
Genome-Wide Atlas of Promoter Expression Reveals Contribution of Transcribed Regulatory Elements to Genetic Control of Disuse-Mediated Atrophy of Skeletal Muscle

 , , ,
, , ,  , ,
, , {kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
Abstract
:Simple Summary
Abstract
1. Introduction
2. Materials and Methods
2.1. Animals
2.2. Experimental Design
2.3. Immunochemistry
2.4. Morphometric and Statistical Analysis
2.5. RNA Isolation
2.6. CAGE-Seq
2.7. Data Analysis
2.7.1. Quality Control
2.7.2. Genome, Annotation, and Indexing
2.7.3. Read Mapping
2.7.4. CTSS Aggregation
2.7.5. TSS Peaks
2.7.6. Enhancers
2.7.7. Annotation of TSS Peaks to Genes and Enhancers
2.7.8. Differentially Expressed Peaks, Genes, and Enhancers
2.7.9. Gene Set Enrichment Analysis (GSEA)
3. Results and Discussion
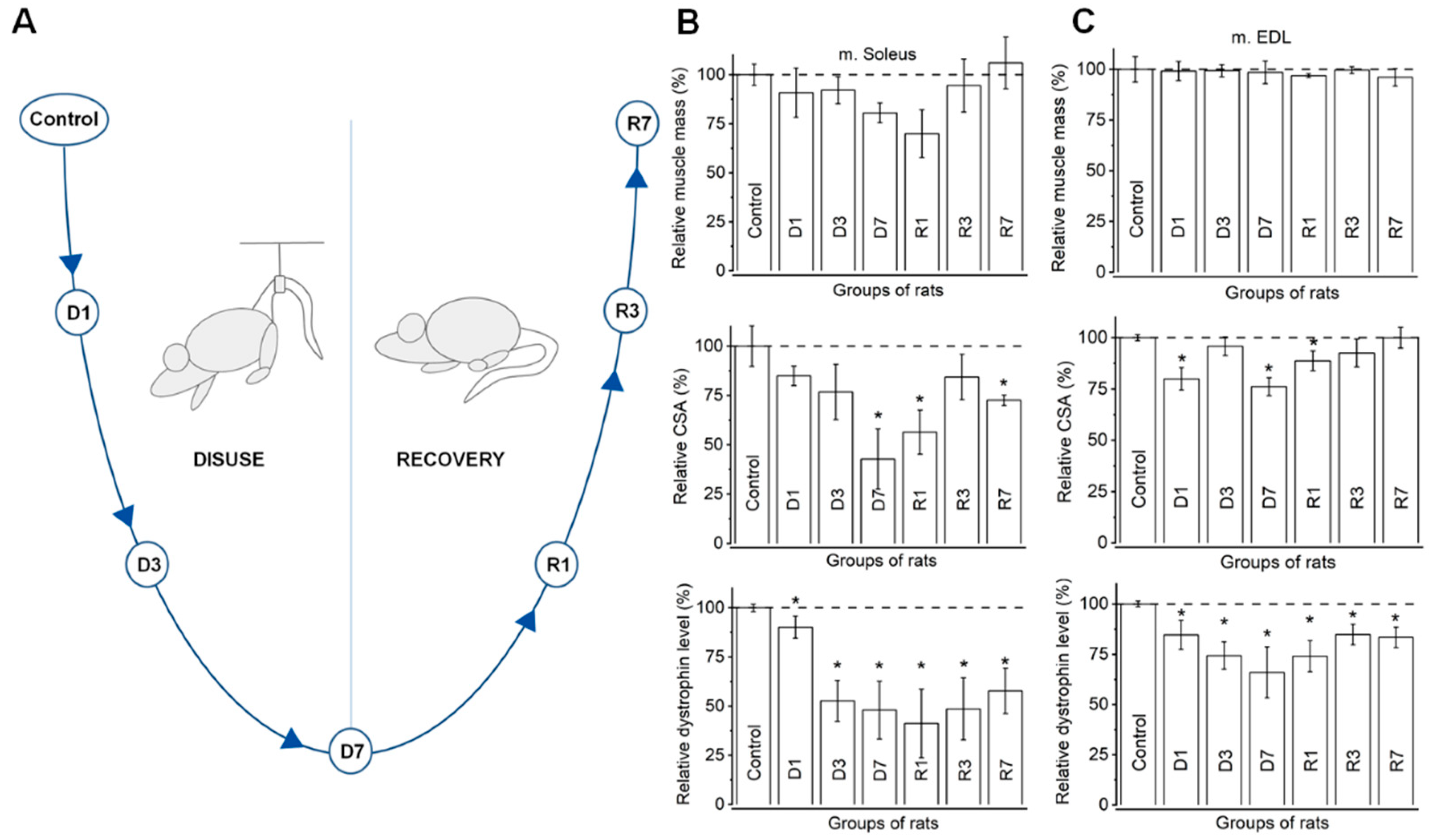
3.1. Morphometric Analysis Confirms Distinct Disuse Associated Remodeling in Slow, but Not Fast Muscles
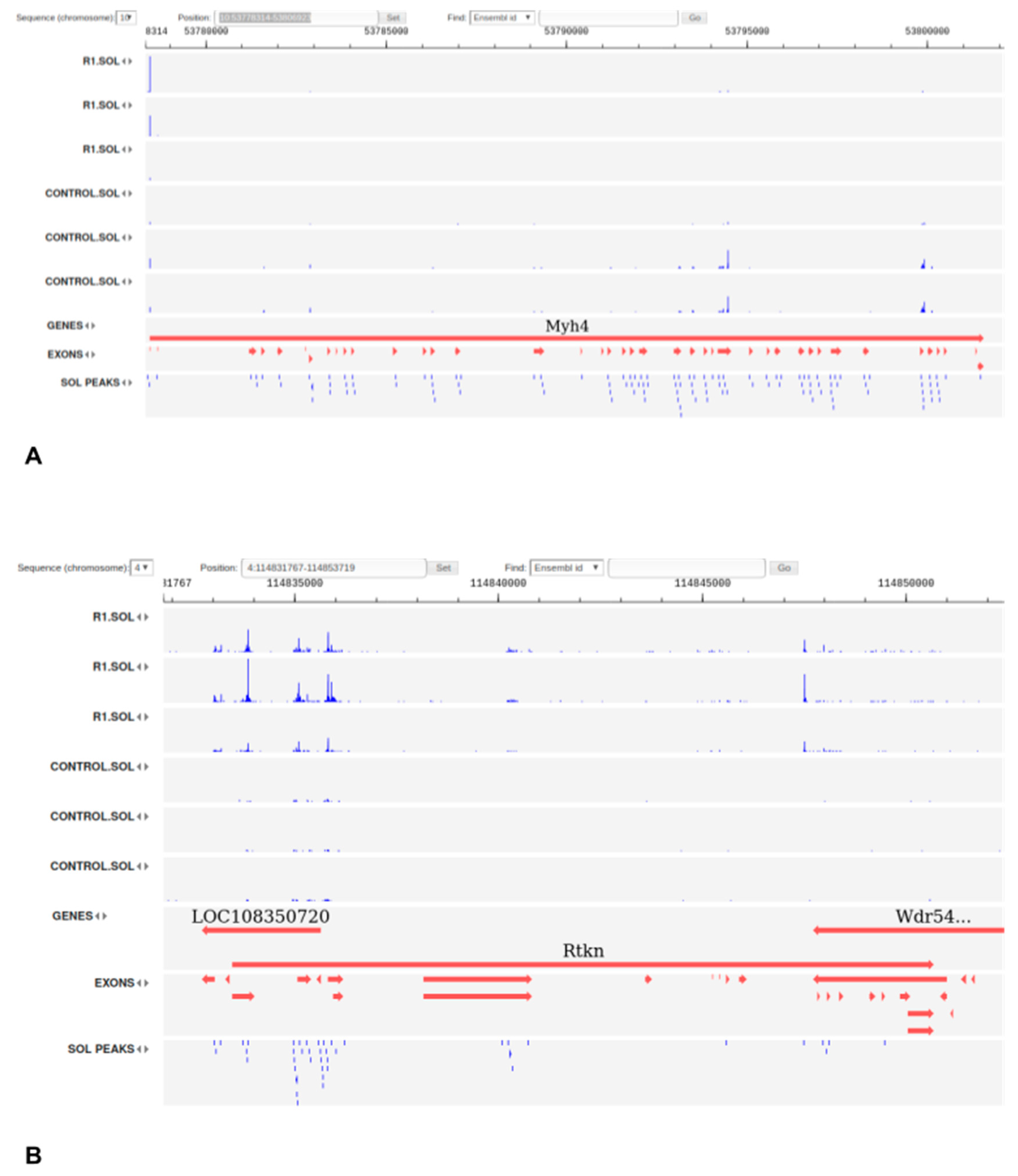
3.2. Integrative Database of Rat’s Regulatory Elements with a Focus on Muscles
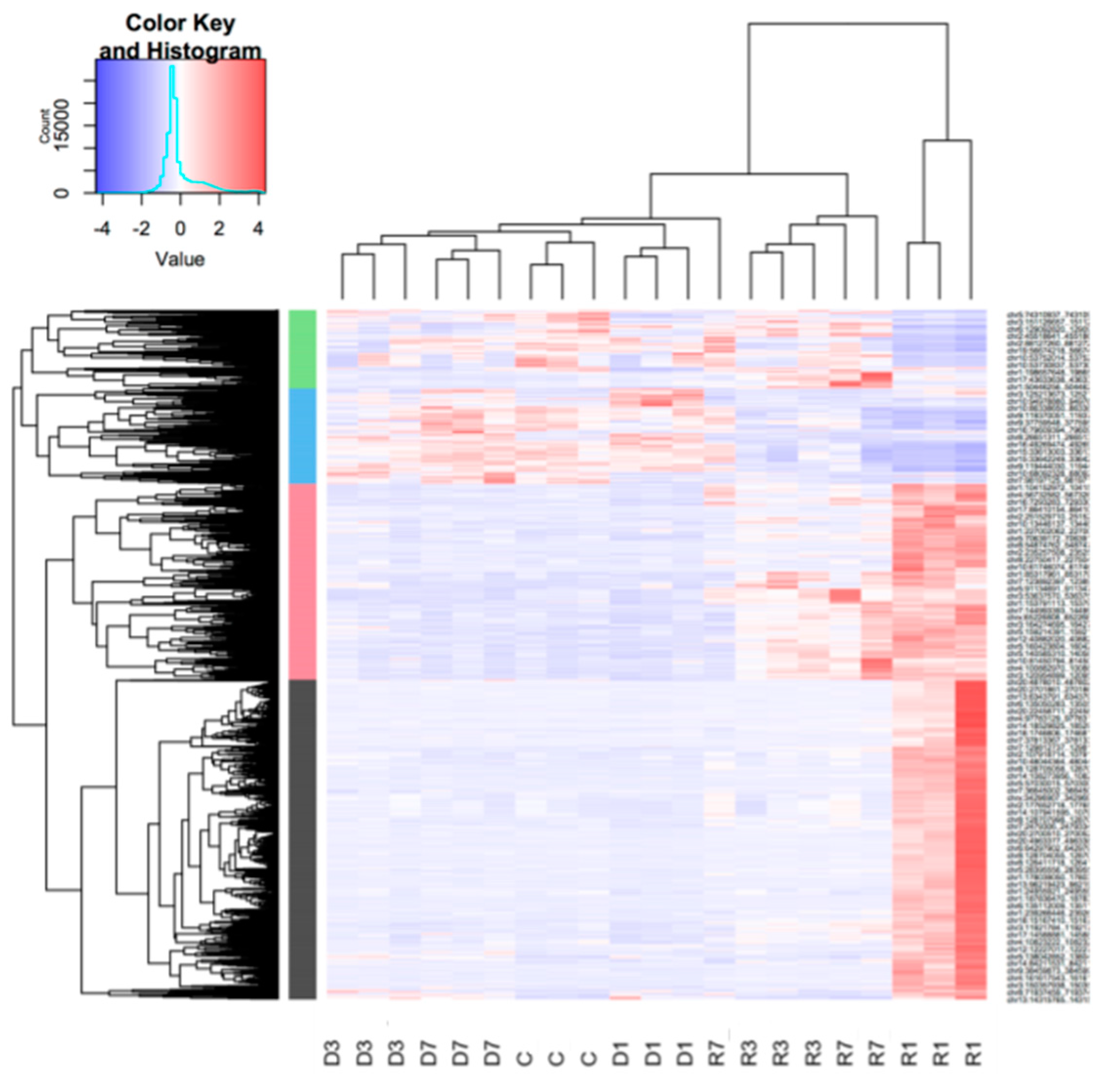
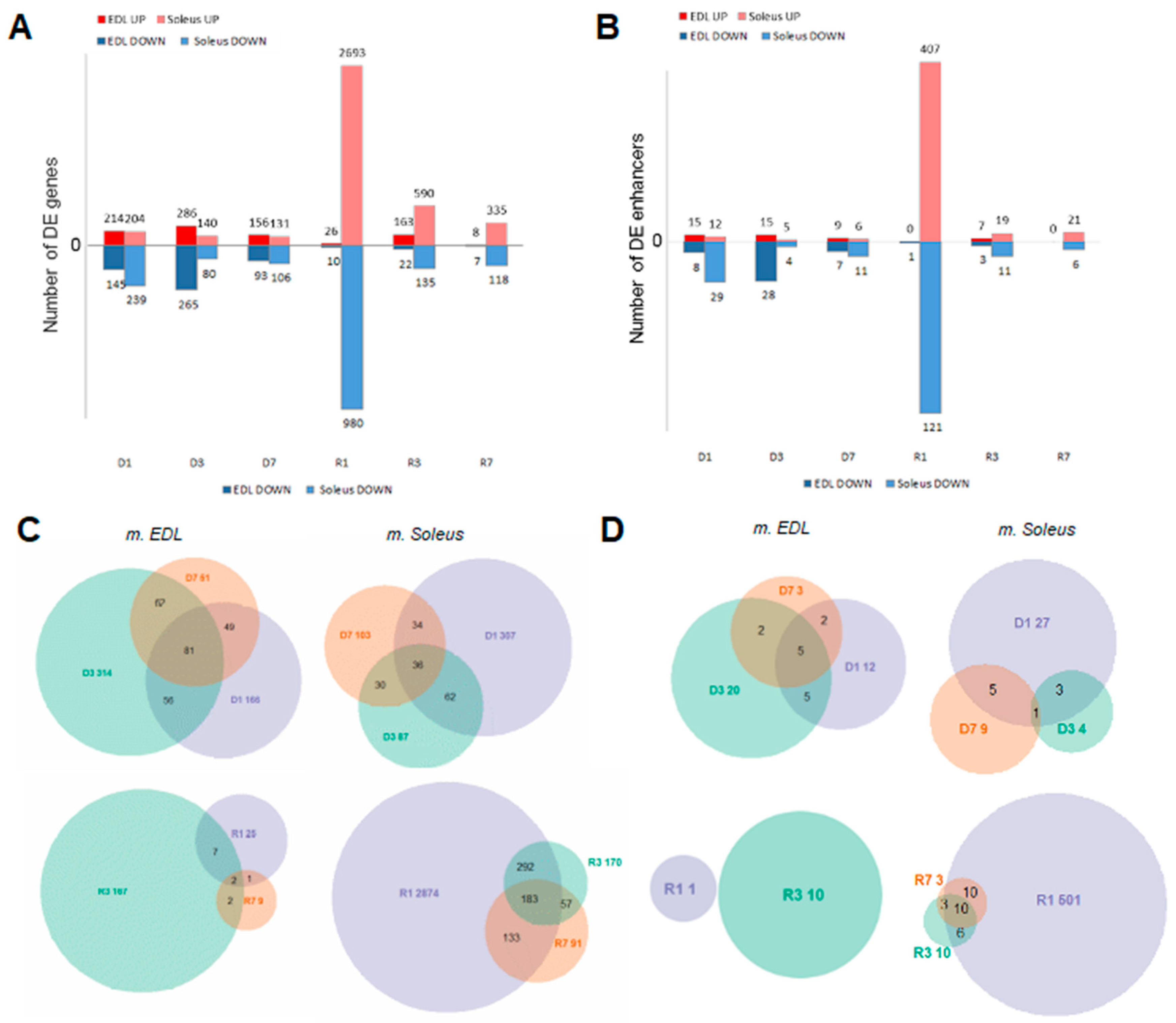
3.3. Muscle Atrophy and Recovery are Associated with Large Changes in the Unique Gene Set Expression Profiles, with Distinct Dynamics on Each Stage
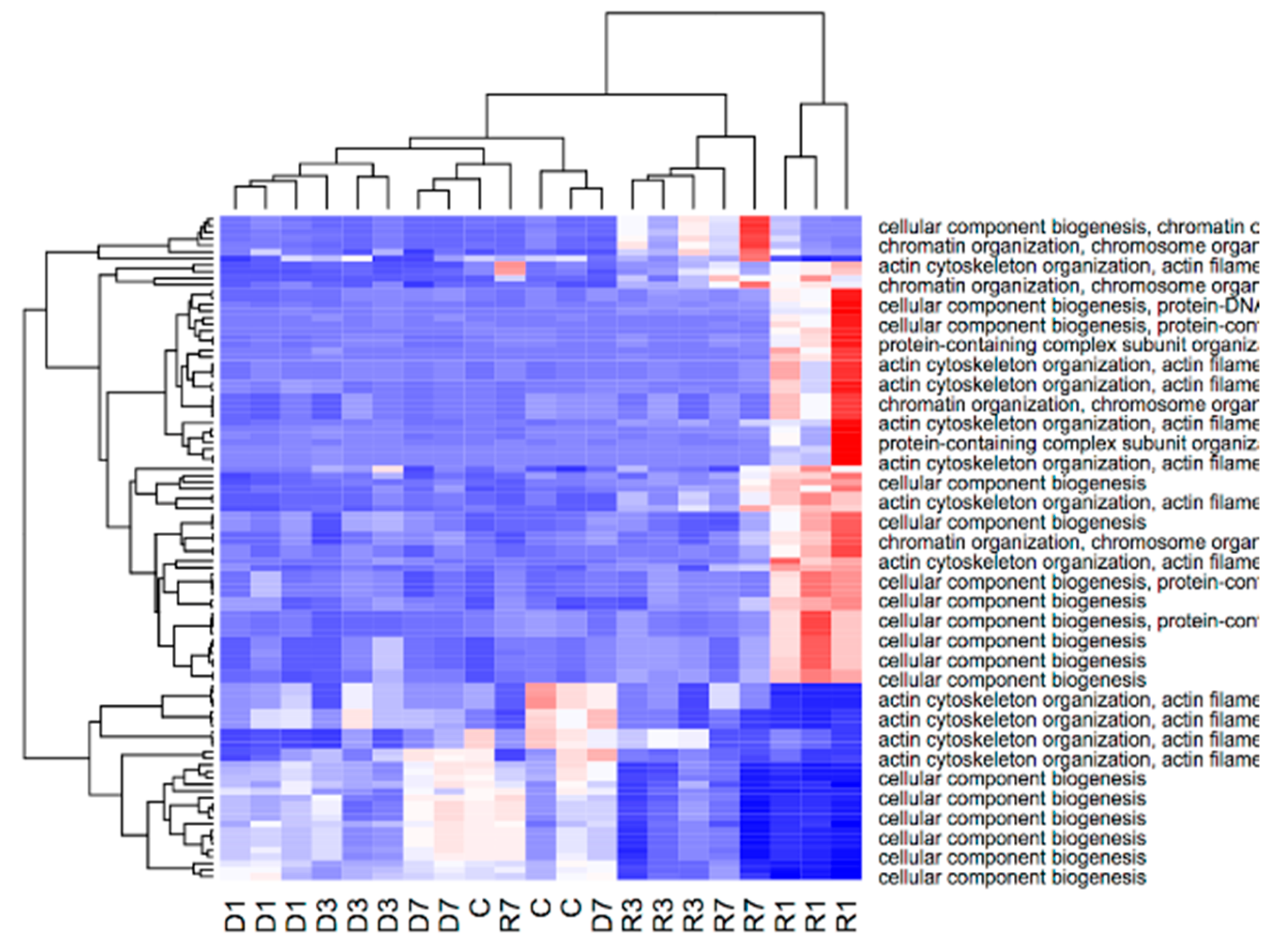
3.4. Gene Enrichment Analysis Reveals that Adaption to Disuse Processes in Slow and Fast Muscles is Mediated by Different Molecular Pathways
3.5. Tissue-Specific Intergenic TSS Show Different Patterns in Slow and Fast Muscles and Widely Co-Express with Coding Genes in Response to Disuse
3.6. Alternative Usage of Dominating Transcription Starting Sites in Differentially Expressed Genes
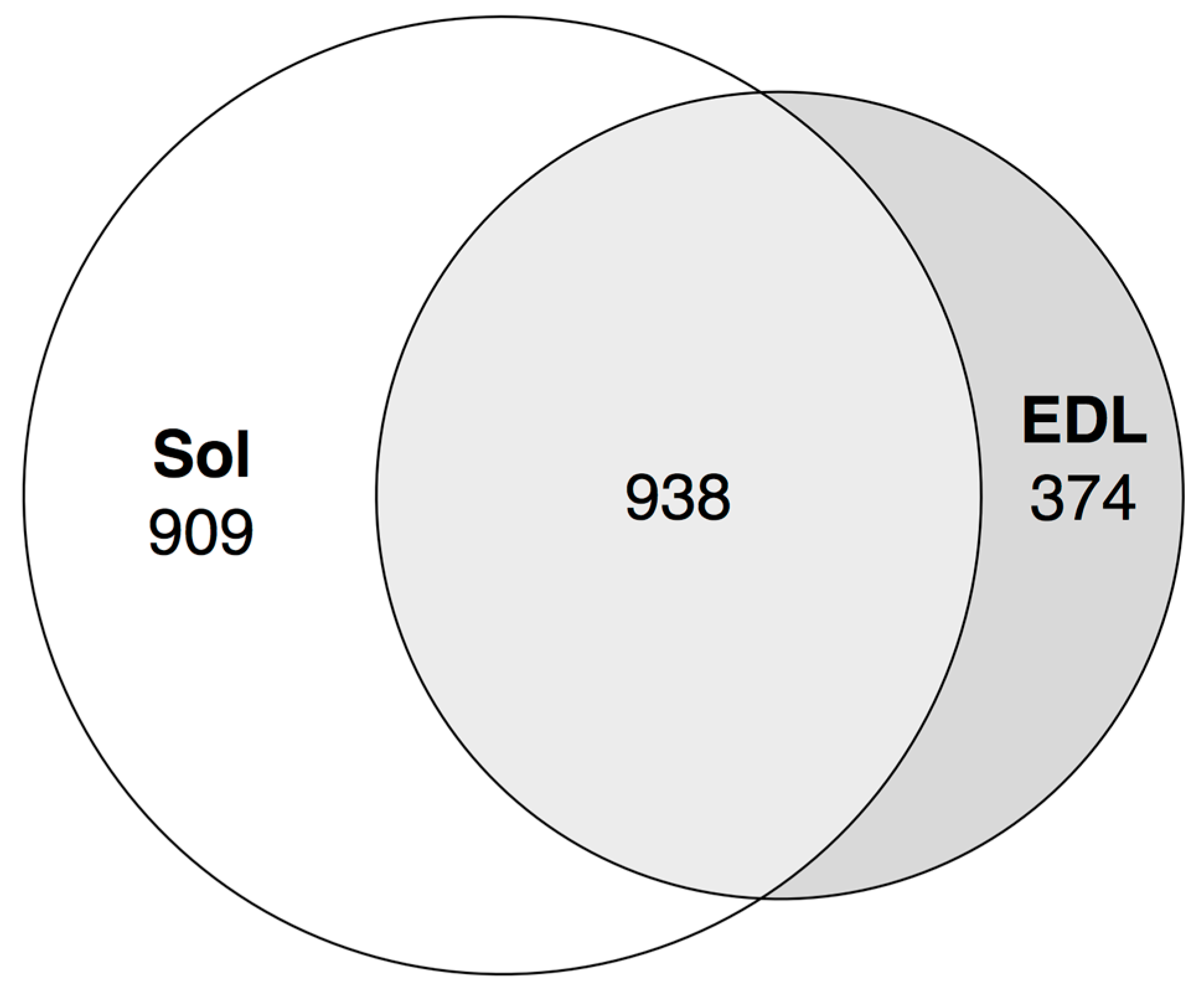
3.7. Expression of Transcribed Enhancers in Rat Skeletal Muscles is Linked to Disuse-Recovery Cycle
3.7.1. Differentially Expressed Enhancers Mediate Muscle-Specific Transcription Program
3.7.2. Switch ON/OFF Enhancers on First Day of Recovery (R1)
4. Conclusions
Supplementary Materials
Author Contributions
Funding
Institutional Review Board Statement
Informed Consent Statement
Data Availability Statement
Conflicts of Interest
Abbreviations
| CAGE | Cap Analysis Gene Expression |
| CSA | Cross-Sectional Area |
| CTSS | CAGE Tag Start Sites |
| DPI1 | Decomposition-Based Peak Identification |
| EDL | Extensor Digitorum Longum |
| eRNA | enhancer RNA |
| GSEA | Gene Set Enrichment Analysis |
| LFC | Log2 Fold Change |
| RFU | Relative Fluorescent Units |
| TSS | Transcription Start Sites |
References
- Wang, Y.; Pessin, J.E. Mechanisms for fiber-type specificity of skeletal muscle atrophy. Curr. Opin. Clin. Nutr. Metab. Care 2013, 16, 243–250. [Google Scholar] [CrossRef]
- Bodine, S.C. Disuse-induced muscle wasting. Int. J. Biochem. Cell Biol. 2013, 45, 2200–2208. [Google Scholar] [CrossRef] [Green Version]
- Ninfali, C.; Siles, L.; Darling, D.S.; Postigo, A. Regulation of muscle atrophy-related genes by the opposing transcriptional activities of ZEB1/CtBP and FOXO3. Nucleic Acids Res. 2018, 46, 10697–10708. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Piekarowicz, K.; Bertrand, A.T.; Azibani, F.; Beuvin, M.; Julien, L.; Machowska, M.; Bonne, G.; Rzepecki, R. A muscle hybrid promoter as a novel tool for gene therapy. Mol. Ther. Methods Clin. Dev. 2019, 15, 157–169. [Google Scholar] [CrossRef] [Green Version]
- Arnold, P.R.; Wells, A.D.; Li, X.C. Diversity and emerging roles of enhancer RNA in regulation of gene expression and cell fate. Front. Cell Dev. Biol. 2019, 7, 377. [Google Scholar] [CrossRef] [Green Version]
- Genin, A.M.; Il’in, A.E.; Kaplanskiĭ, A.S.; Kasatkina, T.B.; Kuznetsova, K.A.; Pestov, I.D.; Smirnova, T.A. Bioethics of research on humans and animals in aviation, space and marine medicine. Aviakosm. Ekolog. Med. 2001, 35, 14–20. [Google Scholar]
- Morey-Holton, E.R.; Globus, R.K. Hindlimb unloading rodent model: Technical aspects. J. Appl. Physiol. 2002, 92, 1367–1377. [Google Scholar] [CrossRef] [PubMed]
- Briguet, A.; Courdier-Fruh, I.; Foster, M.; Meier, T.; Magyar, J.P. Histological parameters for the quantitative assessment of muscular dystrophy in the mdx-mouse. Neuromuscul. Disord. 2004, 14, 675–682. [Google Scholar] [CrossRef]
- Murata, M.; Nishiyori-Sueki, H.; Kojima-Ishiyama, M.; Carninci, P.; Hayashizaki, Y.; Itoh, M. Detecting Expressed Genes Using CAGE. Methods Mol. Biol. 2014, 1164, 67–85. [Google Scholar] [CrossRef] [PubMed]
- Kouno, T.; Moody, J.; Kwon, A.T.-J.; Shibayama, Y.; Kato, S.; Huang, Y.; Böttcher, M.; Motakis, E.; Mendez, M.; Severin, J.; et al. C1 CAGE detects transcription start sites and enhancer activity at single-cell resolution. Nat. Commun. 2019, 10, 360. [Google Scholar] [CrossRef] [Green Version]
- Andersson, R.; Gebhard, C.; Miguel-Escalada, I.; Hoof, I.; Bornholdt, J.; Boyd, M.; Chen, Y.; Zhao, X.; Schmidl, C.; Suzuki, T.; et al. An atlas of active enhancers across human cell types and tissues. Nature 2014, 507, 455–461. [Google Scholar] [CrossRef]
- Schiaffino, S.; Reggiani, C. Fiber types in mammalian skeletal muscles. Physiol. Rev. 2011, 91, 1447–1531. [Google Scholar] [CrossRef] [Green Version]
- Ohira, Y.; Yoshinaga, T.; Nomura, T.; Kawano, F.; Ishihara, A.; Nonaka, I.; Roy, R.R.; Edgerton, V.R. Gravitational unloading effects on muscle fiber size, phenotype and myonuclear number. Adv. Space Res. 2002, 30, 777–781. [Google Scholar] [CrossRef]
- Thomason, D.B.; Booth, F.W. Atrophy of the soleus muscle by hindlimb unweighting. J. Appl. Physiol. 1990, 68, 1–12. [Google Scholar] [CrossRef]
- Globus, R.K.; Morey-Holton, E. Hindlimb unloading: Rodent analog for microgravity. J. Appl. Physiol. 2016, 120, 1196–1206. [Google Scholar] [CrossRef]
- Kolmykov, S.; Yevshin, I.; Kulyashov, M.; Sharipov, R.; Kondrakhin, Y.; Makeev, V.J.; Kulakovskiy, I.V.; Kel, A.; Kolpakov, F. GTRD: An integrated view of transcription regulation. Nucleic Acids Res. 2021, D1, D1–D111. [Google Scholar] [CrossRef]
- Valeev, T.; Yevshin, I.; Kolpakov, F. BioUML genome browser. Virtual Biology 2013, 1, 15–26. [Google Scholar] [CrossRef]
- Kolpakov, F.; Akberdin, I.; Kashapov, T.; Kiselev, L.; Kolmykov, S.; Kondrakhin, Y.; Kutumova, E.; Mandrik, N.; Pintus, S.; Ryabova, A.; et al. BioUML: An integrated environment for systems biology and collaborative analysis of biomedical data. Nucleic Acids Res. 2019, 47, W1, W225–W233. [Google Scholar] [CrossRef] [PubMed]
- Williams, G.P.; Marmion, D.J.; Schonhoff, A.M.; Jurkuvenaite, A.; Won, W.-J.; Standaert, D.G.; Kordower, J.H.; Harms, A.S. T cell infiltration in both human multiple system atrophy and a novel mouse model of the disease. Acta Neuropathol. 2020, 139, 855–874. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Li, Y.; Li, J.; Zhu, J.; Sun, B.; Branca, M.; Tang, Y.; Foster, W.; Xiao, X.; Huard, J. Decorin gene transfer promotes muscle cell differentiation and muscle regeneration. Mol. Ther. 2007, 15, 1616–1622. [Google Scholar] [CrossRef]
- Li, S.; Moore, A.K.; Zhu, J.; Li, X.; Zhou, H.; Lin, J.; He, Y.; Xing, F.; Pan, Y.; Bohler, H.C.; et al. Ggnbp2 is essential for pregnancy success via regulation of mouse trophoblast stem cell proliferation and differentiation. Biol. Reprod. 2016, 94, 41. [Google Scholar] [CrossRef] [Green Version]
- Lin, I.-H.; Chang, J.-L.; Hua, K.; Huang, W.-C.; Hsu, M.-T.; Chen, Y.-F. Skeletal muscle in aged mice reveals extensive transformation of muscle gene expression. BMC Genet. 2018, 19, 55. [Google Scholar] [CrossRef]
- Fiuza-Luces, C.; Santos-Lozano, A.; Llavero, F.; Campo, R.; Nogales-Gadea, G.; Díez-Bermejo, J.; Baladrón, C.; González-Murillo, Á.; Arenas, J.; Martín, M.A.; et al. Muscle molecular adaptations to endurance exercise training are conditioned by glycogen availability: A proteomics-based analysis in the McArdle mouse model. J. Physiol. 2018, 596, 1035–1061. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Osisami, M.; Ali, W.; Frohman, M.A. A role for phospholipase D3 in myotube formation. PLoS ONE 2012, 7, e33341. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Fortuin, F.D.; Morisaki, T.; Holmes, E.W. Subunit composition of AMPD varies in response to changes in AMPD1 and AMPD3 gene expression in skeletal muscle. Proc. Assoc. Am. Physicians 1996, 108, 329–333. [Google Scholar] [PubMed]
- Shiloh, R.; Gilad, Y.; Ber, Y.; Eisenstein, M.; Aweida, D.; Bialik, S.; Cohen, S.; Kimchi, A. Non-canonical activation of DAPK2 by AMPK constitutes a new pathway linking metabolic stress to autophagy. Nat. Commun. 2018, 9, 1759. [Google Scholar] [CrossRef] [Green Version]
- Verma, M.; Shimizu-Motohashi, Y.; Asakura, Y.; Ennen, J.P.; Bosco, J.; Zhou, Z.; Fong, G.-H.; Josiah, S.; Keefe, D.; Asakura, A. Inhibition of FLT1 ameliorates muscular dystrophy phenotype by increased vasculature in a mouse model of Duchenne muscular dystrophy. PLoS Genet. 2019, 15, e1008468. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Sellers, R.S.; Mahmood, S.R.; Perumal, G.S.; Macaluso, F.P.; Kurland, I.J. Phenotypic Modulation of Skeletal Muscle Fibers in LPIN1-Deficient Lipodystrophic (fld) Mice. Vet. Pathol. 2019, 56, 322–331. [Google Scholar] [CrossRef] [PubMed]
- Ghasemizadeh, A.; Christin, E.; Guiraud, A.; Couturier, N.; Risson, V.; Girard, E.; Soler, C.; Laddada, L.; Jagla, K.; Sanchez, C.; et al. Muscle MACF1 maintains myonuclei and mitochondria localization through microtubules to control muscle functionalities. BioRxiv 2019. [Google Scholar] [CrossRef]
- Laforêt, P.; Richard, P.; Said, M.A.; Romero, N.B.; Lacene, E.; Leroy, J.-P.; Baussan, C.; Hogrel, J.-Y.; Lavergne, T.; Wahbi, K.; et al. A new mutation in PRKAG2 gene causing hypertrophic cardiomyopathy with conduction system disease and muscular glycogenosis. Neuromuscul. Disord. 2006, 16, 178–182. [Google Scholar] [CrossRef]
- Otten, C.; van der Ven, P.F.; Lewrenz, I.; Paul, S.; Steinhagen, A.; Busch-Nentwich, E.; Eichhorst, J.; Wiesner, B.; Stemple, D.; Strähle, U.; et al. Xirp proteins mark injured skeletal muscle in zebrafish. PLoS ONE 2012, 7, e31041. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Nelson, B.R.; Wu, F.; Liu, Y.; Anderson, D.M.; McAnally, J.; Lin, W.; Cannon, S.C.; Bassel-Duby, R.; Olson, E.N. Skeletal muscle-specific T-tubule protein STAC3 mediates voltage-induced Ca2+ release and contractility. Proc. Natl Acad Sci USA 2013, 110, 11881–11886. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Glass, D.J. Skeletal muscle hypertrophy and atrophy signaling pathways. Int. J. Biochem. Cell Biol. 2005, 37, 1974–1984. [Google Scholar] [CrossRef]
- Liu, Y.; Shen, H.; Greenbaum, J.; Liu, A.; Su, K.-J.; Zhang, L.-S.; Zhang, L.; Tian, Q.; Hu, H.-G.; He, J.-S.; et al. Gene Expression and RNA Splicing Imputation Identifies Novel Candidate Genes Associated with Osteoporosis. J. Clin. Endocrinol. Metab. 2020, 105. [Google Scholar] [CrossRef] [PubMed]
- Latimer, L.E.; Constantin, D.; Greening, N.J.; Calvert, L.; Menon, M.K.; Steiner, M.C.; Greenhaff, P.L. Impact of transcutaneous neuromuscular electrical stimulation or resistance exercise on skeletal muscle mRNA expression in COPD. Int. J. Chron. Obstruct. Pulmon. Dis. 2019, 14, 1355–1364. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Miyake, N.; Takahashi, H.; Nakamura, K.; Isidor, B.; Hiraki, Y.; Koshimizu, E.; Shiina, M.; Sasaki, K.; Suzuki, H.; Abe, R.; et al. Gain-of-Function MN1 Truncation Variants Cause a Recognizable Syndrome with Craniofacial and Brain Abnormalities. Am. J. Hum. Genet. 2020, 106, 13–25. [Google Scholar] [CrossRef]
- Meester-Smoor, M.A.; Vermeij, M.; van Helmond, M.J.L.; Molijn, A.C.; van Wely, K.H.M.; Hekman, A.C.P.; Vermey-Keers, C.; Riegman, P.H.J.; Zwarthoff, E.C. Targeted disruption of the Mn1 oncogene results in severe defects in development of membranous bones of the cranial skeleton. Mol. Cell. Biol. 2005, 25, 4229–4236. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Zhang, X.; Dowd, D.R.; Moore, M.C.; Kranenburg, T.A.; Meester-Smoor, M.A.; Zwarthoff, E.C.; MacDonald, P.N. Meningioma 1 is required for appropriate osteoblast proliferation, motility, differentiation, and function. J. Biol. Chem. 2009, 284, 18174–18183. [Google Scholar] [CrossRef] [Green Version]
- Kowanetz, K.; Crosetto, N.; Haglund, K.; Schmidt, M.; Heldin, C.-H.; Dikic, I. Suppressors of T-cell receptor signaling Sts-1 and Sts-2 bind to Cbl and inhibit endocytosis of receptor tyrosine kinases. J. Biol. Chem. 2004, 279, 32786–32795. [Google Scholar] [CrossRef] [Green Version]
- Wang, Y.X.; Feige, P.; Brun, C.E.; Hekmatnejad, B.; Dumont, N.A.; Renaud, J.-M.; Faulkes, S.; Guindon, D.E.; Rudnicki, M.A. EGFR-Aurka Signaling Rescues Polarity and Regeneration Defects in Dystrophin-Deficient Muscle Stem Cells by Increasing Asymmetric Divisions. Cell Stem Cell 2019, 24, 419–432.e6. [Google Scholar] [CrossRef] [Green Version]
- Waymack, R.; Fletcher, A.; Enciso, G.; Wunderlich, Z. Shadow enhancers can suppress input transcription factor noise through distinct regulatory logic. eLife 2020, 9. [Google Scholar] [CrossRef] [PubMed]






Publisher’s Note: MDPI stays neutral with regard to jurisdictional claims in published maps and institutional affiliations. |
© 2021 by the authors. Licensee MDPI, Basel, Switzerland. This article is an open access article distributed under the terms and conditions of the Creative Commons Attribution (CC BY) license (https://creativecommons.org/licenses/by/4.0/).
Share and Cite
Pintus, S.S.; Akberdin, I.R.; Yevshin, I.; Makhnovskii, P.; Tyapkina, O.; Nigmetzyanov, I.; Nurullin, L.; Devyatiyarov, R.; Shagimardanova, E.; Popov, D.; et al. Genome-Wide Atlas of Promoter Expression Reveals Contribution of Transcribed Regulatory Elements to Genetic Control of Disuse-Mediated Atrophy of Skeletal Muscle. Biology 2021, 10, 557. https://doi.org/10.3390/biology10060557
Pintus SS, Akberdin IR, Yevshin I, Makhnovskii P, Tyapkina O, Nigmetzyanov I, Nurullin L, Devyatiyarov R, Shagimardanova E, Popov D, et al. Genome-Wide Atlas of Promoter Expression Reveals Contribution of Transcribed Regulatory Elements to Genetic Control of Disuse-Mediated Atrophy of Skeletal Muscle. Biology. 2021; 10(6):557. https://doi.org/10.3390/biology10060557
Chicago/Turabian StylePintus, Sergey S., Ilya R. Akberdin, Ivan Yevshin, Pavel Makhnovskii, Oksana Tyapkina, Islam Nigmetzyanov, Leniz Nurullin, Ruslan Devyatiyarov, Elena Shagimardanova, Daniil Popov, and et al. 2021. "Genome-Wide Atlas of Promoter Expression Reveals Contribution of Transcribed Regulatory Elements to Genetic Control of Disuse-Mediated Atrophy of Skeletal Muscle" Biology 10, no. 6: 557. https://doi.org/10.3390/biology10060557
APA StylePintus, S. S., Akberdin, I. R., Yevshin, I., Makhnovskii, P., Tyapkina, O., Nigmetzyanov, I., Nurullin, L., Devyatiyarov, R., Shagimardanova, E., Popov, D., Kolpakov, F. A., Gusev, O., & Gazizova, G. R. (2021). Genome-Wide Atlas of Promoter Expression Reveals Contribution of Transcribed Regulatory Elements to Genetic Control of Disuse-Mediated Atrophy of Skeletal Muscle. Biology, 10(6), 557. https://doi.org/10.3390/biology10060557