Klagsbrun, M.; Eriksson, U.; Alitalo, K. Differential Binding of Vascular Endothelial Growth Factor B Splice and Proteolytic Isoforms to Neuropilin-1. J. Biol. Chem. 1999, 274, 21217–21222. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Olofsson, B.; Pajusola, K.; von Euler, G.; Chilov, D.; Alitalo, K.; Eriksson, U. Genomic Organization of the Mouse and Human Genes for Vascular Endothelial Growth Factor B (VEGF-B) and Characterization of a Second Splice Isoform. J. Biol. Chem. 1996, 271, 19310–19317. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Rawlings, N.D.; Barrett, A.J.; Thomas, P.D.; Huang, X.; Bateman, A.; Finn, R.D. The MEROPS Database of Proteolytic Enzymes, Their Substrates and Inhibitors in 2017 and a Comparison with Peptidases in the PANTHER Database. Nucleic Acids Res. 2018, 46, D624–D632. [Google Scholar] [CrossRef]
- Yamazaki, Y.; Morita, T. Molecular and Functional Diversity of Vascular Endothelial Growth Factors. Mol. Divers. 2006, 10, 515–527. [Google Scholar] [CrossRef]
- Stenberg, L.M.; Brown, M.A.; Nilsson, E.; Ljungberg, O.; Stenflo, J. A Functional Prothrombin Gene Product Is Synthesized by Human Kidney Cells. Biochem. Biophys. Res. Commun. 2001, 280, 1036–1041. [Google Scholar] [CrossRef] [PubMed]
- Joukov, V.; Sorsa, T.; Kumar, V.; Jeltsch, M.; Claesson-Welsh, L.; Cao, Y.; Saksela, O.; Kalkkinen, N.; Alitalo, K. Proteolytic Processing Regulates Receptor Specificity and Activity of VEGF-C. EMBO J. 1997, 16, 3898–3911. [Google Scholar] [CrossRef] [Green Version]
- McColl, B.K.; Paavonen, K.; Karnezis, T.; Harris, N.C.; Davydova, N.; Rothacker, J.; Nice, E.C.; Harder, K.W.; Roufail, S.; Hibbs, M.L.; et al. Proprotein Convertases Promote Processing of VEGF-D, a Critical Step for Binding the Angiogenic Receptor VEGFR-2. FASEB J. 2007, 21, 1088–1098. [Google Scholar] [CrossRef] [PubMed]
- Siegfried, G.; Basak, A.; Cromlish, J.A.; Benjannet, S.; Marcinkiewicz, J.; Chrétien, M.; Seidah, N.G.; Khatib, A.-M. The Secretory Proprotein Convertases Furin, PC5, and PC7 Activate VEGF-C to Induce Tumorigenesis. J. Clin. Investig. 2003, 111, 1723–1732. [Google Scholar] [CrossRef] [Green Version]
- Bui, H.M.; Enis, D.; Robciuc, M.R.; Nurmi, H.J.; Cohen, J.; Chen, M.; Yang, Y.; Dhillon, V.; Johnson, K.; Zhang, H.; et al. Proteolytic Activation Defines Distinct Lymphangiogenic Mechanisms for VEGFC and VEGFD. J. Clin. Investig. 2016, 126, 2167–2180. [Google Scholar] [CrossRef] [Green Version]
- Janssen, L.; Dupont, L.; Bekhouche, M.; Noel, A.; Leduc, C.; Voz, M.; Peers, B.; Cataldo, D.; Apte, S.S.; Dubail, J.; et al. ADAMTS3 Activity Is Mandatory for Embryonic Lymphangiogenesis and Regulates Placental Angiogenesis. Angiogenesis 2015, 1–13. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Jeltsch, M.; Jha, S.K.; Tvorogov, D.; Anisimov, A.; Leppänen, V.-M.; Holopainen, T.; Kivelä, R.; Ortega, S.; Kärpanen, T.; Alitalo, K. CCBE1 Enhances Lymphangiogenesis via A Disintegrin and Metalloprotease With Thrombospondin Motifs-3–Mediated Vascular Endothelial Growth Factor-C Activation. Circulation 2014, 129, 1962–1971. [Google Scholar] [CrossRef] [Green Version]
- McColl, B.K.; Baldwin, M.E.; Roufail, S.; Freeman, C.; Moritz, R.L.; Simpson, R.J.; Alitalo, K.; Stacker, S.A.; Achen, M.G. Plasmin Activates the Lymphangiogenic Growth Factors VEGF-C and VEGF-D. J. Exp. Med. 2003, 198, 863–868. [Google Scholar] [CrossRef]
- Jha, S.K.; Rauniyar, K.; Chronowska, E.; Mattonet, K.; Maina, E.W.; Koistinen, H.; Stenman, U.-H.; Alitalo, K.; Jeltsch, M. KLK3/PSA and Cathepsin D Activate VEGF-C and VEGF-D. eLife 2019, 8, e44478. [Google Scholar] [CrossRef] [PubMed]
- Lim, L.; Bui, H.; Farrelly, O.; Yang, J.; Li, L.; Enis, D.; Ma, W.; Chen, M.; Oliver, G.; Welsh, J.D.; et al. Hemostasis Stimulates Lymphangiogenesis through Release and Activation of VEGFC. Blood 2019, 134, 1764–1775. [Google Scholar] [CrossRef]
- Kärpänen, T.; Heckman, C.A.; Keskitalo, S.; Jeltsch, M.; Ollila, H.; Neufeld, G.; Tamagnone, L.; Alitalo, K. Functional Interaction of VEGF-C and VEGF-D with Neuropilin Receptors. FASEB J. 2006, 20, 1462–1472. [Google Scholar] [CrossRef] [Green Version]
- Wartiovaara, U.; Salven, P.; Mikkola, H.; Lassila, R.; Kaukonen, J.; Joukov, V.; Orpana, A.; Ristimäki, A.; Heikinheimo, M.; Joensuu, H.; et al. Peripheral Blood Platelets Express VEGF-C and VEGF Which Are Released during Platelet Activation. Thromb. Haemost. 1998, 80, 171–175. [Google Scholar] [CrossRef] [PubMed]
- Akgül, A.; Cirak, M.; Birinci, T. Applications of Platelet-Rich Plasma in Lymphedema. Lymphat. Res. Biol. 2016, 14, 206–209. [Google Scholar] [CrossRef] [PubMed]
- Margolis, D.J.; Kantor, J.; Santanna, J.; Strom, B.L.; Berlin, J.A. Effectiveness of Platelet Releasate for the Treatment of Diabetic Neuropathic Foot Ulcers. Diabetes Care 2001, 24, 483–488. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Lee, H.; Chae, S.; Park, J.; Bae, J.; Go, E.-B.; Kim, S.-J.; Kim, H.; Hwang, D.; Lee, S.-W.; Lee, S.-Y. Comprehensive Proteome Profiling of Platelet Identified a Protein Profile Predictive of Responses to An Antiplatelet Agent Sarpogrelate. Mol. Cell. Proteom. 2016, 15, 3461–3472. [Google Scholar] [CrossRef] [Green Version]
- Yadav, D.B.; Reyes, A.E.; Gupta, P.; Vernes, J.-M.; Meng, Y.G.; Schweiger, M.G.; Stainton, S.L.; Fuh, G.; Fielder, P.J.; Kamath, A.V.; et al. Complex Formation of Anti-VEGF-C with VEGF-C Released during Blood Coagulation Resulted in an Artifact in Its Serum Pharmacokinetics. Pharmacol. Res. Perspect. 2020, 8, e00573. [Google Scholar] [CrossRef] [Green Version]
- Baldwin, M.E.; Halford, M.M.; Roufail, S.; Williams, R.A.; Hibbs, M.L.; Grail, D.; Kubo, H.; Stacker, S.A.; Achen, M.G. Vascular Endothelial Growth Factor D Is Dispensable for Development of the Lymphatic System. Mol. Cell. Biol. 2005, 25, 2441–2449. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Lee, J.; Gray, A.; Yuan, J.; Luoh, S.M.; Avraham, H.; Wood, W.I. Vascular Endothelial Growth Factor-Related Protein: A Ligand and Specific Activator of the Tyrosine Kinase Receptor Flt4. Proc. Natl. Acad. Sci. USA 1996, 93, 1988–1992. [Google Scholar] [CrossRef] [Green Version]
- Bugge, T.H.; Flick, M.J.; Daugherty, C.C.; Degen, J.L. Plasminogen Deficiency Causes Severe Thrombosis but Is Compatible with Development and Reproduction. Genes Dev. 1995, 9, 794–807. [Google Scholar] [CrossRef] [Green Version]
- Kikuchi, S.; Yamanouchi, Y.; Li, L.; Kobayashi, K.; Ijima, H.; Miyazaki, R.; Tsuchiya, S.; Hamaguchi, H. Plasminogen with Type-I Mutation Is Polymorphic in the Japanese Population. Hum. Genet. 1992, 90, 7–11. [Google Scholar] [CrossRef] [PubMed]
- Alders, M.; Hogan, B.M.; Gjini, E.; Salehi, F.; Al-Gazali, L.; Hennekam, E.A.; Holmberg, E.E.; Mannens, M.M.A.M.; Mulder, M.F.; Offerhaus, G.J.A.; et al. Mutations in CCBE1 Cause Generalized Lymph Vessel Dysplasia in Humans. Nat. Genet. 2009, 41, 1272–1274. [Google Scholar] [CrossRef]
- Hennekam, R.C.M.; Geerdink, R.A.; Hamel, B.C.J.; Hennekam, F.A.M.; Kraus, P.; Rammeloo, J.A.; Tillemans, A.A.W. Autosomal Recessive Intestinal Lymphangiectasia and Lymphedema, with Facial Anomalies and Mental Retardation. Am. J. Med. Genet. 1989, 34, 593–600. [Google Scholar] [CrossRef] [PubMed]
- Hogan, B.M.; Bos, F.L.; Bussmann, J.; Witte, M.; Chi, N.C.; Duckers, H.J.; Schulte-Merker, S. Ccbe1 Is Required for Embryonic Lymphangiogenesis and Venous Sprouting. Nat. Genet. 2009, 41, 396–398. [Google Scholar] [CrossRef]
- Bos, F.L.; Caunt, M.; Peterson-Maduro, J.; Planas-Paz, L.; Kowalski, J.; Karpanen, T.; Van Impel, A.; Tong, R.; Ernst, J.A.; Korving, J.; et al. CCBE1 Is Essential for Mammalian Lymphatic Vascular Development and Enhances the Lymphangiogenic Effect of Vascular Endothelial Growth Factor-C In Vivo. Circ. Res. 2011, 109, 486–491. [Google Scholar] [CrossRef]
- Fernandes, R.J.; Hirohata, S.; Engle, J.M.; Colige, A.; Cohn, D.H.; Eyre, D.R.; Apte, S.S. Procollagen II Amino Propeptide Processing by ADAMTS-3 INSIGHTS ON DERMATOSPARAXIS. J. Biol. Chem. 2001, 276, 31502–31509. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Brouillard, P.; Dupont, L.; Helaers, R.; Coulie, R.; Tiller, G.E.; Peeden, J.; Colige, A.; Vikkula, M. Loss of ADAMTS3 Activity Causes Hennekam Lymphangiectasia–Lymphedema Syndrome 3. Hum. Mol. Genet. 2017, 26, 4095–4104. [Google Scholar] [CrossRef] [Green Version]
- Jha, S.K.; Rauniyar, K.; Karpanen, T.; Leppänen, V.-M.; Brouillard, P.; Vikkula, M.; Alitalo, K.; Jeltsch, M. Efficient Activation of the Lymphangiogenic Growth Factor VEGF-C Requires the C-Terminal Domain of VEGF-C and the N-Terminal Domain of CCBE1. Sci. Rep. 2017, 7, 4916. [Google Scholar] [CrossRef] [PubMed]
- Le Guen, L.; Karpanen, T.; Schulte, D.; Harris, N.C.; Koltowska, K.; Roukens, G.; Bower, N.I.; van Impel, A.; Stacker, S.A.; Achen, M.G.; et al. Ccbe1 Regulates Vegfc-Mediated Induction of Vegfr3 Signaling during Embryonic Lymphangiogenesis. Development 2014, 141, 1239–1249. [Google Scholar] [CrossRef] [Green Version]
- Wang, G.; Muhl, L.; Padberg, Y.; Dupont, L.; Peterson-Maduro, J.; Stehling, M.; le Noble, F.; Colige, A.; Betsholtz, C.; Schulte-Merker, S.; et al. Specific Fibroblast Subpopulations and Neuronal Structures Provide Local Sources of Vegfc-Processing Components during Zebrafish Lymphangiogenesis. Nat. Commun. 2020, 11, 2724. [Google Scholar] [CrossRef] [PubMed]
- Bower, N.I.; Vogrin, A.J.; Guen, L.L.; Chen, H.; Stacker, S.A.; Achen, M.G.; Hogan, B.M. Vegfd Modulates Both Angiogenesis and Lymphangiogenesis during Zebrafish Embryonic Development. Development 2017, 144, 507–518. [Google Scholar] [CrossRef] [Green Version]
- Astin, J.W.; Haggerty, M.J.L.; Okuda, K.S.; Le Guen, L.; Misa, J.P.; Tromp, A.; Hogan, B.M.; Crosier, K.E.; Crosier, P.S. Vegfd Can Compensate for Loss of Vegfc in Zebrafish Facial Lymphatic Sprouting. Development 2014, 141, 2680–2690. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Baldwin, M.E.; Catimel, B.; Nice, E.C.; Roufail, S.; Hall, N.E.; Stenvers, K.L.; Karkkainen, M.J.; Alitalo, K.; Stacker, S.A.; Achen, M.G. The Specificity of Receptor Binding by Vascular Endothelial Growth Factor-D Is Different in Mouse and Man. J. Biol. Chem. 2001, 276, 19166–19171. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Vogrin, A.J.; Bower, N.I.; Gunzburg, M.J.; Roufail, S.; Okuda, K.S.; Paterson, S.; Headey, S.J.; Stacker, S.A.; Hogan, B.M.; Achen, M.G. Evolutionary Differences in the Vegf/Vegfr Code Reveal Organotypic Roles for the Endothelial Cell Receptor Kdr in Developmental Lymphangiogenesis. Cell Rep. 2019, 28, 2023–2036.e4. [Google Scholar] [CrossRef] [Green Version]
- Nurmi, H.; Saharinen, P.; Zarkada, G.; Zheng, W.; Robciuc, M.R.; Alitalo, K. VEGF-C Is Required for Intestinal Lymphatic Vessel Maintenance and Lipid Absorption. EMBO Mol. Med. 2015, 7, 1418–1425. [Google Scholar] [CrossRef]
- Fang, S.; Chen, S.; Nurmi, H.; Leppänen, V.-M.; Jeltsch, M.; Scadden, D.T.; Silberstein, L.; Mikkola, H.; Alitalo, K. VEGF-C Protects the Integrity of Bone Marrow Perivascular Niche. Blood 2020, 136, 1871–1883. [Google Scholar] [CrossRef] [PubMed]
- Fang, S.; Nurmi, H.; Heinolainen, K.; Chen, S.; Salminen, E.; Saharinen, P.; Mikkola, H.K.A.; Alitalo, K. Critical Requirement of VEGF-C in Transition to Fetal Erythropoiesis. Blood 2016, 128, 710–720. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Hasselhof, V.; Sperling, A.; Buttler, K.; Ströbel, P.; Becker, J.; Aung, T.; Felmerer, G.; Wilting, J. Morphological and Molecular Characterization of Human Dermal Lymphatic Collectors. PLOS ONE 2016, 11, e0164964. [Google Scholar] [CrossRef]
- Facucho-Oliveira, J.; Bento, M.; Belo, J.-A. Ccbe1 Expression Marks the Cardiac and Lymphatic Progenitor Lineages during Early Stages of Mouse Development. Int. J. Dev. Biol. 2011, 55, 1007–1014. [Google Scholar] [CrossRef]
- Schoppmann, S.F.; Birner, P.; Stöckl, J.; Kalt, R.; Ullrich, R.; Caucig, C.; Kriehuber, E.; Nagy, K.; Alitalo, K.; Kerjaschki, D. Tumor-Associated Macrophages Express Lymphatic Endothelial Growth Factors and Are Related to Peritumoral Lymphangiogenesis. Am. J. Pathol. 2002, 161, 947–956. [Google Scholar] [CrossRef] [Green Version]
- Machnik, A.; Neuhofer, W.; Jantsch, J.; Dahlmann, A.; Tammela, T.; Machura, K.; Park, J.-K.; Beck, F.-X.; Müller, D.N.; Derer, W.; et al. Macrophages Regulate Salt-Dependent Volume and Blood Pressure by a Vascular Endothelial Growth Factor-C–Dependent Buffering Mechanism. Nat. Med. 2009, 15, 545–552. [Google Scholar] [CrossRef] [PubMed]
- Harvey, N.L.; Gordon, E.J. Deciphering the Roles of Macrophages in Developmental and Inflammation Stimulated Lymphangiogenesis. Vasc. Cell 2012, 4, 15. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Heino, T.I.; Kärpänen, T.; Wahlström, G.; Pulkkinen, M.; Eriksson, U.; Alitalo, K.; Roos, C. The Drosophila VEGF Receptor Homolog Is Expressed in Hemocytes. Mech. Dev. 2001, 109, 69–77. [Google Scholar] [CrossRef]
- Seipel, K.; Eberhardt, M.; Müller, P.; Pescia, E.; Yanze, N.; Schmid, V. Homologs of Vascular Endothelial Growth Factor and Receptor, VEGF and VEGFR, in the Jellyfish Podocoryne carnea. Dev. Dyn. 2004, 231, 303–312. [Google Scholar] [CrossRef] [PubMed]
- Tarsitano, M.; Falco, S.D.; Colonna, V.; McGhee, J.D.; Persico, M.G. The C. Elegans Pvf-1 Gene Encodes a PDGF/VEGF-like Factor Able to Bind Mammalian VEGF Receptors and to Induce Angiogenesis. FASEB J. 2006, 20, 227–233. [Google Scholar] [CrossRef]
- Case, S.T.; Cox, C.; Bell, W.C.; Hoffman, R.T.; Martin, J.; Hamilton, R. Extraordinary Conservation of Cysteines Among Homologous Chironomus Silk Proteins Sp185 and Sp220. J. Mol. Evol. 1997, 44, 452–462. [Google Scholar] [CrossRef]
- Johns, S.C.; Yin, X.; Jeltsch, M.; Bishop, J.R.; Schuksz, M.; Ghazal, R.E.; Wilcox-Adelman, S.A.; Alitalo, K.; Fuster, M.M. Functional Importance of a Proteoglycan Co-Receptor in Pathologic Lymphangiogenesis. Circ. Res. 2016, 119, 210–221. [Google Scholar] [CrossRef] [PubMed]
- Keskitalo, S.; Tammela, T.; Lyytikka, J.; Karpanen, T.; Jeltsch, M.; Markkanen, J.; Yla-Herttuala, S.; Alitalo, K. Enhanced Capillary Formation Stimulated by a Chimeric Vascular Endothelial Growth Factor/Vascular Endothelial Growth Factor-C Silk Domain Fusion Protein. Circ. Res. 2007, 100, 1460–1467. [Google Scholar] [CrossRef] [Green Version]
- Tammela, T.; He, Y.; Lyytikkä, J.; Jeltsch, M.; Markkanen, J.; Pajusola, K.; Ylä-Herttuala, S.; Alitalo, K. Distinct Architecture of Lymphatic Vessels Induced by Chimeric Vascular Endothelial Growth Factor-C/Vascular Endothelial Growth Factor Heparin-Binding Domain Fusion Proteins. Circ. Res. 2007, 100, 1468–1475. [Google Scholar] [CrossRef]
- Hyytiäinen, M.; Penttinen, C.; Keski-Oja, J. Latent TGF-Beta Binding Proteins: Extracellular Matrix Association and Roles in TGF-Beta Activation. Crit. Rev. Clin. Lab. Sci. 2004, 41, 233–264. [Google Scholar] [CrossRef] [PubMed]
- Stacker, S.A.; Stenvers, K.; Caesar, C.; Vitali, A.; Domagala, T.; Nice, E.; Roufail, S.; Simpson, R.J.; Moritz, R.; Karpanen, T.; et al. Biosynthesis of Vascular Endothelial Growth Factor-D Involves Proteolytic Processing Which Generates Non-Covalent Homodimers. J. Biol. Chem. 1999, 274, 32127–32136. [Google Scholar] [CrossRef] [Green Version]
- Leppanen, V.-M.; Jeltsch, M.; Anisimov, A.; Tvorogov, D.; Aho, K.; Kalkkinen, N.; Toivanen, P.; Ylä-Herttuala, S.; Ballmer-Hofer, K.; Alitalo, K. Structural Determinants of Vascular Endothelial Growth Factor-D Receptor Binding and Specificity. Blood 2011, 117, 1507–1515. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Rissanen, T.T.; Markkanen, J.E.; Gruchala, M.; Heikura, T.; Puranen, A.; Kettunen, M.I.; Kholová, I.; Kauppinen, R.A.; Achen, M.G.; Stacker, S.A.; et al. VEGF-D Is the Strongest Angiogenic and Lymphangiogenic Effector Among VEGFs Delivered Into Skeletal Muscle via Adenoviruses. Circ. Res. 2003, 92, 1098–1106. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Paquet-Fifield, S.; Levy, S.M.; Sato, T.; Shayan, R.; Karnezis, T.; Davydova, N.; Nowell, C.J.; Roufail, S.; Ma, G.Z.-M.; Zhang, Y.-F.; et al. Vascular Endothelial Growth Factor-d Modulates Caliber and Function of Initial Lymphatics in the Dermis. J. Investig. Dermatol. 2013, 133, 2074–2084. [Google Scholar] [CrossRef] [Green Version]
- Matsumura, M.; Bhatt, A.S.; Andress, D.; Clegg, N.; Takayama, T.K.; Craik, C.S.; Nelson, P.S. Substrates of the Prostate-Specific Serine Protease Prostase/KLK4 Defined by Positional-Scanning Peptide Libraries. Prostate 2005, 62, 1–13. [Google Scholar] [CrossRef]
- Gupta, N.; Sudhakar, D.V.S.; Gangwar, P.K.; Sankhwar, S.N.; Gupta, N.J.; Chakraborty, B.; Thangaraj, K.; Gupta, G.; Rajender, S. Mutations in the Prostate Specific Antigen (PSA/KLK3) Correlate with Male Infertility. Sci. Rep. 2017, 7. [Google Scholar] [CrossRef] [Green Version]
- Jonsson, M.; Linse, S.; Frohm, B.; Lundwall, Å.; Malm, J. Semenogelins I and II Bind Zinc and Regulate the Activity of Prostate-Specific Antigen. Biochem. J. 2005, 387, 447–453. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Brown, L.F.; Yeo, K.-T.; Berse, B.; Morgentaler, A.; Dvorak, H.F.; Rosen, S. Vascular Permeability Factor (Vascular Endothelial Growth Factor) Is Strongly Expressed in the Normal Male Genital Tract and Is Present in Substantial Quantities in Semen. J. Urol. 1995, 154, 576–579. [Google Scholar] [CrossRef]
- Obermair, A.; Obruca, A.; Pöhl, M.; Kaider, A.; Vales, A.; Leodolter, S.; Wojta, J.; Feichtinger, W. Vascular Endothelial Growth Factor and Its Receptors in Male Fertility. Fertil. Steril. 1999, 72, 269–275. [Google Scholar] [CrossRef]
- Iyibozkurt, A.C.; Balcik, P.; Bulgurcuoglu, S.; Arslan, B.K.; Attar, R.; Attar, E. Effect of Vascular Endothelial Growth Factor on Sperm Motility and Survival. Reprod. Biomed. Online 2009, 19, 784–788. [Google Scholar] [CrossRef] [Green Version]
- Saaristo, A.; Tammela, T.; Timonen, J.; Yla-Herttuala, S.; Tukiainen, E.; Asko-Seljavaara, S.; Alitalo, K. Vascular Endothelial Growth Factor-C Gene Therapy Restores Lymphatic Flow across Incision Wounds. FASEB J. 2004, 18, 1707–1709. [Google Scholar] [CrossRef]
- Saaristo, A.; Tammela, T.; Färkkilä, A.; Kärkkäinen, M.; Suominen, E.; Yla-Herttuala, S.; Alitalo, K. Vascular Endothelial Growth Factor-C Accelerates Diabetic Wound Healing. Am. J. Pathol. 2006, 169, 1080–1087. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Lawrence, M.G.; Lai, J.; Clements, J.A. Kallikreins on Steroids: Structure, Function, and Hormonal Regulation of Prostate-Specific Antigen and the Extended Kallikrein Locus. Endocr. Rev. 2010, 31, 407–446. [Google Scholar] [CrossRef]
- Joukov, V.; Pajusola, K.; Kaipainen, A.; Chilov, D.; Lahtinen, I.; Kukk, E.; Saksela, O.; Kalkkinen, N.; Alitalo, K. A Novel Vascular Endothelial Growth Factor, VEGF-C, Is a Ligand for the Flt4 (VEGFR-3) and KDR (VEGFR-2) Receptor Tyrosine Kinases. EMBO J. 1996, 15, 290–298. [Google Scholar] [CrossRef]
- Jeltsch, M.; Kaipainen, A.; Joukov, V.; Meng, X.; Lakso, M.; Rauvala, H.; Swartz, M.; Fukumura, D.; Jain, R.K.; Alitalo, K. Hyperplasia of Lymphatic Vessels in VEGF-C Transgenic Mice. Science 1997, 276, 1423–1425. [Google Scholar] [CrossRef] [Green Version]
- Anderson, J.; Schiffer, C.; Lee, S.-K.; Swanstrom, R. Viral Protease Inhibitors. In Antiviral Strategies; Kräusslich, H.-G., Bartenschlager, R., Eds.; Handbook of Experimental Pharmacology; Springer: Berlin/Heidelberg, Germany, 2009; pp. 85–110. ISBN 978-3-540-79086-0. [Google Scholar]
- Polverino, E.; Rosales-Mayor, E.; Dale, G.E.; Dembowsky, K.; Torres, A. The Role of Neutrophil Elastase Inhibitors in Lung Diseases. Chest 2017, 152, 249–262. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- López-Sendón, J.; Swedberg, K.; McMurray, J.; Tamargo, J.; Maggioni, A.P.; Dargie, H.; Tendera, M.; Waagstein, F.; Kjekshus, J.; Lechat, P.; et al. Expert Consensus Document on Angiotensin Converting Enzyme Inhibitors in Cardiovascular Disease: The Task Force on ACE-Inhibitors of the European Society of Cardiology. Eur. Heart J. 2004, 25, 1454–1470. [Google Scholar] [CrossRef]
- Wardlaw, J.M.; Murray, V.; Berge, E.; del Zoppo, G.; Sandercock, P.; Lindley, R.L.; Cohen, G. Recombinant Tissue Plasminogen Activator for Acute Ischaemic Stroke: An Updated Systematic Review and Meta-Analysis. Lancet 2012, 379, 2364–2372. [Google Scholar] [CrossRef] [Green Version]
- Krebs, R.; Jeltsch, M. Die lymphangiogenen Wachstumsfaktoren VEGF-C und VEGF-D. Teil 2. Die Rolle von VEGF-C und VEGF-D bei Krankheiten des Lymphgefäßsystems. Lymphol. Forsch. Prax. 2013, 17, 96–104. [Google Scholar] [CrossRef]
- Hartiala, P.; Suominen, S.; Suominen, E.; Kaartinen, I.; Kiiski, J.; Viitanen, T.; Alitalo, K.; Saarikko, A.M. Phase 1 LymfactinⓇ Study: Short-Term Safety of Combined Adenoviral VEGF-C and Lymph Node Transfer Treatment for Upper Extremity Lymphedema. J. Plast. Reconstr. Aesthet. Surg. 2020, 73, 1612–1621. [Google Scholar] [CrossRef] [PubMed]
- Jeltsch, M.; Karpanen, T.; Strandin, T.; Aho, K.; Lankinen, H.; Alitalo, K. Vascular Endothelial Growth Factor (VEGF)/VEGF-C Mosaic Molecules Reveal Specificity Determinants and Feature Novel Receptor Binding Patterns. J. Biol. Chem. 2006, 281, 12187–12195. [Google Scholar] [CrossRef] [Green Version]
- Chiu, J.; Wong, J.W.H.; Gerometta, M.; Hogg, P.J. Mechanism of Dimerization of a Recombinant Mature Vascular Endothelial Growth Factor C. Biochemistry 2014, 53, 7–9. [Google Scholar] [CrossRef]
- Suh, S.H.; Choe, K.; Hong, S.P.; Jeong, S.; Mäkinen, T.; Kim, K.S.; Alitalo, K.; Surh, C.D.; Koh, G.Y.; Song, J.-H. Gut Microbiota Regulates Lacteal Integrity by Inducing VEGF-C in Intestinal Villus Macrophages. EMBO Rep. 2019, 20. [Google Scholar] [CrossRef]
- Antila, S.; Karaman, S.; Nurmi, H.; Airavaara, M.; Voutilainen, M.H.; Mathivet, T.; Chilov, D.; Li, Z.; Koppinen, T.; Park, J.-H.; et al. Development and Plasticity of Meningeal Lymphatic Vessels. J. Exp. Med. 2017, 214, 3645–3667. [Google Scholar] [CrossRef] [PubMed]
- Breslin, J.W.; Gaudreault, N.; Watson, K.D.; Reynoso, R.; Yuan, S.Y.; Wu, M.H. Vascular Endothelial Growth Factor-C Stimulates the Lymphatic Pump by a VEGF Receptor-3-Dependent Mechanism. Am. J. Physiol.-Heart Circ. Physiol. 2007, 293, H709–H718. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Karkkainen, M.J.; Saaristo, A.; Jussila, L.; Karila, K.A.; Lawrence, E.C.; Pajusola, K.; Bueler, H.; Eichmann, A.; Kauppinen, R.; Kettunen, M.I.; et al. A Model for Gene Therapy of Human Hereditary Lymphedema. Proc. Natl. Acad. Sci. USA 2001, 98, 12677–12682. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Martens, R.; Permanyer, M.; Werth, K.; Yu, K.; Braun, A.; Halle, O.; Halle, S.; Patzer, G.E.; Bošnjak, B.; Kiefer, F.; et al. Efficient Homing of T Cells via Afferent Lymphatics Requires Mechanical Arrest and Integrin-Supported Chemokine Guidance. Nat. Commun. 2020, 11, 1114. [Google Scholar] [CrossRef] [PubMed]
- Jackson, D.G. Leucocyte Trafficking via the Lymphatic Vasculature— Mechanisms and Consequences. Front. Immunol. 2019, 10. [Google Scholar] [CrossRef] [PubMed]
- Karpanen, T.; Egeblad, M.; Karkkainen, M.J.; Kubo, H.; Ylä-Herttuala, S.; Jäättelä, M.; Alitalo, K. Vascular Endothelial Growth Factor C Promotes Tumor Lymphangiogenesis and Intralymphatic Tumor Growth. Cancer Res. 2001, 61, 1786–1790. [Google Scholar]
- Mandriota, S.J.; Jussila, L.; Jeltsch, M.; Compagni, A.; Baetens, D.; Prevo, R.; Banerji, S.; Huarte, J.; Montesano, R.; Jackson, D.G.; et al. Vascular Endothelial Growth Factor-C-Mediated Lymphangiogenesis Promotes Tumour Metastasis. EMBO J. 2001, 20, 672–682. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Skobe, M.; Hawighorst, T.; Jackson, D.G.; Prevo, R.; Janes, L.; Velasco, P.; Riccardi, L.; Alitalo, K.; Claffey, K.; Detmar, M. Induction of Tumor Lymphangiogenesis by VEGF-C Promotes Breast Cancer Metastasis. Nat. Med. 2001, 7, 192–198. [Google Scholar] [CrossRef]
- Farnsworth, R.H.; Achen, M.G.; Stacker, S.A. The Evolving Role of Lymphatics in Cancer Metastasis. Curr. Opin. Immunol. 2018, 53, 64–73. [Google Scholar] [CrossRef] [PubMed]
- Egeblad, M.; Werb, Z. New Functions for the Matrix Metalloproteinases in Cancer Progression. Nat. Rev. Cancer 2002, 2, 161–174. [Google Scholar] [CrossRef]
- Danø, K.; Behrendt, N.; Høyer-Hansen, G.; Johnsen, M.; Lund, L.R.; Ploug, M.; Rømer, J. Plasminogen Activation and Cancer. Thromb. Haemost. 2005, 93, 676–681. [Google Scholar] [CrossRef]
- Harris, N.C.; Paavonen, K.; Davydova, N.; Roufail, S.; Sato, T.; Zhang, Y.-F.; Karnezis, T.; Stacker, S.A.; Achen, M.G. Proteolytic Processing of Vascular Endothelial Growth Factor-D Is Essential for Its Capacity to Promote the Growth and Spread of Cancer. FASEB J. 2011, 25, 2615–2625. [Google Scholar] [CrossRef]
- Alitalo, A.; Detmar, M. Interaction of Tumor Cells and Lymphatic Vessels in Cancer Progression. Oncogene 2012, 31, 4499–4508. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Michaelsen, S.R.; Staberg, M.; Pedersen, H.; Jensen, K.E.; Majewski, W.; Broholm, H.; Nedergaard, M.K.; Meulengracht, C.; Urup, T.; Villingshøj, M.; et al. VEGF-C Sustains VEGFR2 Activation under Bevacizumab Therapy and Promotes Glioblastoma Maintenance. Neuro-Oncol. 2018, 20, 1462–1474. [Google Scholar] [CrossRef] [Green Version]
- Bergers, G.; Hanahan, D. Modes of Resistance to Anti-Angiogenic Therapy. Nat. Rev. Cancer 2008, 8, 592–603. [Google Scholar] [CrossRef] [Green Version]
- Ndiaye, P.D.; Dufies, M.; Giuliano, S.; Douguet, L.; Grépin, R.; Durivault, J.; Lenormand, P.; Glisse, N.; Mintcheva, J.; Vouret-Craviari, V.; et al. VEGFC Acts as a Double-Edged Sword in Renal Cell Carcinoma Aggressiveness. Theranostics 2019, 9, 661–675. [Google Scholar] [CrossRef] [PubMed]
- Fankhauser, M.; Broggi, M.A.S.; Potin, L.; Bordry, N.; Jeanbart, L.; Lund, A.W.; Costa, E.D.; Hauert, S.; Rincon-Restrepo, M.; Tremblay, C.; et al. Tumor Lymphangiogenesis Promotes T Cell Infiltration and Potentiates Immunotherapy in Melanoma. Sci. Transl. Med. 2017, 9, eaal4712. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Song, E.; Mao, T.; Dong, H.; Boisserand, L.S.B.; Antila, S.; Bosenberg, M.; Alitalo, K.; Thomas, J.-L.; Iwasaki, A. VEGF-C-Driven Lymphatic Drainage Enables Immunosurveillance of Brain Tumours. Nature 2020, 577, 689–694. [Google Scholar] [CrossRef] [PubMed]
- Grada, A.A.; Phillips, T.J. Lymphedema: Pathophysiology and Clinical Manifestations. J. Am. Acad. Dermatol. 2017, 77, 1009–1020. [Google Scholar] [CrossRef]
- Mattonet, K.; Wilting, J.; Jeltsch, M. Die genetischen Ursachen des primären Lymphödems. In Erkrankungen des Lymphgefäßsystems; Weissleder, H., Schuchhardt, C., Eds.; Viavital Verlag: Cologne, Germany, 2015; pp. 210–229. [Google Scholar]
- Gordon, K.; Varney, R.; Keeley, V.; Riches, K.; Jeffery, S.; Zanten, M.V.; Mortimer, P.; Ostergaard, P.; Mansour, S. Update and Audit of the St George’s Classification Algorithm of Primary Lymphatic Anomalies: A Clinical and Molecular Approach to Diagnosis. J. Med. Genet. 2020, 57, 653–659. [Google Scholar] [CrossRef]
- Newman, B.; Lose, F.; Kedda, M.-A.; Francois, M.; Ferguson, K.; Janda, M.; Yates, P.; Spurdle, A.B.; Hayes, S.C. Possible Genetic Predisposition to Lymphedema after Breast Cancer. Lymphat. Res. Biol. 2012, 10, 2–13. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Finegold, D.N.; Schacht, V.; Kimak, M.A.; Lawrence, E.C.; Foeldi, E.; Karlsson, J.M.; Baty, C.J.; Ferrell, R.E. HGF and MET Mutations in Primary and Secondary Lymphedema. Lymphat. Res. Biol. 2008, 6, 65–68. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Finegold, D.N.; Baty, C.J.; Knickelbein, K.Z.; Perschke, S.; Noon, S.E.; Campbell, D.; Karlsson, J.M.; Huang, D.; Kimak, M.A.; Lawrence, E.C.; et al. Connexin 47 Mutations Increase Risk for Secondary Lymphedema Following Breast Cancer Treatment. Clin. Cancer Res. 2012, 18, 2382–2390. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Brouillard, P.; Boon, L.; Vikkula, M. Genetics of Lymphatic Anomalies. J. Clin. Investig. 2014, 124, 898–904. [Google Scholar] [CrossRef] [PubMed]
- Gordon, K.; Spiden, S.L.; Connell, F.C.; Brice, G.; Cottrell, S.; Short, J.; Taylor, R.; Jeffery, S.; Mortimer, P.S.; Mansour, S.; et al. FLT4/VEGFR3 and Milroy Disease: Novel Mutations, a Review of Published Variants and Database Update. Hum. Mutat. 2013, 34, 23–31. [Google Scholar] [CrossRef] [Green Version]
- Gordon, K.; Schulte, D.; Brice, G.; Simpson, M.A.; Roukens, M.G.; van Impel, A.; Connell, F.; Kalidas, K.; Jeffery, S.; Mortimer, P.S.; et al. Mutation in Vascular Endothelial Growth Factor-C, a Ligand for Vascular Endothelial Growth Factor Receptor-3, Is Associated With Autosomal Dominant Milroy-Like Primary Lymphedema. Novelty and Significance. Circ. Res. 2013, 112, 956–960. [Google Scholar] [CrossRef]
- Balboa-Beltran, E.; Fernández-Seara, M.J.; Pérez-Muñuzuri, A.; Lago, R.; García-Magán, C.; Couce, M.L.; Sobrino, B.; Amigo, J.; Carracedo, A.; Barros, F. A Novel Stop Mutation in the Vascular Endothelial Growth Factor-C Gene (VEGFC) Results in Milroy-like Disease. J. Med. Genet. 2014, 51, 475–478. [Google Scholar] [CrossRef] [PubMed]
- Connell, F.; Kalidas, K.; Ostergaard, P.; Brice, G.; Homfray, T.; Roberts, L.; Bunyan, D.J.; Mitton, S.; Mansour, S.; Mortimer, P.; et al. Linkage and Sequence Analysis Indicate That CCBE1 Is Mutated in Recessively Inherited Generalised Lymphatic Dysplasia. Hum. Genet. 2010, 127, 231–241. [Google Scholar] [CrossRef]
- Betterman, K.L.; Sutton, D.L.; Secker, G.A.; Kazenwadel, J.; Oszmiana, A.; Lim, L.; Miura, N.; Sorokin, L.; Hogan, B.M.; Kahn, M.L.; et al. Atypical Cadherin FAT4 Orchestrates Lymphatic Endothelial Cell Polarity in Response to Flow. J. Clin. Investig. 2020, 130, 3315–3328. [Google Scholar] [CrossRef] [Green Version]
- Katoh, Y.; Katoh, M. Comparative Integromics on VEGF Family Members. Int. J. Oncol. 2006, 28, 1585–1589. [Google Scholar] [CrossRef] [Green Version]
- Rauniyar, K.; Jha, S.K.; Jeltsch, M. Biology of Vascular Endothelial Growth Factor C in the Morphogenesis of Lymphatic Vessels. Front. Bioeng. Biotechnol. 2018, 6, 7. [Google Scholar] [CrossRef] [Green Version]
- El Khoury, L.; Posthumus, M.; Collins, M.; Handley, C.J.; Cook, J.; Raleigh, S.M. Polymorphic Variation within the ADAMTS2, ADAMTS14, ADAMTS5, ADAM12 and TIMP2 Genes and the Risk of Achilles Tendon Pathology: A Genetic Association Study. J. Sci. Med. Sport 2013, 16, 493–498. [Google Scholar] [CrossRef]
- Koistinen, H.K.; Stenman, U.-H. PSA (Prostate-Specific Antigen) and other Kallikrein-related Peptidases in Prostate Cancer. In Kallikrein-Related Peptidase. Novel Cancer Related Biomarkers; Magdolen, V., Sommerhoff, C., Fritz, H., Schmitt, M., Eds.; deGruyter: Berlin, Germany, 2012; Volume 2, pp. 61–81. ISBN 978-3-11-030366-7. [Google Scholar]
- Siintola, E.; Partanen, S.; Strömme, P.; Haapanen, A.; Haltia, M.; Maehlen, J.; Lehesjoki, A.-E.; Tyynelä, J. Cathepsin D Deficiency Underlies Congenital Human Neuronal Ceroid-Lipofuscinosis. Brain 2006, 129, 1438–1445. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Seligsohn, U.; Lubetsky, A. Genetic Susceptibility to Venous Thrombosis. N. Engl. J. Med. 2001, 344, 1222–1231. [Google Scholar] [CrossRef] [PubMed]
- Tsopanoglou, N.E.; Maragoudakis, M.E. Role of Thrombin in Angiogenesis and Tumor Progression. Semin. Thromb. Hemost. 2004, 30, 63–69. [Google Scholar] [CrossRef]
- Schuster, V.; Seregard, S. Ligneous Conjunctivitis. Surv. Ophthalmol. 2003, 48, 369–388. [Google Scholar] [CrossRef]
- Petrova, T.V.; Karpanen, T.; Norrmén, C.; Mellor, R.; Tamakoshi, T.; Finegold, D.; Ferrell, R.; Kerjaschki, D.; Mortimer, P.; Ylä-Herttuala, S.; et al. Defective Valves and Abnormal Mural Cell Recruitment Underlie Lymphatic Vascular Failure in Lymphedema Distichiasis. Nat. Med. 2004, 10, 974–981. [Google Scholar] [CrossRef] [PubMed]
- Norrmén, C.; Ivanov, K.I.; Cheng, J.; Zangger, N.; Delorenzi, M.; Jaquet, M.; Miura, N.; Puolakkainen, P.; Horsley, V.; Hu, J.; et al. FOXC2 Controls Formation and Maturation of Lymphatic Collecting Vessels through Cooperation with NFATc1. J. Cell Biol. 2009, 185, 439–457. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Ferrell, R.E.; Baty, C.J.; Kimak, M.A.; Karlsson, J.M.; Lawrence, E.C.; Franke-Snyder, M.; Meriney, S.D.; Feingold, E.; Finegold, D.N. GJC2 Missense Mutations Cause Human Lymphedema. Am. J. Hum. Genet. 2010, 86, 943–948. [Google Scholar] [CrossRef] [Green Version]
- Cappello, S.; Gray, M.J.; Badouel, C.; Lange, S.; Einsiedler, M.; Srour, M.; Chitayat, D.; Hamdan, F.F.; Jenkins, Z.A.; Morgan, T.; et al. Mutations in Genes Encoding the Cadherin Receptor-Ligand Pair DCHS1 and FAT4 Disrupt Cerebral Cortical Development. Nat. Genet. 2013, 45, 1300–1308. [Google Scholar] [CrossRef]
- Frangoul, H.; Altshuler, D.; Cappellini, M.D.; Chen, Y.-S.; Domm, J.; Eustace, B.K.; Foell, J.; de la Fuente, J.; Grupp, S.; Handgretinger, R.; et al. CRISPR-Cas9 Gene Editing for Sickle Cell Disease and β-Thalassemia. N. Engl. J. Med. 2020. [Google Scholar] [CrossRef]
- Ledford, H. CRISPR Treatment Inserted Directly into the Body for First Time. Nature 2020, 579, 185. [Google Scholar] [CrossRef] [Green Version]
- Kiseleva, R.Y.; Glassman, P.M.; Greineder, C.F.; Hood, E.D.; Shuvaev, V.V.; Muzykantov, V.R. Targeting Therapeutics to Endothelium: Are We There Yet? Drug Deliv. Transl. Res. 2018, 8, 883–902. [Google Scholar] [CrossRef] [PubMed]







{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
| Gene | Protein | Human Disease (OMIM) | Remarks |
|---|---|---|---|
| Genes within the VEGF-C/VEGFR-3 signaling pathway; for clinical details see [130], and for molecular details [141] | |||
| VEGFC | Vascular Endothelial Growth Factor-C (VEGF-C) | Hereditary lymphedema type 1D (615907) | VEGF-C is the primary growth factor for lymphatic endothelial cells. |
| FLT4 | Vascular Endothelial Growth Factor Receptor-3 (VEGFR-3) | Hereditary lymphedema type 1A (Milroy disease, 153100) | VEGFR-3 is the primary receptor of VEGF-C. |
| CCBE1 | Collagen and calcium-binding EGF domain-containing protein 1 | Hennekam lymphangiectasia-lymphedema syndrome type 1 (235510) | Enhances the processing of VEGF-C by ADAMTS3 and KLK3. |
| ADAMTS3 | A disintegrin and metalloproteinase with thrombospondin motifs 3 | Hennekam lymphangiectasia-lymphedema syndrome type 3 (618154) | ADAMTS3 catalyzes the final step in the activation of VEGF-C. |
| Genes coding for proteases that can activate VEGF-C and/or VEGF-D (no lymphatic phenotype reported) | |||
| ADAMTS14 | A disintegrin and metalloproteinase with thrombospondin motifs 14 | Association with age of onset in tendinopathy [142] | ADAMTS14 can activate VEGF-C in vitro [65]. |
| KLK3 | Kallikrein-like peptidase 3, Prostate-specific antigen (PSA) | Association with human fertility [91] | KLK3/PSA is most commonly known as a prostate cancer marker [143]. |
| CTSD | Cathepsin D | Neuronal Ceroid Lipofuscinosis type 10 (610127) | CTSD deficiency causes a neurodegenerative disorder [144]. |
| F2 | Prothrombin/Thrombin | Hereditary thrombophilia type 1 (188050) | Specific F2 mutations increase the risk of venous thromboembolism [145]. Thrombin potentiates vascular endothelial growth factor- (VEGF-) induced endothelial cell proliferation [146]. |
| PLG | Plasminogen/Plasmin | Plasminogen deficiency type 1 (217090) | PLG deficiency leads to pathological fibrin deposition but no increased risk of thrombosis [147]. |
| Selected other “lymphedema” genes; for a comprehensive listing, see [130] | |||
| FOXC2 | Forkhead box protein C2 | Lymphedema distichiasis syndrome (153400) | The maturation of lymphatic vessels and the formation of lymphatic valves requires FOXC2 [148,149]. |
| GJC2 | Connexin 47 | Hereditary lymphedema type 1C (613480) | CJC2 is a gap junction protein that enables communication between lymphatic endothelial cells [150]. |
| FAT4 | Protocadherin Fat 4 | Hennekam lymphangiectasia-lymphedema syndrome type 2 (616006) | FAT4 is required for lymphatic endothelial cell polarity and might influence VEGFR-3 signaling [139]. |
| Van Maldergem syndrome type 2 (615546) | Van Maldergem syndrome 2 has overlapping features with Hennekam syndrome type 2 but none or only infrequent lymphatic involvement [151]. | ||
Publisher’s Note: MDPI stays neutral with regard to jurisdictional claims in published maps and institutional affiliations. |
© 2021 by the authors. Licensee MDPI, Basel, Switzerland. This article is an open access article distributed under the terms and conditions of the Creative Commons Attribution (CC BY) license (http://creativecommons.org/licenses/by/4.0/).
Share and Cite
Künnapuu, J.; Bokharaie, H.; Jeltsch, M. Proteolytic Cleavages in the VEGF Family: Generating Diversity among Angiogenic VEGFs, Essential for the Activation of Lymphangiogenic VEGFs. Biology 2021, 10, 167. https://doi.org/10.3390/biology10020167
Künnapuu J, Bokharaie H, Jeltsch M. Proteolytic Cleavages in the VEGF Family: Generating Diversity among Angiogenic VEGFs, Essential for the Activation of Lymphangiogenic VEGFs. Biology. 2021; 10(2):167. https://doi.org/10.3390/biology10020167
Chicago/Turabian StyleKünnapuu, Jaana, Honey Bokharaie, and Michael Jeltsch. 2021. "Proteolytic Cleavages in the VEGF Family: Generating Diversity among Angiogenic VEGFs, Essential for the Activation of Lymphangiogenic VEGFs" Biology 10, no. 2: 167. https://doi.org/10.3390/biology10020167
APA StyleKünnapuu, J., Bokharaie, H., & Jeltsch, M. (2021). Proteolytic Cleavages in the VEGF Family: Generating Diversity among Angiogenic VEGFs, Essential for the Activation of Lymphangiogenic VEGFs. Biology, 10(2), 167. https://doi.org/10.3390/biology10020167