
Identification of Key Proteins from the Alternative Lengthening of Telomeres-Associated Promyelocytic Leukemia Nuclear Bodies Pathway

 ,
,  , ,
, ,  and
and {kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
Abstract
:Simple Summary
Abstract
1. Introduction
2. Materials and Methods
2.1. Gene/Protein Set
2.2. TCGA Pan-Cancer Studies Frequencies and OncoPrint of Genomic and Proteomic Alterations
2.3. Protein–Protein Interaction Network
2.4. Functional Enrichment Analysis
2.5. Correlation between ALT and APBs in the Pan-Cancer Studies
3. Results
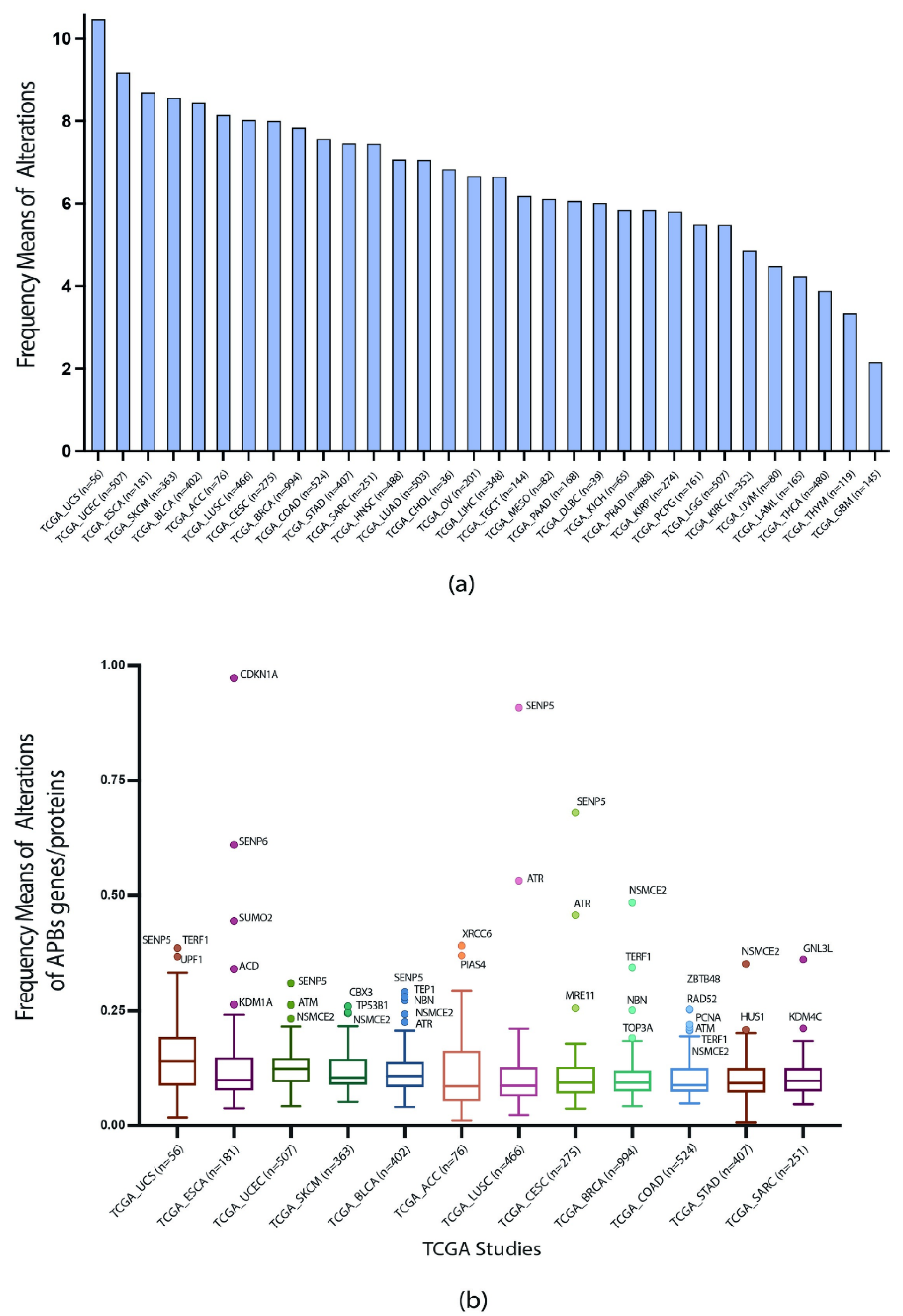
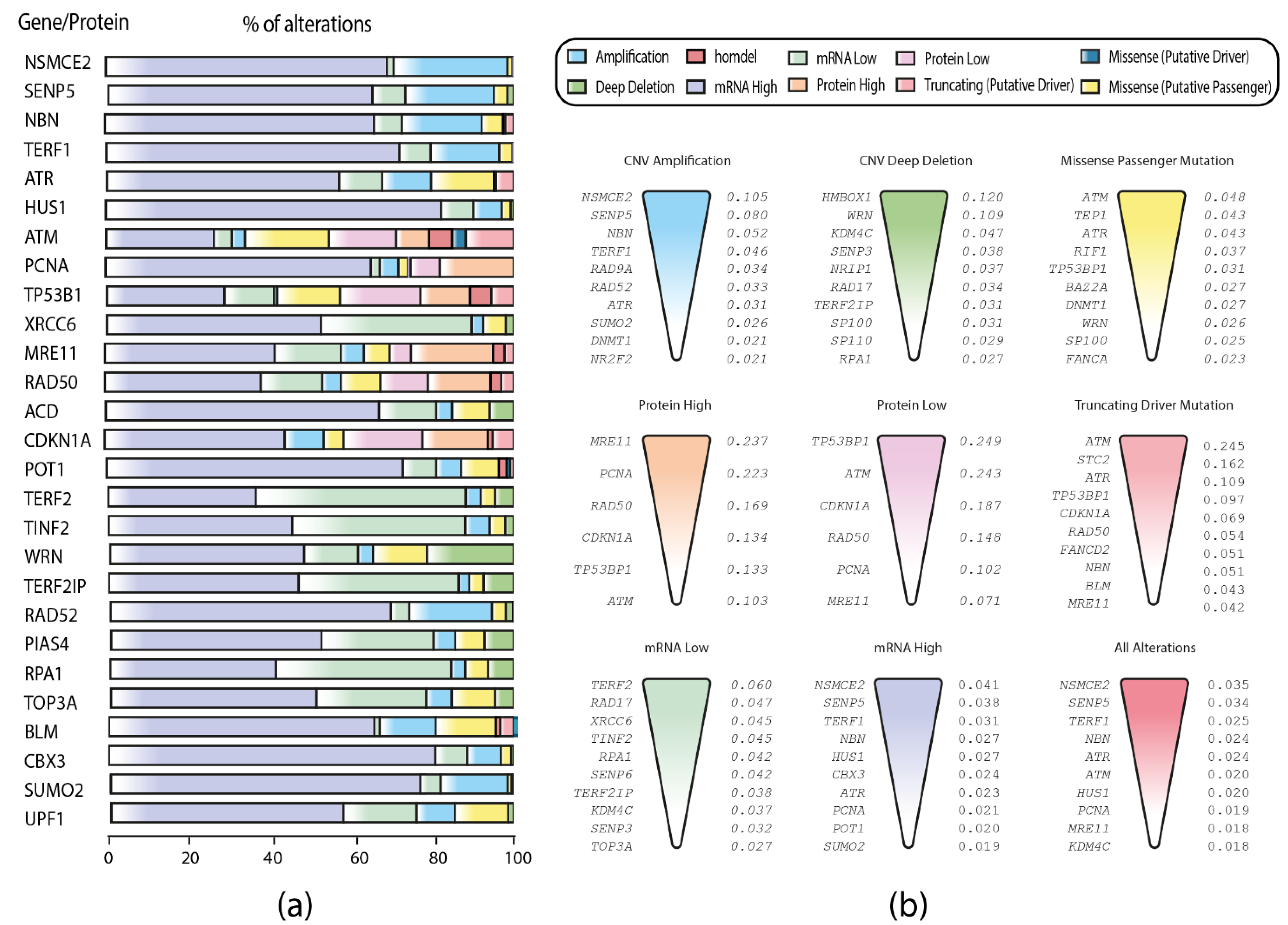
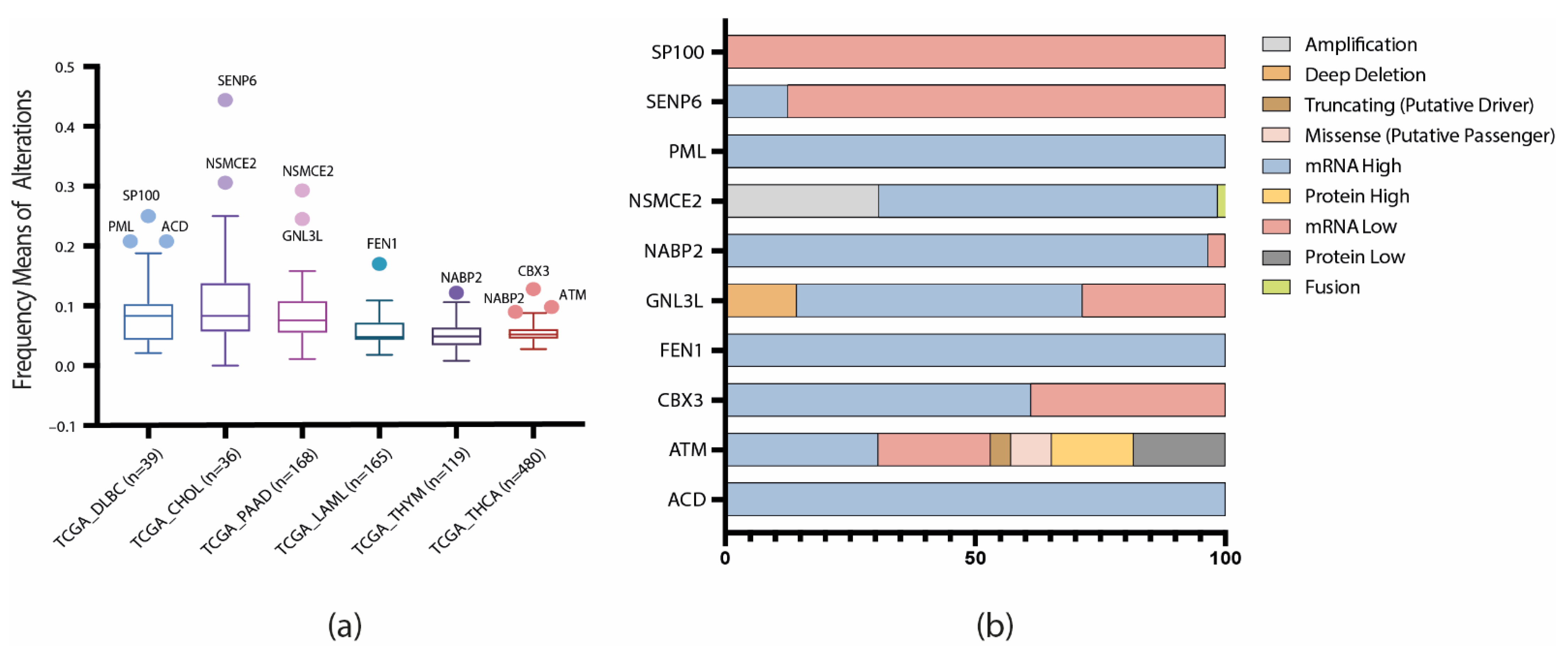
3.1. Gene Set and Genomic, Transcriptomic, and Proteomic Alterations
3.2. TCGA Pan-Cancer Studies Frequencies and OncoPrint of Genomic Transcriptomic and Proteomic Alterations
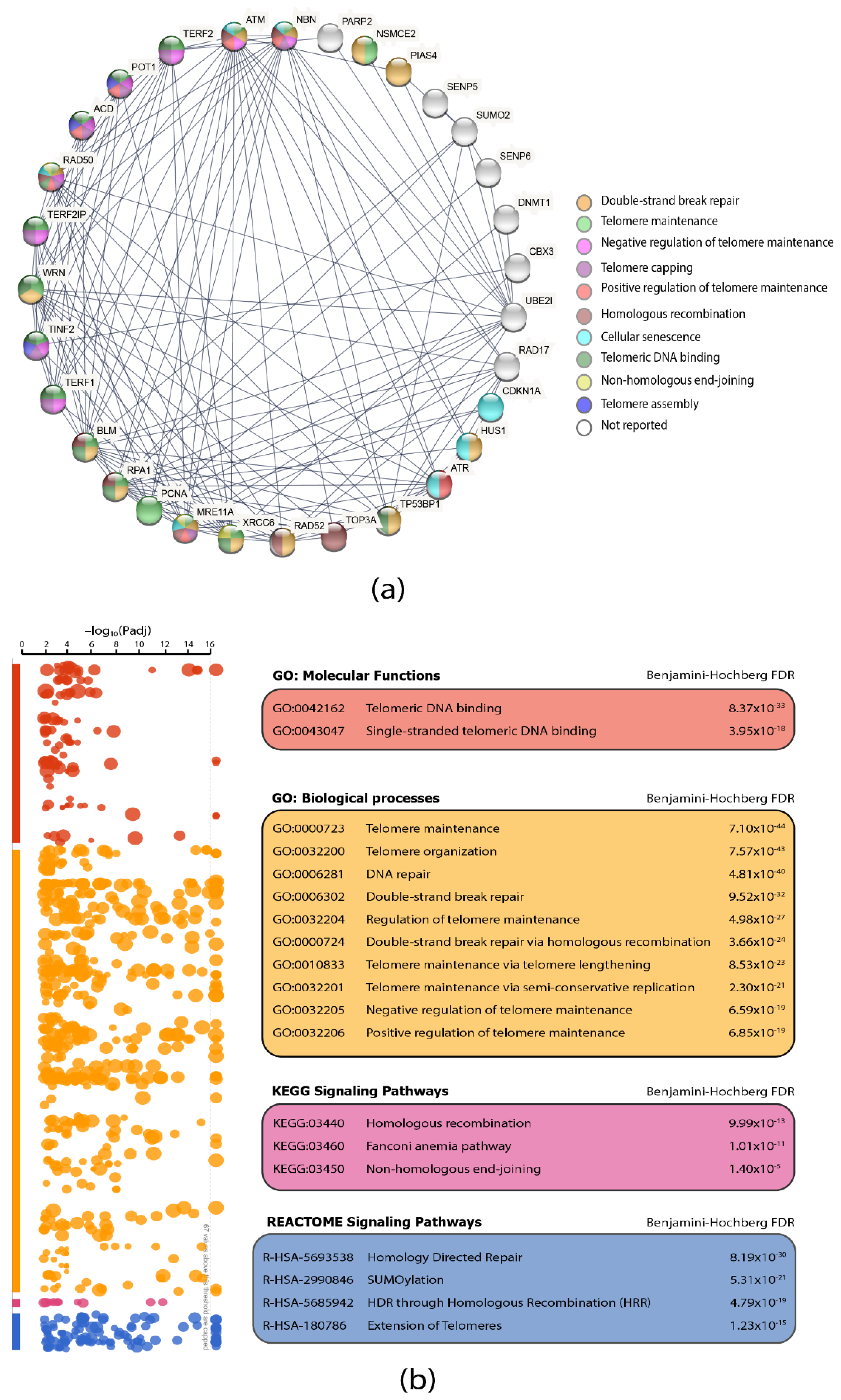
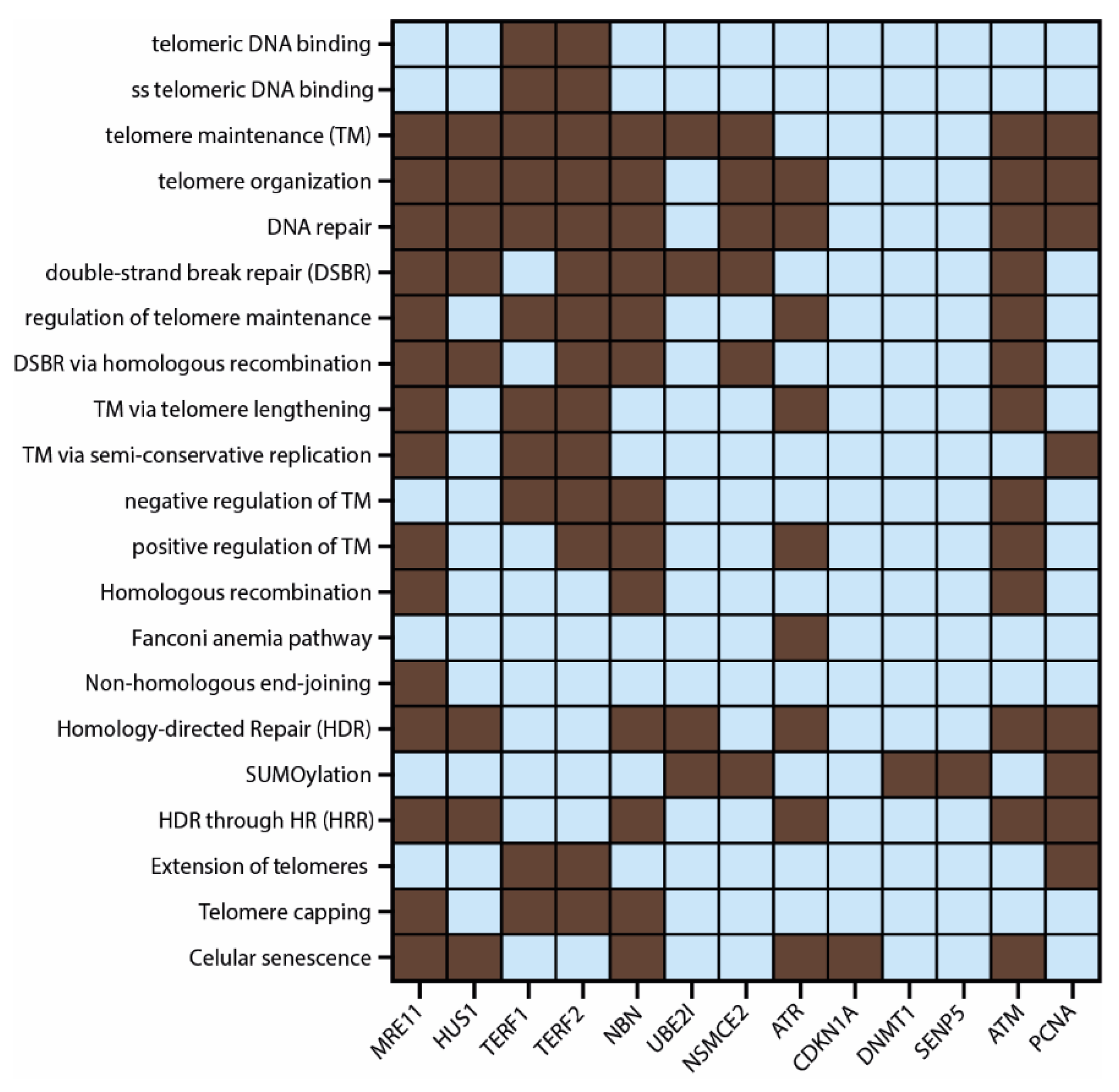
3.3. Protein–Protein Interaction (PPi) Network and Functional Enrichment Analysis
3.4. Correlation between ALT and APBs in TCGA PCA Studies
4. Discussion
5. Conclusions
Supplementary Materials
Author Contributions
Funding
Institutional Review Board Statement
Informed Consent Statement
Data Availability Statement
Conflicts of Interest
References
- Mentegari, E.; Bertoletti, F.; Kissova, M.; Zucca, E.; Galli, S.; Tagliavini, G.; Garbelli, A.; Maffia, A.; Bione, S.; Ferrari, E.; et al. A Role for Human DNA Polymerase λ in Alternative Lengthening of Telomeres. Int. J. Mol. Sci. 2021, 22, 2365. [Google Scholar] [CrossRef]
- Yang, C.-W.; Hsieh, M.-H.; Sun, H.-J.; Teng, S.-C. Nuclear envelope tethering inhibits the formation of ALT-associated PML bodies in ALT cells. Aging 2021, 13, 10490–10516. [Google Scholar] [CrossRef] [PubMed]
- Zhang, H.; Zhao, R.; Tones, J.; Liu, M.; Dilley, R.L.; Chenoweth, D.M.; Greenberg, R.A.; Lampson, M.A. Nuclear body phase separation drives telomere clustering in ALT cancer cells. Mol. Biol. Cell 2020, 31, 2048–2056. [Google Scholar] [CrossRef] [PubMed]
- Jamiruddin, M.R.; Kaitsuka, T.; Hakim, F.; Fujimura, A.; Wei, F.-Y.; Saitoh, H.; Tomizawa, K. HDAC9 regulates the alternative lengthening of telomere (ALT) pathway via the formation of ALT-associated PML bodies. Biochem. Biophys. Res. Commun. 2016, 481, 25–30. [Google Scholar] [CrossRef]
- Li, Y.; Ma, X.; Wu, W.; Chen, Z.; Meng, G. PML Nuclear Body Biogenesis, Carcinogenesis, and Targeted Therapy. Trends Cancer 2020, 6, 889–906. [Google Scholar] [CrossRef]
- Graham, M.K.; Kim, J.; Da, J.; Brosnan-Cashman, J.A.; Rizzo, A.; Baena Del Valle, J.A.; Chia, L.; Rubenstein, M.; Davis, C.; Zheng, Q.; et al. Functional Loss of ATRX and TERC Activates Alternative Lengthening of Telomeres (ALT) in LAPC4 Prostate Cancer Cells. Mol. Cancer Res. 2019, 17, 2480–2491. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Min, J.; Wright, W.E.; Shay, J.W. Clustered telomeres in phase-separated nuclear condensates engage mitotic DNA synthesis through BLM and RAD52. Genes Dev. 2019, 33, 814–827. [Google Scholar] [CrossRef]
- Fonin, A.V.; Silonov, S.A.; Shpironok, O.G.; Antifeeva, I.A.; Petukhov, A.V.; Romanovich, A.E.; Kuznetsova, I.M.; Uversky, V.N.; Turoverov, K.K. The Role of Non-Specific Interactions in Canonical and ALT-Associated PML-Bodies Formation and Dynamics. Int. J. Mol. Sci. 2021, 22, 5821. [Google Scholar] [CrossRef]
- Salmina, K.; Bojko, A.; Inashkina, I.; Staniak, K.; Dudkowska, M.; Podlesniy, P.; Rumnieks, F.; Vainshelbaum, N.M.; Pjanova, D.; Sikora, E.; et al. “Mitotic Slippage” and Extranuclear DNA in Cancer Chemoresistance: A Focus on Telomeres. Int. J. Mol. Sci. 2020, 21, 2779. [Google Scholar] [CrossRef] [Green Version]
- Zhang, J.-M.; Yadav, T.; Ouyang, J.; Lan, L.; Zou, L. Alternative Lengthening of Telomeres through Two Distinct Break-Induced Replication Pathways. Cell Rep. 2019, 26, 955–968.e3. [Google Scholar] [CrossRef] [Green Version]
- Armendáriz-Castillo, I.; López-Cortés, A.; García-Cárdenas, J.; Guevara-Ramírez, P.; Leone, P.E.; Pérez-Villa, A.; Yumiceba, V.; Zambrano, A.K.; Guerrero, S.; Paz-Y-Miño, C. TCGA Pan-Cancer Genomic Analysis of Alternative Lengthening of Telomeres (ALT) Related Genes. Genes 2020, 11, 834. [Google Scholar] [CrossRef] [PubMed]
- Braun, D.M.; Chung, I.; Kepper, N.; Deeg, K.I.; Rippe, K. TelNet—A database for human and yeast genes involved in telomere maintenance. BMC Genet. 2018, 19, 1–10. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Hoadley, K.A.; Yau, C.; Hinoue, T.; Wolf, D.M.; Lazar, A.J.; Drill, E.; Shen, R.; Taylor, A.M.; Cherniack, A.D.; Thorsson, V.; et al. Cell-of-Origin Patterns Dominate the Molecular Classification of 10,000 Tumors from 33 Types of Cancer. Cell 2018, 173, 291–304.e6. [Google Scholar] [CrossRef] [Green Version]
- Huang, K.; Mashl, R.J.; Wu, Y.; Ritter, D.I.; Wang, J.; Oh, C.; Paczkowska, M.; Reynolds, S.; Wyczalkowski, M.A.; Oak, N.; et al. Pathogenic Germline Variants in 10,389 Adult Cancers. Cell 2018, 173, 355–370.e14. [Google Scholar] [CrossRef] [Green Version]
- Gao, J.; Aksoy, B.A.; Dogrusoz, U.; Dresdner, G.; Gross, B.; Sumer, S.O.; Sun, Y.; Jacobsen, A.; Sinha, R.; Larsson, E.; et al. Integrative analysis of complex cancer genomics and clinical profiles using the cBioPortal. Sci. Signal. 2013, 6, pl1. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Cerami, E.; Gao, J.; Dogrusoz, U.; Gross, B.E.; Sumer, S.O.; Aksoy, B.A.; Jacobsen, A.; Byrne, C.J.; Heuer, M.L.; Larsson, E.; et al. The cBio Cancer Genomics Portal: An Open Platform for Exploring Multidimensional Cancer Genomics Data. Cancer Discov. 2012, 2, 401–404. [Google Scholar] [CrossRef] [Green Version]
- Berger, A.C.; Korkut, A.; Kanchi, R.S.; Hegde, A.M.; Lenoir, W.; Liu, W.; Liu, Y.; Fan, H.; Shen, H.; Ravikumar, V.; et al. A Comprehensive Pan-Cancer Molecular Study of Gynecologic and Breast Cancers. Cancer Cell 2018, 33, 690–705.e9. [Google Scholar] [CrossRef] [Green Version]
- Liu, Y.; Sethi, N.S.; Hinoue, T.; Schneider, B.G.; Cherniack, A.D.; Sanchez-Vega, F.; Seoane, J.A.; Farshidfar, F.; Bowlby, R.; Islam, M.; et al. Comparative Molecular Analysis of Gastrointestinal Adenocarcinomas. Cancer Cell 2018, 33, 721–735.e8. [Google Scholar] [CrossRef] [Green Version]
- Campbell, J.D.; Yau, C.; Bowlby, R.; Liu, Y.; Brennan, K.; Fan, H.; Taylor, A.M.; Wang, C.; Walter, V.; Akbani, R.; et al. Genomic, Pathway Network, and Immunologic Features Distinguishing Squamous Carcinomas. Cell Rep. 2018, 23, 194–212.e6. [Google Scholar] [CrossRef] [Green Version]
- Ricketts, C.J.; De Cubas, A.A.; Fan, H.; Smith, C.C.; Lang, M.; Reznik, E.; Bowlby, R.; Gibb, E.A.; Akbani, R.; Beroukhim, R.; et al. The Cancer Genome Atlas Comprehensive Molecular Characterization of Renal Cell Carcinoma. Cell Rep. 2018, 23, 313–326.e5. [Google Scholar] [CrossRef] [Green Version]
- Bailey, M.H.; Tokheim, C.; Porta-Pardo, E.; Sengupta, S.; Bertrand, D.; Weerasinghe, A.; Colaprico, A.; Wendl, M.C.; Kim, J.; Reardon, B.; et al. Comprehensive Characterization of Cancer Driver Genes and Mutations. Cell 2018, 173, 371–385.e18. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Gao, Q.; Liang, W.-W.; Foltz, S.M.; Mutharasu, G.; Jayasinghe, R.G.; Cao, S.; Liao, W.-W.; Reynolds, S.M.; Wyczalkowski, M.A.; Yao, L.; et al. Driver Fusions and Their Implications in the Development and Treatment of Human Cancers. Cell Rep. 2018, 23, 227–238.e3. [Google Scholar] [CrossRef] [Green Version]
- Liu, J.; Lichtenberg, T.; Hoadley, K.A.; Poisson, L.M.; Lazar, A.J.; Cherniack, A.D.; Kovatich, A.J.; Benz, C.C.; Levine, D.A.; Lee, A.V.; et al. An Integrated TCGA Pan-Cancer Clinical Data Resource to Drive High-Quality Survival Outcome Analytics. Cell 2018, 173, 400–416.e11. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Sanchez-Vega, F.; Mina, M.; Armenia, J.; Chatila, W.K.; Luna, A.; La, K.C.; Dimitriadoy, S.; Liu, D.L.; Kantheti, H.S.; Saghafinia, S.; et al. Oncogenic Signaling Pathways in The Cancer Genome Atlas. Cell 2018, 173, 321–337.e10. [Google Scholar] [CrossRef] [Green Version]
- Mermel, C.H.; Schumacher, S.E.; Hill, B.; Meyerson, M.L.; Beroukhim, R.; Getz, G. GISTIC2.0 facilitates sensitive and confident localization of the targets of focal somatic copy-number alteration in human cancers. Genome Biol. 2011, 12, R41. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Li, B.; Dewey, C.N. RSEM: Accurate transcript quantification from RNA-Seq data with or without a reference genome. BMC Bioinform. 2011, 12, 323. [Google Scholar] [CrossRef] [Green Version]
- Boellner, S.; Becker, K.-F. Reverse Phase Protein Arrays-Quantitative Assessment of Multiple Biomarkers in Biopsies for Clinical Use. Microarrays 2015, 4, 98–114. [Google Scholar] [CrossRef] [Green Version]
- López-Cortés, A.; Paz-y-Miño, C.; Guerrero, S.; Cabrera-Andrade, A.; Barigye, S.J.; Munteanu, C.R.; González-Díaz, H.; Pazos, A.; Pérez-Castillo, Y.; Tejera, E. OncoOmics approaches to reveal essential genes in breast cancer: A panoramic view from pathogenesis to precision medicine. Sci. Rep. 2020, 10, 5285. [Google Scholar] [CrossRef] [Green Version]
- Szklarczyk, D.; Franceschini, A.; Wyder, S.; Forslund, K.; Heller, D.; Huerta-Cepas, J.; Simonovic, M.; Roth, A.; Santos, A.; Tsafou, K.P.; et al. STRING v10: Protein-protein interaction networks, integrated over the tree of life. Nucleic Acids Res. 2015, 43, D447–D452. [Google Scholar] [CrossRef]
- Guerrero, S.; López-Cortés, A.; García-Cárdenas, J.M.; Armendáriz-Castillo, I.; Zambrano, A.K.; Indacochea, A.; Pérez-Villa, A.; Yumiceba, V.; Guevara-Ramírez, P.; Andrea-Jácome-Alvarado; et al. In silico analyses reveal new putative Breast Cancer RNA-binding proteins. bioRxiv 2020. [Google Scholar] [CrossRef]
- Reimand, J.; Isserlin, R.; Voisin, V.; Kucera, M.; Tannus-Lopes, C.; Rostamianfar, A.; Wadi, L.; Meyer, M.; Wong, J.; Xu, C.; et al. Pathway enrichment analysis and visualization of omics data using g:Profiler, GSEA, Cytoscape and EnrichmentMap. Nat. Protoc. 2019, 14, 482–517. [Google Scholar] [CrossRef]
- Raudvere, U.; Kolberg, L.; Kuzmin, I.; Arak, T.; Adler, P.; Peterson, H.; Vilo, J. g:Profiler: A web server for functional enrichment analysis and conversions of gene lists (2019 update). Nucleic Acids Res. 2019, 47, W191–W198. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- López-Cortés, A.; Abarca, E.; Silva, L.; Velastegui, E.; León-Sosa, A.; Karolys, G.; Cabrera, F.; Caicedo, A. Identification of key proteins in the signaling crossroads between wound healing and cancer hallmark phenotypes. Sci. Rep. 2021, 11, 17245. [Google Scholar] [CrossRef]
- López-Cortés, A.; Guevara-Ramírez, P.; Guerrero, S.; Ortiz-Prado, E.; García-Cárdenas, J.M.; Zambrano, A.K.; Armendáriz-Castillo, I.; Pérez-Villa, A.; Yumiceba, V.; Varela, N.; et al. Metastatic signaling of hypoxia-related genes across TCGA Pan-Cancer types. bioRxiv 2020. [Google Scholar] [CrossRef]
- García-Cárdenas, J.M.; Guerrero, S.; López-Cortés, A.; Armendáriz-Castillo, I.; Guevara-Ramírez, P.; Pérez-Villa, A.; Yumiceba, V.; Zambrano, A.K.; Leone, P.E.; Paz-y-Miño, C. Post-transcriptional Regulation of Colorectal Cancer: A Focus on RNA-Binding Proteins. Front. Mol. Biosci. 2019, 6, 65. [Google Scholar] [CrossRef] [Green Version]
- Fabregat, A.; Sidiropoulos, K.; Garapati, P.; Gillespie, M.; Hausmann, K.; Haw, R.; Jassal, B.; Jupe, S.; Korninger, F.; McKay, S.; et al. The Reactome pathway Knowledgebase. Nucleic Acids Res. 2016, 44, D481–D487. [Google Scholar] [CrossRef] [Green Version]
- Lovejoy, C.A.; Takai, K.; Huh, M.S.; Picketts, D.J.; de Lange, T. ATRX affects the repair of telomeric DSBs by promoting cohesion and a DAXX-dependent activity. PLoS Biol. 2020, 18, e3000594. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Chang, H.R.; Munkhjargal, A.; Kim, M.-J.; Park, S.Y.; Jung, E.; Ryu, J.-H.; Yang, Y.; Lim, J.-S.; Kim, Y. The functional roles of PML nuclear bodies in genome maintenance. Mutat. Res. 2018, 809, 99–107. [Google Scholar] [CrossRef] [PubMed]
- Wee, Y.; Liu, Y.; Lu, J.; Li, X.; Zhao, M. Identification of novel prognosis-related genes associated with cancer using integrative network analysis. Sci. Rep. 2018, 8, 3233. [Google Scholar] [CrossRef]
- Shao, X.; Lv, N.; Liao, J.; Long, J.; Xue, R.; Ai, N.; Xu, D.; Fan, X. Copy number variation is highly correlated with differential gene expression: A pan-cancer study. BMC Med. Genet. 2019, 20, 175. [Google Scholar] [CrossRef]
- Sobinoff, A.P.; Pickett, H.A. Mechanisms that drive telomere maintenance and recombination in human cancers. Curr. Opin. Genet. Dev. 2020, 60, 25–30. [Google Scholar] [CrossRef]
- De Vitis, M.; Berardinelli, F.; Sgura, A. Telomere length maintenance in cancer: At the crossroad between telomerase and alternative lengthening of telomeres (ALT). Int. J. Mol. Sci. 2018, 19, 606. [Google Scholar] [CrossRef] [Green Version]
- Hu, Y.; Shi, G.; Zhang, L.; Li, F.; Jiang, Y.; Jiang, S.; Ma, W.; Zhao, Y.; Songyang, Z.; Huang, J. Switch telomerase to ALT mechanism by inducing telomeric DNA damages and dysfunction of ATRX and DAXX. Sci. Rep. 2016, 6, 1–10. [Google Scholar] [CrossRef]
- Alhendi, A.S.N.; Royle, N.J. The absence of (TCAGGG)(n) repeats in some telomeres, combined with variable responses to NR2F2 depletion, suggest that this nuclear receptor plays an indirect role in the alternative lengthening of telomeres. Sci. Rep. 2020, 10, 20597. [Google Scholar] [CrossRef] [PubMed]
- Zhang, J.-M.; Zou, L. Alternative lengthening of telomeres: From molecular mechanisms to therapeutic outlooks. Cell Biosci. 2020, 10, 1–9. [Google Scholar] [CrossRef] [Green Version]
- Lovejoy, C.A.; Li, W.; Reisenweber, S.; Thongthip, S.; Bruno, J.; de Lange, T.; De, S.; Petrini, J.H.J.; Sung, P.A.; Jasin, M.; et al. Loss of ATRX, genome instability, and an altered DNA damage response are hallmarks of the alternative lengthening of Telomeres pathway. PLoS Genet. 2012, 8, 12–15. [Google Scholar] [CrossRef] [PubMed]
- Osterwald, S.; Deeg, K.I.; Chung, I.; Parisotto, D.; Worz, S.; Rohr, K.; Erfle, H.; Rippe, K. PML induces compaction, TRF2 depletion and DNA damage signaling at telomeres and promotes their alternative lengthening. J. Cell Sci. 2015, 128, 1887–1900. [Google Scholar] [CrossRef] [Green Version]
- Dejardin, J.; Kingston, R.E. Purification of proteins associated with specific genomic Loci. Cell 2009, 136, 175–186. [Google Scholar] [CrossRef] [Green Version]
- Fu, D.; Collins, K. Purification of Human Telomerase Complexes Identifies Factors Involved in Telomerase Biogenesis and Telomere Length Regulation. Mol. Cell 2007, 28, 773–785. [Google Scholar] [CrossRef] [Green Version]
- Zhu, Q.; Meng, L.; Hsu, J.K.; Lin, T.; Teishima, J.; Tsai, R.Y.L. GNL3L stabilizes the TRF1 complex and promotes mitotic transition. J. Cell Biol. 2009, 185, 827–839. [Google Scholar] [CrossRef] [Green Version]
- Spinella, J.-F.; Cassart, P.; Garnier, N.; Rousseau, P.; Drullion, C.; Richer, C.; Ouimet, M.; Saillour, V.; Healy, J.; Autexier, C.; et al. A novel somatic mutation in ACD induces telomere lengthening and apoptosis resistance in leukemia cells. BMC Cancer 2015, 15, 621. [Google Scholar] [CrossRef] [Green Version]
- Hussain, S.S.; Majumdar, R.; Moore, G.M.; Narang, H.; Buechelmaier, E.S.; Bazil, M.J.; Ravindran, P.T.; Leeman, J.E.; Li, Y.; Jalan, M.; et al. Measuring nonhomologous end-joining, homologous recombination and alternative end-joining simultaneously at an endogenous locus in any transfectable human cell. Nucleic Acids Res. 2021, 49, e74. [Google Scholar] [CrossRef]
- Zhang, J.-M.; Genois, M.-M.; Ouyang, J.; Lan, L.; Zou, L. Alternative lengthening of telomeres is a self-perpetuating process in ALT-associated PML bodies. Mol. Cell 2021, 81, 1027–1042.e4. [Google Scholar] [CrossRef] [PubMed]
- Meng, Y.; Li, X. Expression and Prognosis Analysis of SUMOylation Regulators in Oral Squamous Cell Carcinoma Based on High-Throughput Sequencing. Front. Genet. 2021, 12, 671392. [Google Scholar] [CrossRef] [PubMed]
- Yang, H.; Gao, S.; Chen, J.; Lou, W. UBE2I promotes metastasis and correlates with poor prognosis in hepatocellular carcinoma. Cancer Cell Int. 2020, 20, 234. [Google Scholar] [CrossRef] [PubMed]
- Jiang, W.-Q.; Zhong, Z.-H.; Henson, J.D.; Reddel, R.R. Identification of candidate alternative lengthening of telomeres genes by methionine restriction and RNA interference. Oncogene 2007, 26, 4635–4647. [Google Scholar] [CrossRef] [Green Version]
- Yu, J.; Lan, J.; Wang, C.; Wu, Q.; Zhu, Y.; Lai, X.; Sun, J.; Jin, C.; Huang, H. PML3 interacts with TRF1 and is essential for ALT-associated PML bodies assembly in U2OS cells. Cancer Lett. 2010, 291, 177–186. [Google Scholar] [CrossRef] [PubMed]
- Feng, X.; Luo, Z.; Jiang, S.; Li, F.; Han, X.; Hu, Y.; Wang, D.; Zhao, Y.; Ma, W.; Liu, D.; et al. The telomere-associated homeobox-containing protein TAH1/HMBOX1 participates in telomere maintenance in ALT cells. J. Cell Sci. 2013, 126, 3982–3989. [Google Scholar] [CrossRef] [Green Version]
- Hsu, J.K.; Lin, T.; Tsai, R.Y.L. Nucleostemin prevents telomere damage by promoting PML-IV recruitment to SUMOylated TRF1. J. Cell Biol. 2012, 197, 613–624. [Google Scholar] [CrossRef] [Green Version]
- Ho, A.; Wilson, F.R.; Peragine, S.L.; Jeyanthan, K.; Mitchell, T.R.H.; Zhu, X.-D. TRF1 phosphorylation on T271 modulates telomerase-dependent telomere length maintenance as well as the formation of ALT-associated PML bodies. Sci. Rep. 2016, 6, 36913. [Google Scholar] [CrossRef] [Green Version]
- Wang, C.; Songyang, Z.; Huang, Y. TRIM28 inhibits alternative lengthening of telomere phenotypes by protecting SETDB1 from degradation. Cell Biosci. 2021, 11, 149. [Google Scholar] [CrossRef]
- Wilson, F.R.; Ho, A.; Walker, J.R.; Zhu, X.-D. Cdk-dependent phosphorylation regulates TRF1 recruitment to PML bodies and promotes C-circle production in ALT cells. J. Cell Sci. 2016, 129, 2559–2572. [Google Scholar] [CrossRef] [Green Version]
- Garcia-Exposito, L.; Bournique, E.; Bergoglio, V.; Bose, A.; Barroso-Gonzalez, J.; Zhang, S.; Roncaioli, J.L.; Lee, M.; Wallace, C.T.; Watkins, S.C.; et al. Proteomic Profiling Reveals a Specific Role for Translesion DNA Polymerase η in the Alternative Lengthening of Telomeres. Cell Rep. 2016, 17, 1858–1871. [Google Scholar] [CrossRef] [Green Version]
- Min, J.; Wright, W.E.; Shay, J.W. Alternative Lengthening of Telomeres Mediated by Mitotic DNA Synthesis Engages Break-Induced Replication Processes. Mol. Cell. Biol. 2017, 37, e00226-17. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Tan, J.; Wang, X.; Hwang, B.-J.; Gonzales, R.; Konen, O.; Lan, L.; Lu, A.-L. An ordered assembly of MYH glycosylase, SIRT6 protein deacetylase, and Rad9-Rad1-Hus1 checkpoint clamp at oxidatively damaged telomeres. Aging 2020, 12, 17761–17785. [Google Scholar] [CrossRef] [PubMed]
- Loe, T.K.; Li, J.S.Z.; Zhang, Y.; Azeroglu, B.; Boddy, M.N.; Denchi, E.L. Telomere length heterogeneity in ALT cells is maintained by PML-dependent localization of the BTR complex to telomeres. Genes Dev. 2020, 34, 650–662. [Google Scholar] [CrossRef]
- Shen, M.; Young, A.; Autexier, C. PCNA, a focus on replication stress and the alternative lengthening of telomeres pathway. DNA Repair 2021, 100, 103055. [Google Scholar] [CrossRef] [PubMed]
- Xu, M.; Qin, J.; Wang, L.; Lee, H.-J.; Kao, C.-Y.; Liu, D.; Songyang, Z.; Chen, J.; Tsai, M.-J.; Tsai, S.Y. Nuclear receptors regulate alternative lengthening of telomeres through a novel noncanonical FANCD2 pathway. Sci. Adv. 2019, 5, eaax6366. [Google Scholar] [CrossRef] [Green Version]







Publisher’s Note: MDPI stays neutral with regard to jurisdictional claims in published maps and institutional affiliations. |
© 2022 by the authors. Licensee MDPI, Basel, Switzerland. This article is an open access article distributed under the terms and conditions of the Creative Commons Attribution (CC BY) license (https://creativecommons.org/licenses/by/4.0/).
Share and Cite
Armendáriz-Castillo, I.; Hidalgo-Fernández, K.; Pérez-Villa, A.; García-Cárdenas, J.M.; López-Cortés, A.; Guerrero, S. Identification of Key Proteins from the Alternative Lengthening of Telomeres-Associated Promyelocytic Leukemia Nuclear Bodies Pathway. Biology 2022, 11, 185. https://doi.org/10.3390/biology11020185
Armendáriz-Castillo I, Hidalgo-Fernández K, Pérez-Villa A, García-Cárdenas JM, López-Cortés A, Guerrero S. Identification of Key Proteins from the Alternative Lengthening of Telomeres-Associated Promyelocytic Leukemia Nuclear Bodies Pathway. Biology. 2022; 11(2):185. https://doi.org/10.3390/biology11020185
Chicago/Turabian StyleArmendáriz-Castillo, Isaac, Katherine Hidalgo-Fernández, Andy Pérez-Villa, Jennyfer M. García-Cárdenas, Andrés López-Cortés, and Santiago Guerrero. 2022. "Identification of Key Proteins from the Alternative Lengthening of Telomeres-Associated Promyelocytic Leukemia Nuclear Bodies Pathway" Biology 11, no. 2: 185. https://doi.org/10.3390/biology11020185
APA StyleArmendáriz-Castillo, I., Hidalgo-Fernández, K., Pérez-Villa, A., García-Cárdenas, J. M., López-Cortés, A., & Guerrero, S. (2022). Identification of Key Proteins from the Alternative Lengthening of Telomeres-Associated Promyelocytic Leukemia Nuclear Bodies Pathway. Biology, 11(2), 185. https://doi.org/10.3390/biology11020185