
Insulin and Its Key Role for Mitochondrial Function/Dysfunction and Quality Control: A Shared Link between Dysmetabolism and Neurodegeneration


Abstract
:Simple Summary
Abstract
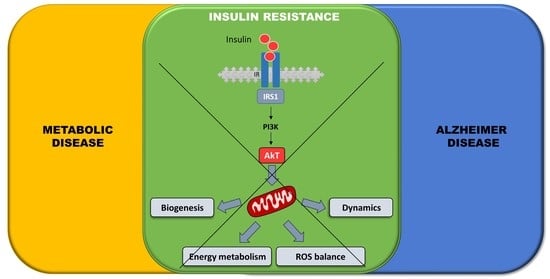
{kind=link}
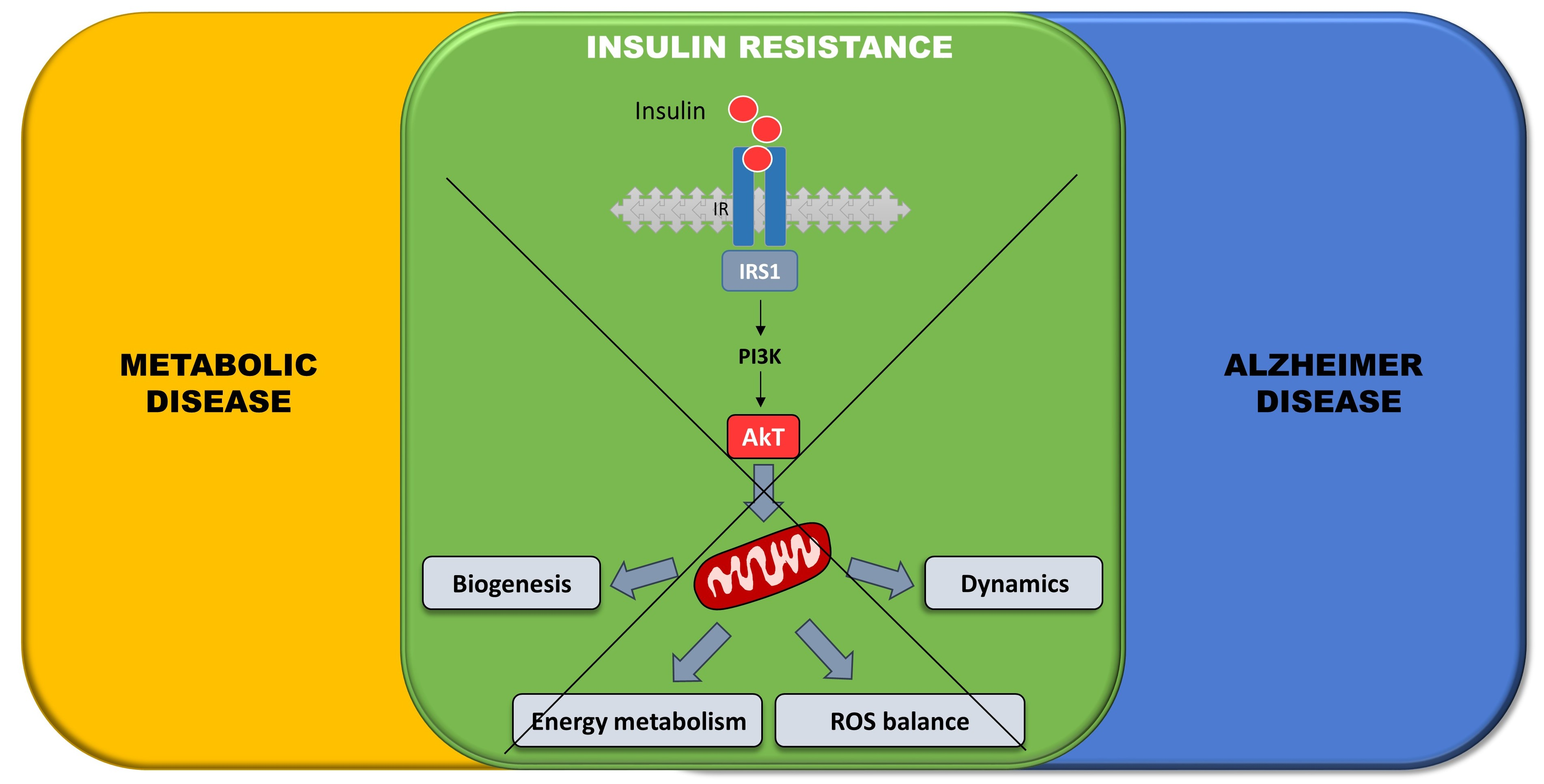
{kind=link}
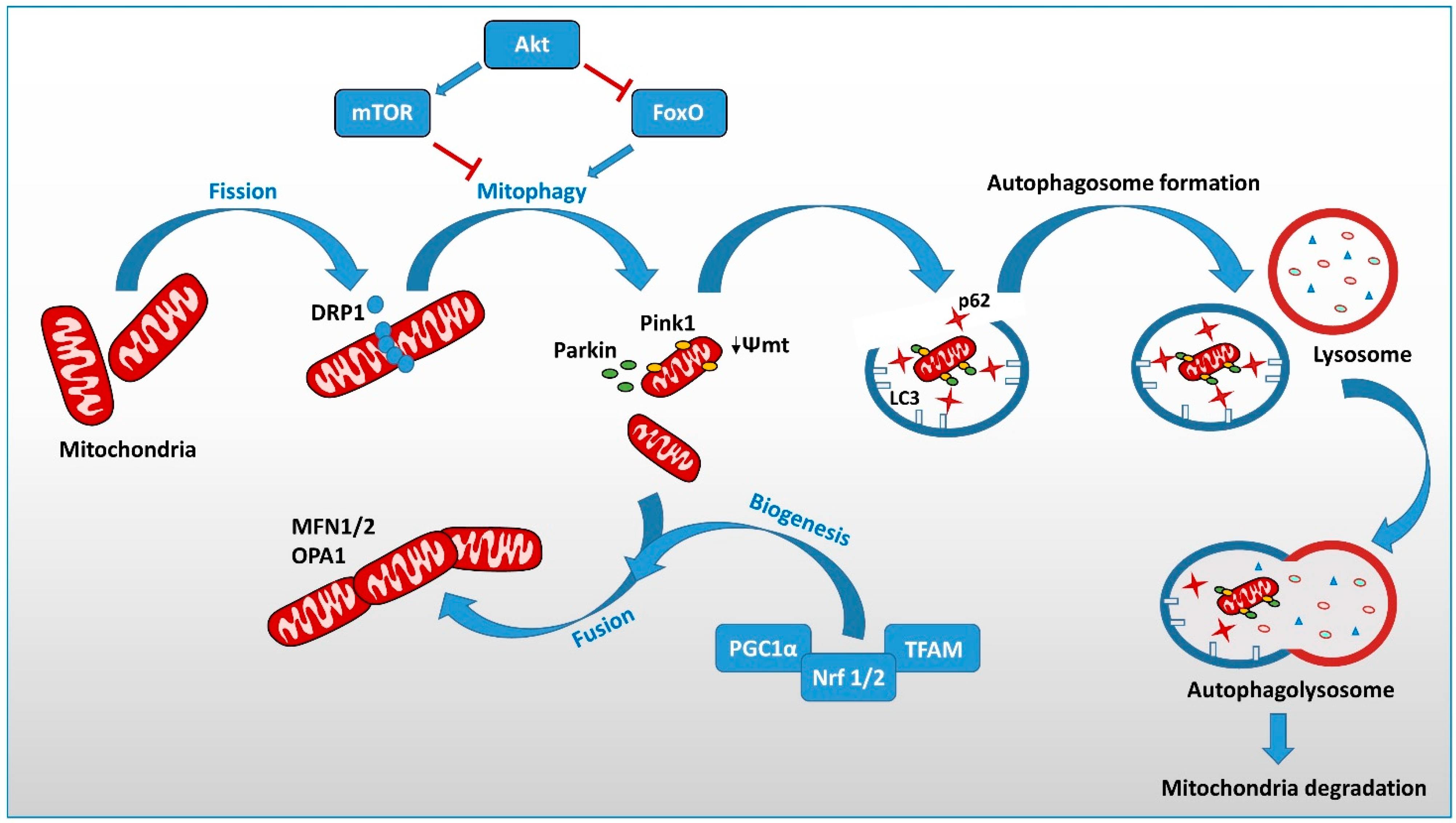
{kind=link}
1. Introduction
2. Insulin Tales
3. Insulin Synthesis and Secretion
4. Insulin Receptors and Insulin Binding
5. Insulin Signaling
6. Insulin in the Brain
7. Brain Insulin Resistance
8. Alzheimer Disease and Insulin
9. Intranasal Insulin as an AD Therapeutic Agent
10. Insulin, Insulin Resistance, and Mitochondrial Dysfunction
11. Mitochondrial Dynamics
12. Mitochondrial Dynamics and Insulin
13. Mitophagy
14. Mitophagy and Insulin
15. Mitochondrial Biogenesis
16. AMPK
17. SIRT1
18. Mitochondrial Biogenesis and Insulin
19. Mitochondrial Therapy: Targeting Mitochondria to Break down the Damage
20. Conclusions
Author Contributions
Funding
Institutional Review Board Statement
Informed Consent Statement
Data Availability Statement
Acknowledgments
Conflicts of Interest
References
- Zhao, W.; Chen, H.; Xu, H.; Moore, E.; Meiri, N.; Quon, M.J.; Alkon, D.L. Brain insulin receptors and spatial memory. Correlated changes in gene expression, tyrosine phosphorylation, and signaling molecules in the hippocampus of water maze trained rats. J. Biol. Chem. 1999, 274, 34893–34902. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Banks, W.A.; Owen, J.B.; Erickson, M.A. Insulin in the brain: There and back again. Pharmacol. Ther. 2012, 136, 82–93. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Ghasemi, R.; Haeri, A.; Dargahi, L.; Mohamed, Z.; Ahmadiani, A. Insulin in the Brain: Sources, Localization and Functions. Mol. Neurobiol. 2013, 47, 145–171. [Google Scholar] [CrossRef] [PubMed]
- Salas, I.H.; De Strooper, B. Diabetes and Alzheimer’s Disease: A Link not as Simple as it Seems. Neurochem. Res. 2018, 44, 1271–1278. [Google Scholar] [CrossRef] [Green Version]
- Craft, S. Insulin resistance and AD—Extending the translational path. Nat. Rev. Neurol. 2012, 8, 360–362. [Google Scholar] [CrossRef]
- Nuzzo, D.; Picone, P.; Baldassano, S.; Caruana, L.; Messina, E.; Gammazza, M.A.; Cappello, F.; Mulè, F.; Carlo, M. Insulin Resistance as Common Molecular Denominator Linking Obesity to Alzheimer’s Disease. Curr. Alzheimer Res. 2015, 12, 723–735. [Google Scholar] [CrossRef] [Green Version]
- de M Lyra E Silva, N.; Gonçalves, R.A.; Boehnke, S.E.; Forny-Germano, L.; Munoz, D.P.; De Felice, F.G. Understanding the link between insulin resistance and Alzheimer’s disease: Insights from animal models. Exp. Neurol. 2019, 316, 1–11. [Google Scholar] [CrossRef]
- De Felice, F.G.; Ferreira, S.T. Inflammation, Defective Insulin Signaling, and Mitochondrial Dysfunction as Common Molecular Denominators Connecting Type 2 Diabetes to Alzheimer Disease. Diabetes 2014, 63, 2262–2272. [Google Scholar] [CrossRef] [Green Version]
- Ma, J.; Zhang, W.; Wang, H.-F.; Wang, Z.-X.; Jiang, T.; Tan, M.-S.; Yu, J.-T.; Tan, L. Peripheral Blood Adipokines and Insulin Levels in Patients with Alzheimer’s Disease: A Replication Study and Meta-Analysis. Curr. Alzheimer Res. 2016, 13, 223. [Google Scholar] [CrossRef]
- Sergi, D.; Naumovski, N.; Heilbronn, L.K.; Abeywardena, M.; O’Callaghan, N.; Lionetti, L.; Luscombe-Marsh, N. Mitochondrial (Dys)function and Insulin Resistance: From Pathophysiological Molecular Mechanisms to the Impact of Diet. Front. Physiol. 2019, 10, 532. [Google Scholar] [CrossRef]
- Picone, P.; Nuzzo, D.; Caruana, L.; Scafidi, V.; Di Carlo, M. Mitochondrial Dysfunction: Different Routes to Alzheimer’s Disease Therapy. Oxidative Med. Cell. Longev. 2014, 2014, 780179. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Van der Bliek, A.M.; Shen, Q.; Kawajiri, S. Mechanisms of Mitochondrial Fission and Fusion. Cold Spring Harb. Lab. Perspect. Biol. 2013, 5, a011072. [Google Scholar] [CrossRef] [PubMed]
- Fisher-Wellman, K.H.; Neufer, P.D. Linking mitochondrial bioenergetics to insulin resistance via redox biology. Trends Endocrinol. Metab. 2012, 23, 142–153. [Google Scholar] [CrossRef] [Green Version]
- Morino, K.; Petersen, K.F.; Dufour, S.; Befroy, D.; Frattini, J.; Shatzkes, N.; Neschen, S.; White, M.F.; Bilz, S.; Sono, S.; et al. Reduced mitochondrial density and increased IRS-1 serine phosphorylation in muscle of insulin-resistant offspring of type 2 diabetic parents. J. Clin. Investig. 2005, 115, 3587–3593. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Bliss, M. The History of Insulin. Diabetes Care 1993, 16 (Suppl. S3), 4–7. [Google Scholar] [CrossRef] [PubMed]
- Banting, F.G.; Best, C.H. The internal secretion of the pancreas. J. Lab. Clin. Med. 1922, 7, 251–266. [Google Scholar]
- Rydén, L.; Lindsten, J. The history of the Nobel prize for the discovery of insulin. Diabetes Res. Clin. Pract. 2021, 175, 108819. [Google Scholar] [CrossRef] [PubMed]
- Celeste, C.; Cheikh, Q.J. History of insulin. J. Community Hosp. Intern. Med. Perspect. 2012, 2, 18701. [Google Scholar] [CrossRef] [Green Version]
- Honka, M.J.; Latva-Rasku, A.; Bucci, M.; Virtanen, K.A.; Hannukainen, J.C.; Kalliokoski, K.K.; Nuutila, P. Insulin-stimulated glucose uptake in skeletal muscle, adipose tissue and liver: A positron emission tomography study. Eur. J. Endocrinol. 2018, 178, 523–531. [Google Scholar] [CrossRef]
- Weiss, M.A. Proinsulin and the genetics of diabetes mellitus. J. Biol. Chem. 2009, 284, 19159–19163. [Google Scholar] [CrossRef] [Green Version]
- Wilcox, G. Insulin and Insulin Resistance. Clin. Biochem. Rev. 2005, 26, 19–39. [Google Scholar] [PubMed]
- Simon, C.; Brandenberger, G. Ultradian Oscillations of Insulin Secretion in Humans. Diabetes 2002, 51, S258–S261. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Pørksen, N.; Hollingdal, M.; Juhl, C.; Butler, P.; Veldhuis, J.D.; Schmitz, O. Pulsatile Insulin Secretion: Detection, Regulation, and Role in Diabetes. Diabetes 2002, 51 (Suppl. S1), S245–S254. [Google Scholar] [CrossRef] [Green Version]
- Palumbo, P.; De Gaetano, A. An islet population model of the endocrine pancreas. J. Math. Biol. 2009, 61, 171–205. [Google Scholar] [CrossRef] [PubMed]
- Contardi, M.; Kossyvaki, D.; Picone, P.; Summa, M.; Guo, X.; Heredia-Guerrero, J.A.; Giacomazza, D.; Carzino, R.; Goldoni, L.; Scoponi, G.; et al. Electrospun polyvinylpyrrolidone (PVP) hydrogels containing hydroxycinnamic acid derivatives as potential wound dressings. Chem. Eng. J. 2021, 409, 128144. [Google Scholar] [CrossRef]
- Contardi, M.; Summa, M.; Picone, P.; Brancato, O.R.; Di Carlo, M.; Bertorelli, R.; Athanassiou, A. Evaluation of a Multifunctional Polyvinylpyrrolidone/Hyaluronic Acid-Based Bilayer Film Patch with Anti-Inflammatory Properties as an Enhancer of the Wound Healing Process. Pharmaceutics 2022, 14, 483. [Google Scholar] [CrossRef] [PubMed]
- Kido, Y.; Nakae, J.; Accili, M. The Insulin Receptor and Its Cellular Targets 1. J. Clin. Endocrinol. Metab. 2001, 86, 972–979. [Google Scholar] [CrossRef]
- Van Obberghen, E.; Baron, V.; Delahaye, L.; Emanuelli, B.; Filippa, N.; Giorgetti-Peraldi, S.; Lebrun, P.; Mothe-Satney, I.; Peraldi, P.; Rocchi, S.; et al. Surfing the insulin signaling web. Eur. J. Clin. Investig. 2001, 31, 966–977. [Google Scholar] [CrossRef]
- Lizcano, J.M.; Alessi, D.R. The insulin signalling pathway. Curr. Biol. 2002, 12, R236–R238. [Google Scholar] [CrossRef] [Green Version]
- Taniguchi, C.M.; Emanuelli, B.; Kahn, C.R. Critical nodes in signalling pathways: Insights into insulin action. Nat. Rev. Mol. Cell Biol. 2006, 7, 85–96. [Google Scholar] [CrossRef]
- Dyer, A.H.; Vahdatpour, C.; Sanfeliu, A.; Tropea, D. The role of Insulin-Like Growth Factor 1 (IGF-1) in brain development, maturation and neuroplasticity. Neuroscience 2016, 325, 89–99. [Google Scholar] [CrossRef] [PubMed]
- Jethwa, N.; Chung, G.H.C.; Lete, M.; Alonso, A.; Byrne, R.D.; Calleja, V.; Larijani, B. Endomembrane PtdIns(3,4,5)P3 activates the PI3K/Akt pathway. J. Cell Sci. 2015, 128, 3456–3465. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Alessi, D.R.; James, S.R.; Downes, C.; Holmes, A.B.; Gaffney, P.R.; Reese, C.B.; Cohen, P. Characterization of a 3-phosphoinositide-dependent protein kinase which phosphorylates and activates protein kinase Bα. Curr. Biol. 1997, 7, 261–269. [Google Scholar] [CrossRef] [Green Version]
- Chang, F.; Lee, J.T.; Navolanic, P.M.; Steelman, L.S.; Shelton, J.G.; Blalock, W.L.; Franklin, R.A.; McCubrey, J.A. Involvement of PI3K/Akt pathway in cell cycle progression, apoptosis, and neoplastic transformation: A target for cancer chemotherapy. Leukemia 2003, 17, 590–603. [Google Scholar] [CrossRef] [Green Version]
- Frame, S.; Cohen, P. GSK3 takes centre stage more than 20 years after its discovery. Biochem. J. 2001, 359, 1–16. [Google Scholar] [CrossRef]
- Saltiel, A.R.; Kahn, C.R. Insulin signalling and the regulation of glucose and lipid metabolism. Nature 2001, 414, 799–806. [Google Scholar] [CrossRef]
- Accili, D.; Arden, K.C. FoxOs at the Crossroads of Cellular Metabolism, Differentiation, and Transformation. Cell 2004, 117, 421–426. [Google Scholar] [CrossRef] [Green Version]
- Brunet, A.; Sweeney, L.B.; Sturgill, J.F.; Chua, K.F.; Greer, P.L.; Lin, Y.; Tran, H.; Ross, S.E.; Mostoslavsky, R.; Cohen, H.Y.; et al. Stress-Dependent Regulation of FOXO Transcription Factors by the SIRT1 Deacetylase. Science 2004, 303, 2011–2015. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Greer, E.L.; Brunet, A. FOXO transcription factors at the interface between longevity and tumor suppression. Oncogene 2005, 24, 7410–7425. [Google Scholar] [CrossRef] [Green Version]
- Perluigi, M.; Di Domenico, F.; Butterfield, D.A. mTOR signaling in aging and neurodegeneration: At the crossroad between metabolism dysfunction and impairment of autophagy. Neurobiol. Dis. 2015, 84, 39–49. [Google Scholar] [CrossRef]
- Romanelli, R.J.; LeBeau, A.P.; Fulmer, C.G.; Lazzarino, D.A.; Hochberg, A.; Wood, T. Insulin-like Growth Factor Type-I Receptor Internalization and Recycling Mediate the Sustained Phosphorylation of Akt. J. Biol. Chem. 2007, 282, 22513–22524. [Google Scholar] [CrossRef] [Green Version]
- Wullschleger, S.; Loewith, R.; Hall, M.N. TOR Signaling in Growth and Metabolism. Cell 2006, 124, 471–484. [Google Scholar] [CrossRef] [Green Version]
- Zhang, W.; Liu, H.T. MAPK signal pathways in the regulation of cell proliferation in mammalian cells. Cell Res. 2002, 12, 9–18. [Google Scholar] [CrossRef]
- Rafaelsen, O.J. Action of Insulin on Carbohydrate Uptake of Isolated Rat Spinal Cord. J. Neurochem. 1961, 7, 33–44. [Google Scholar] [CrossRef]
- Rafaelsen, O.J. Action of insulin on glucose uptake of rat brain slices and isolated rat cerebellum. J. Neurochem. 1961, 7, 45–51. [Google Scholar] [CrossRef]
- Butterfield, W.; Sells, R.; Abrams, M.; Sterky, G.; Whichelow, M. Insulin sensitivity of the human brain. Lancet 1966, 287, 557–560. [Google Scholar] [CrossRef]
- Margolis, R.U.; Altszuler, N. Insulin in the Cerebrospinal Fluid. Nature 1967, 215, 1375–1376. [Google Scholar] [CrossRef]
- Havrankova, J.; Schmechel, D.; Roth, J.; Brownstein, M. Identification of insulin in rat brain. Proc. Natl. Acad. Sci. USA 1978, 75, 5737–5741. [Google Scholar] [CrossRef] [Green Version]
- Havrankova, J.; Roth, J.; Brownstein, M. Insulin receptors are widely distributed in the central nervous system of the rat. Nature 1978, 272, 827–829. [Google Scholar] [CrossRef]
- Havrankova, J.; Roth, J.; Brownstein, M.J. Concentrations of insulin and of insulin receptors in the brain are independent of peripheral insulin levels. Studies of obese and streptozotocin-treated rodents. J. Clin. Investig. 1979, 64, 636–642. [Google Scholar] [CrossRef]
- Mehran, A.E.; Templeman, N.M.; Brigidi, S.; Lim, G.; Chu, K.-Y.; Hu, X.; Botezelli, J.D.; Asadi, A.; Hoffman, B.G.; Kieffer, T.J.; et al. Hyperinsulinemia Drives Diet-Induced Obesity Independently of Brain Insulin Production. Cell Metab. 2012, 16, 723–737. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Takano, K.; Koarashi, K.; Kawabe, K.; Itakura, M.; Nakajima, H.; Moriyama, M.; Nakamura, Y. Insulin expression in cultured astrocytes and the decrease by amyloid β. Neurochem. Int. 2018, 119, 171–177. [Google Scholar] [CrossRef] [PubMed]
- Kuwabara, T.; Kagalwala, M.N.; Onuma, Y.; Ito, Y.; Warashina, M.; Terashima, K.; Sanosaka, T.; Nakashima, K.; Gage, F.H.; Asashima, M. Insulin biosynthesis in neuronal progenitors derived from adult hippocampus and the olfactory bulb. EMBO Mol. Med. 2011, 3, 742–754. [Google Scholar] [CrossRef]
- Arnold, S.E.; Arvanitakis, Z.; Macauley-Rambach, S.L.; Koenig, A.M.; Wang, H.-Y.; Ahima, R.S.; Craft, S.; Gandy, S.; Buettner, C.; Stoeckel, L.E.; et al. Brain insulin resistance in type 2 diabetes and Alzheimer disease: Concepts and conundrums. Nat. Rev. Neurol. 2018, 14, 168–181. [Google Scholar] [CrossRef]
- Beattie, E.C.; Carroll, R.C.; Yu, X.; Morishita, W.; Yasuda, H.; Von Zastrow, M.; Malenka, R.C. Regulation of AMPA receptor endocytosis by a signaling mechanism shared with LTD. Nat. Neurosci. 2000, 3, 1291–1300. [Google Scholar] [CrossRef]
- Skeberdis, V.A.; Lan, J.-Y.; Zheng, X.; Zukin, R.S.; Bennett, M.V.L. Insulin promotes rapid delivery of N-methyl-d-aspartate receptors to the cell surface by exocytosis. Proc. Natl. Acad. Sci. USA 2001, 98, 3561–3566. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Song, J.; Wu, L.; Chen, Z.; Kohanski, R.A.; Pick, L. Axons Guided by Insulin Receptor in Drosophila Visual System. Science 2003, 300, 502–505. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Gu, T.; Zhao, T.; Hewes, R.S. Insulin signaling regulates neurite growth during metamorphic neuronal remodeling. Biol. Open 2014, 3, 81–93. [Google Scholar] [CrossRef] [Green Version]
- Grote, C.W.; Morris, J.K.; Ryals, J.M.; Geiger, P.C.; Wright, D.E. Insulin Receptor Substrate 2 Expression and Involvement in Neuronal Insulin Resistance in Diabetic Neuropathy. Exp. Diabetes Res. 2011, 2011, 212571. [Google Scholar] [CrossRef] [Green Version]
- Roloff, F.; Scheiblich, H.; Dewitz, C.; Dempewolf, S.; Stern, M.; Bicker, G. Enhanced Neurite Outgrowth of Human Model (NT2) Neurons by Small-Molecule Inhibitors of Rho/ROCK Signaling. PLoS ONE 2015, 10, e0118536. [Google Scholar] [CrossRef] [Green Version]
- Cunnane, S.C.; Trushina, E.; Morland, C.; Prigione, A.; Casadesus, G.; Andrews, Z.B.; Beal, M.F.; Bergersen, L.H.; Brinton, R.D.; de la Monte, S.; et al. Brain energy rescue: An emerging therapeutic concept for neurodegenerative disorders of ageing. Nat. Rev. Drug Discov. 2020, 19, 609–633. [Google Scholar] [CrossRef] [PubMed]
- Kleinridders, A.; Pothos, E.N. Impact of Brain Insulin Signaling on Dopamine Function, Food Intake, Reward, and Emotional Behavior. Curr. Nutr. Rep. 2019, 8, 83–91. [Google Scholar] [CrossRef] [PubMed]
- Goldstein, B.J. Insulin resistance as the core defect in type 2 diabetes mellitus. Am. J. Cardiol. 2002, 90, 3–10. [Google Scholar] [CrossRef]
- Mielke, J.G.; Taghibiglou, C.; Liu, L.; Zhang, Y.; Jia, Z.; Adeli, K.; Wang, Y.T. A biochemical and functional characterization of diet-induced brain insulin resistance. J. Neurochem. 2005, 93, 1568–1578. [Google Scholar] [CrossRef] [PubMed]
- Zhao, W.; De Felice, F.G.; Fernandez, S.; Chen, H.; Lambert, M.P.; Quon, M.; Krafft, G.A.; Klein, W.L. Amyloid beta oligomers induce impairment of neuronal insulin receptors. FASEB J. 2007, 22, 246–260. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- De Felice, F.G.; Vieira, M.N.N.; Bomfim, T.R.; Decker, H.; Velasco, P.T.; Lambert, M.P.; Viola, K.L.; Zhao, W.-Q.; Ferreira, S.T.; Klein, W.L. Protection of synapses against Alzheimer’s-linked toxins: Insulin signaling prevents the pathogenic binding of Aβ oligomers. Proc. Natl. Acad. Sci. USA 2009, 106, 1971–1976. [Google Scholar] [CrossRef] [Green Version]
- Picone, P.; Giacomazza, D.; Vetri, V.; Carrotta, R.; Militello, V.; San Biagio, P.L.; Di Carlo, M. Insulin-activated Akt rescues Aβ oxidative stress-induced cell death by orchestrating molecular trafficking. Aging Cell 2011, 10, 832–843. [Google Scholar] [CrossRef] [Green Version]
- Biddinger, S.B.; Kahn, C.R. From Mice to Men: Insights into the Insulin Resistance Syndromes. Annu. Rev. Physiol. 2006, 68, 123–158. [Google Scholar] [CrossRef] [Green Version]
- De La Monte, S. Brain Insulin Resistance and Deficiency as Therapeutic Targets in Alzheimer’s Disease. Curr. Alzheimer Res. 2012, 9, 35–66. [Google Scholar] [CrossRef]
- Arvanitakis, Z.; Wilson, R.S.; Bennett, D.A. Diabetes mellitus, dementia, and cognitive function in older persons. J. Nutr. Health Aging 2006, 10, 287–291. [Google Scholar]
- Tong, M.; de la Monte, S.M. Mechanisms of Ceramide-Mediated Neurodegeneration. J. Alzheimers Dis. 2009, 16, 705–714. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- De la Monte, S.M. Type 3 diabetes is sporadic Alzheimer’s disease: Mini-review. Eur. Neuropsychopharmacol. 2014, 24, 1954–1960. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Cardoso, S.; Seiça, R.; Moreira, P.I. Diabesity and brain energy metabolism: The case of Alzheimer’s disease. In Obesity and Brain Function. Advances in Neurobiology; Letra, L., Seiça, R., Eds.; Springer: Cham, Switzerland, 2017; p. 117. [Google Scholar] [CrossRef]
- De La Monte, S.M. Insulin Resistance and Neurodegeneration: Progress Towards the Development of New Therapeutics for Alzheimer’s Disease. Drugs 2017, 77, 47–65. [Google Scholar] [CrossRef] [Green Version]
- Steen, E.; Terry, B.M.; Rivera, E.J.; Cannon, J.L.; Neely, T.R.; Tavares, R.; Xu, X.J.; Wands, J.R.; de la Monte, S.M. Impaired insulin and insulin-like growth factor expression and signaling mechanisms in Alzheimer’s disease—Is this type 3 diabetes? J. Alzheimers Dis. 2005, 7, 63–80. [Google Scholar] [CrossRef] [Green Version]
- Maciejczyk, M.; Żebrowska, E.; Chabowski, A. Insulin Resistance and Oxidative Stress in the Brain: What’s New? Int. J. Mol. Sci. 2019, 20, 874. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Luchsinger, J.A. Type 2 Diabetes, Related Conditions, in Relation and Dementia: An Opportunity for Prevention? J. Alzheimers Dis. 2010, 20, 723–736. [Google Scholar] [CrossRef]
- Hölscher, C. Brain insulin resistance: Role in neurodegenerative disease and potential for targeting. Expert Opin. Investig. Drugs 2020, 29, 333–348. [Google Scholar] [CrossRef]
- Wang, J.; Gallagher, D.; DeVito, L.M.; Cancino, G.I.; Tsui, D.; He, L.; Keller, G.M.; Frankland, P.W.; Kaplan, D.R.; Miller, F.D. Metformin Activates an Atypical PKC-CBP Pathway to Promote Neurogenesis and Enhance Spatial Memory Formation. Cell Stem Cell 2012, 11, 23–35. [Google Scholar] [CrossRef] [Green Version]
- Chen, Y.; Zhou, K.; Wang, R.; Liu, Y.; Kwak, Y.-D.; Ma, T.; Thompson, R.C.; Zhao, Y.; Smith, L.; Gasparini, L.; et al. Antidiabetic drug metformin (GlucophageR) increases biogenesis of Alzheimer’s amyloid peptides via up-regulating BACE1 transcription. Proc. Natl. Acad. Sci. USA 2009, 106, 3907–3912. [Google Scholar] [CrossRef] [Green Version]
- Beeri, M.S.; Schmeidler, J.; Silverman, J.M.; Gandy, S.; Wysocki, M.; Hannigan, C.M.; Purohit, D.P.; Lesser, G.; Grossman, H.T.; Haroutunian, V. Insulin in combination with other diabetes medication is associated with less Alzheimer neuropathology. Neurology 2008, 71, 750–757. [Google Scholar] [CrossRef] [Green Version]
- Picone, P.; Nuzzo, D.; Caruana, L.; Messina, E.; Barera, A.; Vasto, S.; Di Carlo, M. Metformin increases APP expression and processing via oxidative stress, mitochondrial dysfunction and NF-κB activation: Use of insulin to attenuate metformin’s effect. Biochim. Biophys. Acta 2015, 1853, 1046–1059. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Picone, P.; Vilasi, S.; Librizzi, F.; Contardi, M.; Nuzzo, D.; Caruana, L.; Baldassano, S.; Amato, A.; Mulè, F.; Biagio, P.L.S.; et al. Biological and biophysics aspects of metformin-induced effects: Cortex mitochondrial dysfunction and promotion of toxic amyloid pre-fibrillar aggregates. Aging 2016, 8, 1718–1734. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Shoshan-Barmatz, V.; Anand, U.; Nahon-Crystal, E.; Di Carlo, M.; Shteinfer-Kuzmine, A. Adverse Effects of Metformin from Diabetes to COVID-19, Cancer, Neurodegenerative Diseases, and Aging: Is VDAC1 a Common Target? Front. Physiol. 2021, 12, 730048. [Google Scholar] [CrossRef]
- Ning, P.; Luo, A.; Mu, X.; Xu, Y.; Li, T. Exploring the dual character of metformin in Alzheimer’s disease. Neuropharmacology 2022, 207, 108966. [Google Scholar] [CrossRef]
- McNay, E.C.; Pearson-Leary, J. GluT4: A central player in hippocampal memory and brain insulin resistance. Exp. Neurol. 2019, 323, 113076. [Google Scholar] [CrossRef]
- Nuzzo, D.; Amato, A.; Picone, P.; Terzo, S.; Galizzi, G.; Bonina, F.P.; Mulè, F.; Di Carlo, M. A Natural Dietary Supplement with a Combination of Nutrients Prevents Neurodegeneration Induced by a High Fat Diet in Mice. Nutrients 2018, 10, 1130. [Google Scholar] [CrossRef] [Green Version]
- Nuzzo, D.; Presti, G.; Picone, P.; Galizzi, G.; Gulotta, E.; Giuliano, S.; Mannino, C.; Gambino, V.; Scoglio, S.; Di Carlo, M. Effects of the Aphanizomenon flos-aquae Extract (Klamin®) on a Neurodegeneration Cellular Model. Oxidative Med. Cell. Longev. 2018, 2018, 9089016. [Google Scholar] [CrossRef]
- Nuzzo, D.; Contardi, M.; Kossyvaki, D.; Picone, P.; Cristaldi, L.; Galizzi, G.; Bosco, G.; Scoglio, S.; Athanassiou, A.; Di Carlo, M. Heat-Resistant Aphanizomenon flos-aquae (AFA) Extract (Klamin®) as a Functional Ingredient in Food Strategy for Prevention of Oxidative Stress. Oxidative Med. Cell. Longev. 2019, 2019, 9481390. [Google Scholar] [CrossRef] [Green Version]
- Nuzzo, D.; Galizzi, G.; Amato, A.; Terzo, S.; Picone, P.; Cristaldi, L.; Mulè, F.; Di Carlo, M. Regular Intake of Pistachio Mitigates the Deleterious Effects of a High Fat-Diet in the Brain of Obese Mice. Antioxidants 2020, 9, 317. [Google Scholar] [CrossRef] [Green Version]
- Terzo, S.; Calvi, P.; Nuzzo, D.; Picone, P.; Galizzi, G.; Caruana, L.; Di Carlo, M.; Lentini, L.; Puleio, R.; Mulè, F.; et al. Preventive Impact of Long-Term Ingestion of Chestnut Honey on Glucose Disorders and Neurodegeneration in Obese Mice. Nutrients 2022, 14, 756. [Google Scholar] [CrossRef]
- Caracciolo, B.; Xu, W.; Collins, S.; Fratiglioni, L. Cognitive decline, dietary factors and gut–brain interactions. Mech. Ageing Dev. 2014, 136–137, 59–69. [Google Scholar] [CrossRef] [PubMed]
- James, B.D.; Leurgans, S.E.; Hebert, L.E.; Scherr, P.A.; Yaffe, K.; Bennett, D.A. Contribution of Alzheimer disease to mortality in the United States. Neurology 2014, 82, 1045–1050. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- De Strooper, B. Loss-of-function presenilin mutations in Alzheimer disease. talking point on the role of presenilin mutations in Alzheimer disease. EMBO Rep. 2007, 8, 141–146. [Google Scholar] [CrossRef] [Green Version]
- Walsh, D.M.; Klyubin, I.; Fadeeva, J.V.; Cullen, W.K.; Anwyl, R.; Wolfe, M.S.; Rowan, M.J.; Selkoe, D.J. Naturally secreted oligomers of amyloid β protein potently inhibit hippocampal long-term potentiation in vivo. Nature 2002, 416, 535–539. [Google Scholar] [CrossRef]
- Selkoe, D.J. Toward a Comprehensive Theory for Alzheimer’s Disease. Hypothesis: Alzheimer’s Disease Is Caused by the Cerebral Accumulation and Cytotoxicity of Amyloid β-Protein. Ann. N. Y. Acad. Sci. 2006, 924, 17–25. [Google Scholar] [CrossRef]
- Chiti, F.; Dobson, C.M. Protein misfolding, functional amyloid, and human disease. Annu. Rev. Biochem. 2006, 75, 333–366. [Google Scholar] [CrossRef] [Green Version]
- Lambert, M.P.; Barlow, A.K.; Chromy, B.A.; Edwards, C.; Freed, R.; Liosatos, M.; Morgan, T.E.; Rozovsky, I.; Trommer, B.; Viola, K.L.; et al. Diffusible, nonfibrillar ligands derived from Aβ1-42 are potent central nervous system neurotoxins. Proc. Natl. Acad. Sci. USA 1998, 95, 6448–6453. [Google Scholar] [CrossRef] [Green Version]
- Hardy, J.; Selkoe, D.J. The Amyloid Hypothesis of Alzheimer’s Disease: Progress and Problems on the Road to Therapeutics. Science 2002, 297, 353–356. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Abyadeh, M.; Gupta, V.; Gupta, V.; Chitranshi, N.; Wu, Y.; Amirkhani, A.; Meyfour, A.; Sheriff, S.; Shen, T.; Dhiman, K.; et al. Comparative Analysis of Aducanumab, Zagotenemab and Pioglitazone as Targeted Treatment Strategies for Alzheimer’s Disease. Aging Dis. 2021, 12, 1964. [Google Scholar] [CrossRef]
- Carrotta, R.; Di Carlo, M.; Manno, M.; Montana, G.; Picone, P.; Romancino, D.; Biagio, P.L.S. Toxicity of recombinant β-amyloid prefibrillar oligomers on the morphogenesis of the sea urchin Paracentrotus lividus. FASEB J. 2006, 20, 1916–1917. [Google Scholar] [CrossRef]
- Pellicanò, M.; Picone, P.; Cavalieri, V.; Carrotta, R.; Spinelli, G.; Di Carlo, M. The sea urchin embryo: A model to study Alzheimer’s beta amyloid induced toxicity. Arch. Biochem. Biophys. 2009, 483, 120–126. [Google Scholar] [CrossRef] [PubMed]
- Di Carlo, M. Simple model systems: A challenge for Alzheimer’s disease. Immun. Ageing 2012, 9, 1–8. [Google Scholar] [CrossRef] [Green Version]
- Serrano-Pozo, A.; Frosch, M.P.; Masliah, E.; Hyman, B.T. Neuropathological Alterations in Alzheimer Disease. Cold Spring Harb. Perspect. Med. 2011, 1, a006189. [Google Scholar] [CrossRef]
- Cai, Q.; Tammineni, P. Mitochondrial Aspects of Synaptic Dysfunction in Alzheimer’s Disease. J. Alzheimers Dis. 2017, 57, 1087–1103. [Google Scholar] [CrossRef] [Green Version]
- Swerdlow, R.H. Mitochondria and Mitochondrial Cascades in Alzheimer’s Disease. J. Alzheimers Dis. 2018, 62, 1403–1416. [Google Scholar] [CrossRef] [Green Version]
- Picone, P.; Nuzzo, D.; Giacomazza, D.; Di Carlo, M. β-Amyloid Peptide: The Cell Compartment Multi-faceted Interaction in Alzheimer’s Disease. Neurotox. Res. 2019, 37, 250–263. [Google Scholar] [CrossRef]
- Ott, A.; Stolk, R.P.; Van Harskamp, F.; Pols, H.A.; Hofman, A.; Breteler, M.M. Diabetes mellitus and the risk of dementia: The Rotterdam Study. Neurology 1999, 53, 1937. [Google Scholar] [CrossRef]
- Acosta, J.B.; Nieto, G.G.; Rodríguez, N.; Vega, M.L.B.; García del Barco Herrera, D.; Saez, B.; García Ojalvo, A.J.O.; Valdés Sosa, M.J.; Valdés-Sosa, P.A. Insulin Resistance at the Crossroad of Alzheimer Disease Pathology: A Review. Front. Endocrinol. 2020, 11, 560375. [Google Scholar] [CrossRef]
- Chatterjee, S.; Mudher, A. Alzheimer’s Disease and Type 2 Diabetes: A Critical Assessment of the Shared Pathological Traits. Front. Neurosci. 2018, 12, 383. [Google Scholar] [CrossRef] [Green Version]
- Willette, A.A.; Johnson, S.C.; Birdsill, A.C.; Sager, M.A.; Christian, B.; Baker, L.D.; Craft, S.; Oh, J.; Statz, E.; Hermann, B.P.; et al. Insulin resistance predicts brain amyloid deposition in late middle-aged adults. Alzheimers Dement. 2014, 11, 504–510.e1. [Google Scholar] [CrossRef] [Green Version]
- Di Carlo, M.; Picone, P.; Carrotta, R.; Giacomazza, D.; Biagio, P.L.S. Insulin Promotes Survival of Amyloid-Beta Oligomers Neuroblastoma Damaged Cells via Caspase 9 Inhibition and Hsp70 Upregulation. J. Biomed. Biotechnol. 2010, 2010, 147835. [Google Scholar] [CrossRef]
- Thorne, G.; Pronk, G.J.; Padmanabhan, V.; Frey, W.H., II. Delivery of insulin-like growth factor into the rat brain and spinal cord along olfactory and trigeminal pathways following intranasal administration. Neuroscience 2004, 127, 481–496. [Google Scholar] [CrossRef]
- Dhuria, S.V.; Hanson, L.R.; Frey, W.H., II. Intranasal delivery to the central nervous system: Mechanisms and experimental considerations. J. Pharm. Sci. 2010, 99, 1654–1673. [Google Scholar] [CrossRef]
- Hallschmid, M. Intranasal insulin. J. Neuroendocr. 2021, 33, e12934. [Google Scholar] [CrossRef]
- Craft, S.; Baker, L.D.; Montine, T.J.; Minoshima, S.; Watson, G.S.; Claxton, A.; Arbuckle, M.; Callaghan, M.; Tsai, E.; Plymate, S.R.; et al. Intranasal insulin therapy for Alzheimer disease and amnestic mild cognitive impairment: A pilot clinical trial. Arch. Neurol. 2012, 69, 29–38. [Google Scholar] [CrossRef] [Green Version]
- Kellar, D.; Craft, S. Insulin resistance in Alzheimer’s disease and related disorders: Mechanisms and therapeutic approaches. Lancet Neurol. 2020, 19, 758–766. [Google Scholar] [CrossRef]
- Craft, S.; Raman, R.; Chow, T.W.; Rafii, M.S.; Sun, C.-K.; Rissman, R.A.; Donohue, M.C.; Brewer, J.B.; Jenkins, C.; Harless, K.; et al. Safety, Efficacy, and Feasibility of Intranasal Insulin for the Treatment of Mild Cognitive Impairment and Alzheimer Disease Dementia. JAMA Neurol. 2020, 77, 1099–1109. [Google Scholar] [CrossRef]
- Nolte, M.S.; Taboga, C.; Salamon, E.; Moses, A.; Longenecker, J.; Flier, J.; Karam, J.H. Biological Activity of Nasally Administered Insulin in Normal Subjects. Horm. Metab. Res. 1990, 22, 170–174. [Google Scholar] [CrossRef]
- Cernea, S.; Raz, I. Noninjectable methods of insulin administration. Drugs Today 2006, 42, 405–424. [Google Scholar] [CrossRef]
- Kamei, N.; Takeda-Morishita, M. Brain delivery of insulin boosted by intranasal coadministration with cell-penetrating peptides. J. Control. Release 2015, 197, 105–110. [Google Scholar] [CrossRef]
- Picone, P.; Ditta, L.A.; Sabatino, M.A.; Militello, V.; Biagio, P.L.S.; Di Giacinto, M.L.; Cristaldi, L.; Nuzzo, D.; Dispenza, C.; Giacomazza, D.; et al. Ionizing radiation-engineered nanogels as insulin nanocarriers for the development of a new strategy for the treatment of Alzheimer’s disease. Biomaterials 2016, 80, 179–194. [Google Scholar] [CrossRef]
- Picone, P.; Sabatino, M.A.; Ditta, L.A.; Amato, A.; Biagio, P.L.S.; Mulè, F.; Giacomazza, D.; Dispenza, C.; Di Carlo, M. Nose-to-brain delivery of insulin enhanced by a nanogel carrier. J. Control. Release 2018, 270, 23–36. [Google Scholar] [CrossRef] [PubMed]
- Heni, M.; Kullmann, S.; Preißl, H.; Fritsche, A.; Häring, H.-U. Impaired insulin action in the human brain: Causes and metabolic consequences. Nat. Rev. Endocrinol. 2015, 11, 701–711. [Google Scholar] [CrossRef] [PubMed]
- Kullmann, S.; Heni, M.; Hallschmid, M.; Fritsche, A.; Preissl, H.; Häring, H.-U. Brain Insulin Resistance at the Crossroads of Metabolic and Cognitive Disorders in Humans. Physiol. Rev. 2016, 96, 1169–1209. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Cardoso, S.; Santos, R.X.; Correia, S.C.; Carvalho, C.; Santos, M.S.; Baldeiras, I.; Oliveira, C.R.; Moreira, P.I. Insulin-induced recurrent hypoglycemia exacerbates diabetic brain mitochondrial dysfunction and oxidative imbalance. Neurobiol. Dis. 2013, 49, 1–12. [Google Scholar] [CrossRef]
- Audano, M.; Pedretti, S.; Ligorio, S.; Crestani, M.; Caruso, D.; De Fabiani, E.; Mitro, N. “The Loss of Golden Touch”: Mitochondria-Organelle Interactions, Metabolism, and Cancer. Cells 2020, 9, 2519. [Google Scholar] [CrossRef]
- Liu, Y.J.; McIntyre, R.; Janssens, G.E.; Houtkooper, R.H. Mitochondrial fission and fusion: A dynamic role in aging and potential target for age-related disease. Mech. Ageing Dev. 2020, 186, 111212. [Google Scholar] [CrossRef]
- Scott, I.; Youle, R.J. Mitochondrial fission and fusion. Essays Biochem. 2010, 47, 85–98. [Google Scholar] [CrossRef] [Green Version]
- Błaszczyk, J.W. Energy Metabolism Decline in the Aging Brain—Pathogenesis of Neurodegenerative Disorders. Metabolites 2020, 10, 450. [Google Scholar] [CrossRef]
- Yan, X.; Wang, B.; Hu, Y.; Wang, S.; Zhang, X. Abnormal Mitochondrial Quality Control in Neurodegenerative Diseases. Front. Cell. Neurosci. 2020, 14, 138. [Google Scholar] [CrossRef]
- Roberts, D.J.; Tan-Sah, V.P.; Smith, J.M.; Miyamoto, S. Akt Phosphorylates HK-II at Thr-473 and Increases Mitochondrial HK-II Association to Protect Cardiomyocytes. J. Biol. Chem. 2013, 288, 23798–23806. [Google Scholar]
- Abolhassani, N.; Leon, J.; Sheng, Z.; Oka, S.; Hamasaki, H.; Iwaki, T.; Nakabeppu, Y. Molecular pathophysiology of impaired glucose metabolism, mitochondrial dysfunction, and oxidative DNA damage in Alzheimer’s disease brain. Mech. Ageing Dev. 2017, 161, 95–104. [Google Scholar] [CrossRef] [PubMed]
- Montgomery, M.K.; Turner, N. Mitochondrial dysfunction and insulin resistance: An update. Endocr. Connect. 2015, 4, R1–R15. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Rudich, A.; Tirosh, A.; Potashnik, R.; Hemi, R.; Kanety, H.; Bashan, N. Prolonged oxidative stress impairs insulin-induced GLUT4 translocation in 3T3-L1 adipocytes. Diabetes 1998, 47, 1562–1569. [Google Scholar] [CrossRef] [PubMed]
- Schell, M.; Wardelmann, K.; Kleinridders, A. Untangling the effect of insulin action on brain mitochondria and metabolism. J. Neuroendocr. 2021, 33, e12932. [Google Scholar] [CrossRef]
- Yi, C.; Walter, M.; Gao, Y.; Pitra, S.; Legutko, B.; Kälin, S.; Layritz, C.; García-Cáceres, C.; Bielohuby, M.; Bidlingmaier, M.; et al. TNFα drives mitochondrial stress in POMC neurons in obesity. Nat. Commun. 2017, 8, 15143. [Google Scholar] [CrossRef] [Green Version]
- García-Cáceres, C.; Quarta, C.; Varela, L.; Gao, Y.; Gruber, T.; Legutko, B.; Jastroch, M.; Johansson, P.; Ninkovic, J.; Yi, C.-X.; et al. Astrocytic Insulin Signaling Couples Brain Glucose Uptake with Nutrient Availability. Cell 2016, 166, 867–880. [Google Scholar] [CrossRef] [Green Version]
- Cai, W.; Xue, C.; Sakaguchi, M.; Konishi, M.; Shirazian, A.; Ferris, H.A.; Li, M.E.; Yu, R.; Kleinridders, A.; Pothos, E.N.; et al. Insulin regulates astrocyte gliotransmission and modulates behavior. J. Clin. Investig. 2018, 128, 2914–2926. [Google Scholar] [CrossRef]
- Miyamoto, S.; Murphy, A.N.; Brown, J.H. Akt mediates mitochondrial protection in cardiomyocytes through phosphorylation of mitochondrial hexokinase-II. Cell Death Differ. 2008, 15, 521–529. [Google Scholar] [CrossRef] [Green Version]
- Yang, J.-Y.; Yeh, H.-Y.; Lin, K.; Wang, P.H. Insulin stimulates Akt translocation to mitochondria: Implications on dysregulation of mitochondrial oxidative phosphorylation in diabetic myocardium. J. Mol. Cell. Cardiol. 2009, 46, 919–926. [Google Scholar] [CrossRef] [Green Version]
- Ruegsegger, G.N.; Creo, A.L.; Cortes, T.M.; Dasari, S.; Nair, K.S. Altered mitochondrial function in insulin-deficient and insulin-resistant states. J. Clin. Investig. 2018, 128, 3671–3681. [Google Scholar] [CrossRef] [Green Version]
- Ruegsegger, G.N.; Manjunatha, S.; Summer, P.; Gopala, S.; Zabeilski, P.; Dasari, S.; Vanderboom, P.M.; Lanza, I.R.; Klaus, K.A.; Nair, K.S. Insulin deficiency and intranasal insulin alter brain mitochondrial function: A potential factor for dementia in diabetes. FASEB J. 2018, 33, 4458–4472. [Google Scholar] [CrossRef]
- Ribeiro, M.; Rosenstock, T.R.; Oliveira, A.M.; Oliveira, C.R.; Rego, A.C. Insulin and IGF-1 improve mitochondrial function in a PI-3K/Akt-dependent manner and reduce mitochondrial generation of reactive oxygen species in Huntington’s disease knock-in striatal cells. Free Radic. Biol. Med. 2014, 74, 129–144. [Google Scholar] [CrossRef]
- Galizzi, G.; Palumbo, L.; Amato, A.; Conigliaro, A.; Nuzzo, D.; Terzo, S.; Caruana, L.; Picone, P.; Alessandro, R.; Mulè, F.; et al. Altered insulin pathway compromises mitochondrial function and quality control both in in vitro and in vivo model systems. Mitochondrion 2021, 60, 178–188. [Google Scholar] [CrossRef]
- Tilokani, L.; Nagashima, S.; Paupe, V.; Prudent, J. Mitochondrial dynamics: Overview of molecular mechanisms. Essays Biochem. 2018, 62, 341–360. [Google Scholar] [CrossRef] [Green Version]
- Cao, Y.-L.; Meng, S.; Chen, Y.; Feng, J.-X.; Gu, D.-D.; Yu, B.; Li, Y.-J.; Yang, J.-Y.; Liao, S.; Chan, D.C.; et al. MFN1 structures reveal nucleotide-triggered dimerization critical for mitochondrial fusion. Nature 2017, 542, 372–376. [Google Scholar] [CrossRef]
- Ban, T.; Ishihara, T.; Kohno, H.; Saita, S.; Ichimura, A.; Maenaka, K.; Oka, T.; Mihara, K.; Ishihara, N. Molecular basis of selective mitochondrial fusion by heterotypic action between OPA1 and cardiolipin. Nat. Cell Biol. 2017, 19, 856–863. [Google Scholar] [CrossRef]
- Basso, V.; Marchesan, E.; Peggion, C.; Chakraborty, J.; von Stockum, S.; Giacomello, M.; Ottolini, D.; Debattisti, V.; Caicci, F.; Tasca, E.; et al. Regulation of ER-mitochondria contacts by Parkin via Mfn2. Pharmacol. Res. 2018, 138, 43–56. [Google Scholar] [CrossRef]
- Cohen, S.; Valm, A.M.; Lippincott-Schwartz, J. Interacting organelles. Curr. Opin. Cell Biol. 2018, 53, 84–91. [Google Scholar] [CrossRef]
- Oliver, D.M.A.; Reddy, P.H. Dynamics of Dynamin-Related Protein 1 in Alzheimer’s Disease and Other Neurodegenerative Diseases. Cells 2019, 8, 961. [Google Scholar] [CrossRef] [Green Version]
- Losón, O.C.; Song, Z.; Chen, H.; Chan, D.C. Fis1, Mff, MiD49, and MiD51 mediate Drp1 recruitment in mitochondrial fission. Mol. Biol. Cell 2013, 24, 659–667. [Google Scholar] [CrossRef] [PubMed]
- Liesa, M.; Van der Bliek, A.; Shirihai, O.S. To Fis or not to Fuse? This is the question! EMBO J. 2019, 38, e101839. [Google Scholar] [CrossRef]
- Yu, R.; Jin, S.B.; Lendahl, U.; Nistér, M.; Zhao, J. Human Fis1regulates mitochondrial dynamics through inhibition of the fusion machinery. EMBO J. 2019, 38, e99748. [Google Scholar] [CrossRef] [PubMed]
- Tubbs, E.; Theurey, P.; Vial, G.; Bendridi, N.; Bravard, A.; Chauvin, M.-A.; Ji-Cao, J.; Zoulim, F.; Bartosch, B.; Ovize, M.; et al. Mitochondria-Associated Endoplasmic Reticulum Membrane (MAM) Integrity Is Required for Insulin Signaling and Is Implicated in Hepatic Insulin Resistance. Diabetes 2014, 63, 3279–3294. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Tubbs, E.; Chanon, S.; Robert, M.; Bendridi, N.; Bidaux, G.; Chauvin, M.-A.; Ji-Cao, J.; Durand, C.; Gauvrit-Ramette, D.; Vidal, H.; et al. Disruption of Mitochondria-Associated Endoplasmic Reticulum Membrane (MAM) Integrity Contributes to Muscle Insulin Resistance in Mice and Humans. Diabetes 2018, 67, 636–650. [Google Scholar] [CrossRef] [Green Version]
- Wong, Y.C.; Ysselstein, D.; Krainc, D. Mitochondria–lysosome contacts regulate mitochondrial fission via RAB7 GTP hydrolysis. Nature 2018, 554, 382–386. [Google Scholar] [CrossRef]
- Kulkarni, S.S.; Joffraud, M.; Boutant, M.; Ratajczak, J.; Gao, A.W.; Maclachlan, C.; Hernandez-Alvarez, M.I.; Raymond, F.; Metairon, S.; Descombes, P.; et al. Mfn1 Deficiency in the Liver Protects Against Diet-Induced Insulin Resistance and Enhances the Hypoglycemic Effect of Metformin. Diabetes 2016, 65, 3552–3560. [Google Scholar] [CrossRef] [Green Version]
- Sebastián, D.; Hernández-Alvarez, M.I.; Segalés, J.; Sorianello, E.; Muñoz, J.P.; Sala, D.; Waget, A.; Liesa, M.; Paz, J.C.; Gopalacharyulu, P.; et al. Mitofusin 2 (Mfn2) links mitochondrial and endoplasmic reticulum function with insulin signaling and is essential for normal glucose homeostasis. Proc. Natl. Acad. Sci. USA 2012, 109, 5523–5528. [Google Scholar] [CrossRef] [Green Version]
- Wang, L.; Ishihara, T.; Ibayashi, Y.; Tatsushima, K.; Setoyama, D.; Hanada, Y.; Takeichi, Y.; Sakamoto, S.; Yokota, S.; Mihara, K.; et al. Disruption of mitochondrial fission in the liver protects mice from diet-induced obesity and metabolic deterioration. Diabetologia 2015, 58, 2371–2380. [Google Scholar] [CrossRef] [Green Version]
- Wai, T.; Langer, T. Mitochondrial Dynamics and Metabolic Regulation. Trends Endocrinol. Metab. 2016, 27, 105–117. [Google Scholar] [CrossRef]
- Yu, T.; Robotham, J.L.; Yoon, Y. Increased production of reactive oxygen species in hyperglycemic conditions requires dynamic change of mitochondrial morphology. Proc. Natl. Acad. Sci. USA 2006, 103, 2653–2658. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Park, G.; Lee, J.Y.; Han, H.M.; An, H.S.; Jin, Z.; Jeong, E.A.; Kim, K.E.; Shin, H.J.; Lee, J.; Kang, D.; et al. Ablation of dynamin-related protein 1 promotes diabetes-induced synaptic injury in the hippocampus. Cell Death Dis. 2021, 12, 445. [Google Scholar] [CrossRef] [PubMed]
- Luo, F.; Herrup, K.; Qi, X.; Yang, Y. Inhibition of Drp1 hyper-activation is protective in animal models of experimental multiple sclerosis. Exp. Neurol. 2017, 292, 21–34. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Cao, K.; Xu, J.; Zou, X.; Li, Y.; Chen, C.; Zheng, A.; Li, H.; Li, H.; Szeto, I.M.-Y.; Shi, Y.; et al. Hydroxytyrosol prevents diet-induced metabolic syndrome and attenuates mitochondrial abnormalities in obese mice. Free Radic. Biol. Med. 2014, 67, 396–407. [Google Scholar] [CrossRef]
- Huang, S.; Wang, Y.; Gan, X.; Fang, D.; Zhong, C.; Wu, L.; Hu, G.; Sosunov, A.A.; McKhann, G.M.; Yu, H.; et al. Drp1-Mediated Mitochondrial Abnormalities Link to Synaptic Injury in Diabetes Model. Diabetes 2015, 64, 1728–1742. [Google Scholar] [CrossRef] [Green Version]
- Edwards, J.L.; Quattrini, A.; Lentz, S.I.; Figueroa-Romero, C.; Cerri, F.; Backus, C.; Hong, Y.; Feldman, E.L. Diabetes regulates mitochondrial biogenesis and fission in mouse neurons. Diabetologia 2009, 53, 160–169. [Google Scholar] [CrossRef] [Green Version]
- Andres, A.M.; Stotland, A.; Queliconi, B.B.; Gottlieb, R.A. A time to reap, a time to sow: Mitophagy and biogenesis in cardiac pathophysiology. J. Mol. Cell. Cardiol. 2015, 78, 62–72. [Google Scholar] [CrossRef] [Green Version]
- Wollert, T. Autophagy. Curr. Biol. 2019, 29, R671–R677. [Google Scholar] [CrossRef]
- Murakawa, T.; Yamaguchi, O.; Hashimoto, A.; Hikoso, S.; Takeda, T.O.; Oka, T.; Yasui, H.; Ueda, H.; Akazawa, Y.; Nakayama, H.; et al. Bcl-2-like protein 13 is a mammalian Atg32 homologue that mediates mitophagy and mitochondrial fragmentation. Nat. Commun. 2015, 6, 7527. [Google Scholar] [CrossRef] [Green Version]
- Novak, I.; Kirkin, V.; McEwan, D.G.; Zhang, J.; Wild, P.; Rozenknop, A.; Rogov, V.; Löhr, F.; Popovic, D.; Occhipinti, A.; et al. Nix is a selective autophagy receptor for mitochondrial clearance. EMBO Rep. 2009, 11, 45–51. [Google Scholar] [CrossRef] [Green Version]
- Liu, L.; Feng, D.; Chen, G.; Chen, M.; Zheng, Q.; Song, P.; Ma, Q.; Zhu, C.; Wang, R.; Qi, W.; et al. Mitochondrial outer-membrane protein FUNDC1 mediates hypoxia-induced mitophagy in mammalian cells. Nat. Cell Biol. 2012, 14, 177–185. [Google Scholar] [CrossRef] [PubMed]
- Bader, V.; Winklhofer, K.F. PINK1 and Parkin: Team players in stress-induced mitophagy. Biol. Chem. 2020, 401, 891–899. [Google Scholar] [CrossRef] [PubMed]
- Lampert, M.A.; Orogo, A.M.; Najor, R.H.; Hammerling, B.C.; Leon, L.J.; Wang, B.J.; Kim, T.; Sussman, M.A.; Gustafsson, B. BNIP3L/NIX and FUNDC1-mediated mitophagy is required for mitochondrial network remodeling during cardiac progenitor cell differentiation. Autophagy 2019, 15, 1182–1198. [Google Scholar] [CrossRef] [PubMed]
- Harper, J.W.; Ordureau, A.; Heo, J.-M. Building and decoding ubiquitin chains for mitophagy. Nat. Rev. Mol. Cell Biol. 2018, 19, 93–108. [Google Scholar] [CrossRef]
- Lazarou, M.; Jin, S.M.; Kane, L.A.; Youle, R.J. Role of PINK1 Binding to the TOM Complex and Alternate Intracellular Membranes in Recruitment and Activation of the E3 Ligase Parkin. Dev. Cell 2012, 22, 320–333. [Google Scholar] [CrossRef] [Green Version]
- Pickrell, A.M.; Youle, R.J. The Roles of PINK1, Parkin, and Mitochondrial Fidelity in Parkinson’s Disease. Neuron 2015, 85, 257–273. [Google Scholar] [CrossRef] [Green Version]
- Picca, A.; Guerra, F.; Calvani, R.; Coelho-Junior, H.J.; Bossola, M.; Landi, F.; Bernabei, R.; Bucci, C.; Marzetti, E. Generation and Release of Mitochondrial-Derived Vesicles in Health, Aging and Disease. J. Clin. Med. 2020, 9, 1440. [Google Scholar] [CrossRef]
- Lazarou, M.; Sliter, D.A.; Kane, L.A.; Sarraf, S.A.; Wang, C.; Burman, J.L.; Sideris, D.P.; Fogel, A.I.; Youle, R.J. The ubiquitin kinase PINK1 recruits autophagy receptors to induce mitophagy. Nature 2015, 524, 309–314. [Google Scholar] [CrossRef] [Green Version]
- Ihenacho, U.K.; Meacham, K.A.; Harwig, M.C.; Widlansky, M.E.; Hill, R.B. Mitochondrial Fission Protein 1: Emerging Roles in Organellar Form and Function in Health and Disease. Front. Endocrinol. 2021, 12, 660095. [Google Scholar] [CrossRef]
- Shenouda, S.M.; Widlansky, M.E.; Chen, K.; Xu, G.; Holbrook, M.; Tabit, C.E.; Hamburg, N.M.; Frame, A.A.; Caiano, T.L.; Kluge, M.A.; et al. Altered Mitochondrial Dynamics Contributes to Endothelial Dysfunction in Diabetes Mellitus. Circulation 2011, 124, 444–453. [Google Scholar] [CrossRef] [Green Version]
- Twig, G.; Elorza, A.; Molina, A.J.A.; Mohamed, H.; Wikstrom, J.D.; Walzer, G.; Stiles, L.; Haigh, S.E.; Katz, S.; Las, G.; et al. Fission and selective fusion govern mitochondrial segregation and elimination by autophagy. EMBO J. 2008, 27, 433–446. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Casellas-Díaz, S.; Larramona-Arcas, R.; Riqué-Pujol, G.; Tena-Morraja, P.; Müller-Sánchez, C.; Segarra-Mondejar, M.; Gavaldà-Navarro, A.; Villarroya, F.; Reina, M.; Martínez-Estrada, O.M.; et al. Mfn2 localization in the ER is necessary for its bioenergetic function and neuritic development. EMBO Rep. 2021, 22, e51954. [Google Scholar] [CrossRef] [PubMed]
- Gonzalez, C.; Lee, M.-S.; Marchetti, P.; Pietropaolo, M.; Towns, R.; Vaccaro, M.; Watada, H.; Wiley, J.W. The emerging role of autophagy in the pathophysiology of diabetes mellitus. Autophagy 2011, 7, 2–11. [Google Scholar] [CrossRef] [PubMed]
- Caberlotto, L.; Nguyen, T.-P.; Lauria, M.; Priami, C.; Rimondini, R.; Maioli, S.; Cedazo-Minguez, A.; Sita, G.; Morroni, F.; Corsi, M.; et al. Cross-disease analysis of Alzheimer’s disease and type-2 Diabetes highlights the role of autophagy in the pathophysiology of two highly comorbid diseases. Sci. Rep. 2019, 9, 3965. [Google Scholar] [CrossRef] [Green Version]
- Mammucari, C.; Milan, G.; Romanello, V.; Masiero, E.; Rudolf, R.; Del Piccolo, P.; Burden, S.J.; Di Lisi, R.; Sandri, C.; Zhao, J.; et al. FoxO3 Controls Autophagy in Skeletal Muscle In Vivo. Cell Metab. 2007, 6, 458–471. [Google Scholar] [CrossRef] [PubMed]
- Axe, E.L.; Walker, S.A.; Manifava, M.; Chandra, P.; Roderick, H.L.; Habermann, A.; Griffiths, G.; Ktistakis, N.T. Autophagosome formation from membrane compartments enriched in phosphatidylinositol 3-phosphate and dynamically connected to the endoplasmic reticulum. J. Cell Biol. 2008, 182, 685–701. [Google Scholar] [CrossRef] [Green Version]
- Di Malta, C.; Cinque, L.; Settembre, C. Transcriptional Regulation of Autophagy: Mechanisms and Diseases. Front. Cell Dev. Biol. 2019, 7, 114. [Google Scholar] [CrossRef]
- Menon, M.B.; Dhamija, S. Beclin 1 Phosphorylation–at the Center of Autophagy Regulation. Front. Cell Dev. Biol. 2018, 6, 137. [Google Scholar] [CrossRef] [Green Version]
- Nishida, Y.; Arakawa, S.; Fujitani, K.; Yamaguchi, H.; Mizuta, T.; Kanaseki, T.; Komatsu, M.; Otsu, K.; Tsujimoto, Y.; Shimizu, S. Discovery of Atg5/Atg7-independent alternative macroautophagy. Nature 2009, 461, 654–658. [Google Scholar] [CrossRef]
- Cunningham, J.T.; Rodgers, J.T.; Arlow, D.H.; Vazquez, F.; Mootha, V.K.; Puigserver, P. mTOR controls mitochondrial oxidative function through a YY1–PGC-1α transcriptional complex. Nature 2007, 450, 736–740. [Google Scholar] [CrossRef]
- Schieke, S.M.; Phillips, D.; McCoy, J.P., Jr.; Aponte, A.M.; Shen, R.-F.; Balaban, R.S.; Finkel, T. The Mammalian Target of Rapamycin (mTOR) Pathway Regulates Mitochondrial Oxygen Consumption and Oxidative Capacity. J. Biol. Chem. 2006, 281, 27643–27652. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Cheng, Z.; Guo, S.; Copps, K.; Dong, X.; Kollipara, R.; Rodgers, J.T.; DePinho, R.A.; Puigserver, P.; White, M.F. Foxo1 integrates insulin signaling with mitochondrial function in the liver. Nat. Med. 2009, 15, 1307–1311. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Daitoku, H.; Yamagata, K.; Matsuzaki, H.; Hatta, M.; Fukamizu, A. Regulation of PGC-1 Promoter Activity by Protein Kinase B and the Forkhead Transcription Factor FKHR. Diabetes 2003, 52, 642–649. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Sidarala, V.; Pearson, G.L.; Parekh, V.S.; Thompson, B.; Christen, L.; Gingerich, M.A.; Zhu, J.; Stromer, T.; Ren, J.; Reck, E.C.; et al. Mitophagy protects β cells from inflammatory damage in diabetes. JCI Insight 2020, 5, e141138. [Google Scholar] [CrossRef]
- Jung, H.S.; Chung, K.W.; Kim, J.; Kim, J.; Komatsu, M.; Tanaka, K.; Nguyen, Y.H.; Kang, T.M.; Yoon, K.; Kim, J.; et al. Loss of autophagy diminishes pancreatic beta cell mass and function with resultant hyperglycemia. Cell Metab. 2008, 8, 318–324. [Google Scholar] [CrossRef] [Green Version]
- Zhou, P.; Xie, W.; Meng, X.; Zhai, Y.; Dong, X.; Zhang, X.; Sun, G.; Sun, X. Notoginsenoside R1 Ameliorates Diabetic Retinopathy through PINK1-Dependent Activation of Mitophagy. Cells 2019, 8, 213. [Google Scholar] [CrossRef] [Green Version]
- Santos, R.X.; Correia, S.C.; Alves, M.G.; Oliveira, P.F.; Cardoso, S.; Carvalho, C.; Seiça, R.; Santos, M.S.; Moreira, P.I. Mitochondrial quality control systems sustain brain mitochondrial bioenergetics in early stages of type 2 diabetes. Mol. Cell. Biochem. 2014, 394, 13–22. [Google Scholar] [CrossRef]
- Pickford, F.; Masliah, E.; Britschgi, M.; Lucin, K.; Narasimhan, R.; Jaeger, P.A.; Small, S.; Spencer, B.; Rockenstein, E.; Levine, B.; et al. The autophagy-related protein beclin 1 shows reduced expression in early Alzheimer disease and regulates amyloid β accumulation in mice. J. Clin. Investig. 2008, 118, 2190–2199. [Google Scholar] [CrossRef] [Green Version]
- Cummins, N.; Tweedie, A.; Zuryn, S.; Bertran-Gonzalez, J.; Götz, J. Disease-associated tau impairs mitophagy by inhibiting Parkin translocation to mitochondria. EMBO J. 2018, 38, e99360. [Google Scholar] [CrossRef]
- Rocchi, A.; Yamamoto, S.; Ting, T.; Fan, Y.; Sadleir, K.; Wang, Y.; Zhang, W.; Huang, S.; Levine, B.; Vassar, R.; et al. A Becn1 mutation mediates hyperactive autophagic sequestration of amyloid oligomers and improved cognition in Alzheimer’s disease. PLoS Genet. 2017, 13, e1006962. [Google Scholar] [CrossRef] [Green Version]
- Nilsson, P.; Loganathan, K.; Sekiguchi, M.; Matsuba, Y.; Hui, K.; Tsubuki, S.; Tanaka, M.; Iwata, N.; Saito, T.; Saido, T.C. Aβ Secretion and Plaque Formation Depend on Autophagy. Cell Rep. 2013, 5, 61–69. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Caccamo, A.; Majumder, S.; Richardson, A.; Strong, R.; Oddo, S. Molecular Interplay between Mammalian Target of Rapamycin (mTOR), Amyloid-β, and Tau: Effects on cognitive impairments. J. Biol. Chem. 2010, 285, 13107–13120. [Google Scholar] [CrossRef] [Green Version]
- Tran, M.; Reddy, P.H. Defective Autophagy and Mitophagy in Aging and Alzheimer’s Disease. Front. Neurosci. 2021, 14, 612757. [Google Scholar] [CrossRef] [PubMed]
- Bordi, M.; Berg, M.J.; Mohan, P.S.; Peterhoff, C.M.; Alldred, M.J.; Che, S.; Ginsberg, S.D.; Nixon, R.A. Autophagy flux in CA1 neurons of Alzheimer hippocampus: Increased induction overburdens failing lysosomes to propel neuritic dystrophy. Autophagy 2016, 12, 2467–2483. [Google Scholar] [CrossRef] [PubMed]
- Coffey, E.E.; Beckel, J.M.; Laties, A.M.; Mitchell, C.H. Lysosomal alkalization and dysfunction in human fibroblasts with the Alzheimer’s disease-linked presenilin 1 A246E mutation can be reversed with cAMP. Neuroscience 2014, 263, 111–124. [Google Scholar] [CrossRef] [Green Version]
- Chen, L.; Liu, C.; Gao, J.; Xie, Z.; Chan, L.; Keating, D.; Yang, Y.; Sun, J.; Zhou, F.; Wei, Y.; et al. Inhibition of Miro1 disturbs mitophagy and pancreatic β-cell function interfering insulin release via IRS-Akt-Foxo1 in diabetes. Oncotarget 2017, 8, 90693–90705. [Google Scholar] [CrossRef] [Green Version]
- Yang, S.; Xia, C.; Li, S.; Du, L.; Zhang, L.; Zhou, R. Defective mitophagy driven by dysregulation of rheb and KIF5B contributes to mitochondrial reactive oxygen species (ROS)-induced nod-like receptor 3 (NLRP3) dependent proinflammatory response and aggravates lipotoxicity. Redox Biol. 2014, 3, 63–71. [Google Scholar] [CrossRef] [Green Version]
- Ebato, C.; Uchida, T.; Arakawa, M.; Komatsu, M.; Ueno, T.; Komiya, K.; Azuma, K.; Hirose, T.; Tanaka, K.; Kominami, E.; et al. Autophagy Is Important in Islet Homeostasis and Compensatory Increase of Beta Cell Mass in Response to High-Fat Diet. Cell Metab. 2008, 8, 325–332. [Google Scholar] [CrossRef] [Green Version]
- Scheele, C.; Nielsen, A.R.; Walden, T.B.; Sewell, D.A.; Fischer, C.; Brogan, R.J.; Petrovic, N.; Larsson, O.; Tesch, P.A.; Wennmalm, K.; et al. Altered regulation of the PINK1 locus: A link between type 2 diabetes and neurodegeneration? FASEB J. 2007, 21, 3653–3665. [Google Scholar] [CrossRef]
- Gureev, A.P.; Shaforostova, E.A.; Popov, V.N. Regulation of Mitochondrial Biogenesis as a Way for Active Longevity: Interaction Between the Nrf2 and PGC-1α Signaling Pathways. Front. Genet. 2019, 10, 435. [Google Scholar] [CrossRef] [Green Version]
- Ventura-Clapier, R.; Garnier, A.; Veksler, V. Transcriptional control of mitochondrial biogenesis: The central role of PGC-1 alpha. Cardiovasc. Res. 2008, 79, 208–217. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Tritos, N.A.; Mastaitis, J.W.; Kokkotou, E.G.; Puigserver, P.; Spiegelman, B.M.; Maratos-Flier, E. Characterization of the peroxisome proliferator activated receptor coactivator 1 alpha (PGC 1α) expression in the murine brain. Brain Res. 2003, 961, 255–260. [Google Scholar] [CrossRef]
- Barbera, M.J.; Schluter, A.; Pedraza, N.; Iglesias, R.; Villarroya, F.; Giralt, M. Peroxisome Proliferator-activated Receptorα Activates Transcription of the Brown Fat Uncoupling Protein-1 Gene: A link between regulation of the thermogenic and lipid oxidation pathways in the brown fat cell. J. Biol. Chem. 2001, 276, 1486–1493. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Larson, C.; Opichka, M.; McGlynn, M.L.; Collins, C.W.; Slivka, D. Exercise- and Cold-Induced Human PGC-1α mRNA Isoform Specific Responses. Int. J. Environ. Res. Public Health 2020, 17, 5740. [Google Scholar] [CrossRef] [PubMed]
- Scarpulla, R.C. Metabolic control of mitochondrial biogenesis through the PGC-1 family regulatory network. Biochim. Biophys. Acta 2011, 1813, 1269–1278. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Vernier, M.; Giguère, V. Aging, senescence and mitochondria: The PGC-1/ERR axis. J. Mol. Endocrinol. 2021, 66, R1–R14. [Google Scholar] [CrossRef]
- Vega, R.B.; Huss, J.M.; Kelly, D.P. The Coactivator PGC-1 Cooperates with Peroxisome Proliferator-Activated Receptor α in Transcriptional Control of Nuclear Genes Encoding Mitochondrial Fatty Acid Oxidation Enzymes. Mol. Cell. Biol. 2000, 20, 1868–1876. [Google Scholar] [CrossRef] [Green Version]
- Mootha, V.K.; Lindgren, C.M.; Eriksson, K.-F.; Subramanian, A.; Sihag, S.; Lehar, J.; Puigserver, P.; Carlsson, E.; Ridderstråle, M.; Laurila, E.; et al. PGC-1α-responsive genes involved in oxidative phosphorylation are coordinately downregulated in human diabetes. Nat. Genet. 2003, 34, 267–273. [Google Scholar] [CrossRef]
- Kang, I.; Chu, C.T.; Kaufman, B.A. The mitochondrial transcription factor TFAM in neurodegeneration: Emerging evidence and mechanisms. FEBS Lett. 2018, 592, 793–811. [Google Scholar] [CrossRef] [Green Version]
- Jornayvaz, F.R.; Shulman, G.I. Regulation of mitochondrial biogenesis. Essays Biochem. 2010, 47, 69–84. [Google Scholar] [CrossRef] [Green Version]
- Wu, Z.; Puigserver, P.; Andersson, U.; Zhang, C.; Adelmant, G.; Mootha, V.; Troy, A.; Cinti, S.; Lowell, B.; Scarpulla, R.C.; et al. Mechanisms Controlling Mitochondrial Biogenesis and Respiration through the Thermogenic Coactivator PGC-1. Cell 1999, 98, 115–124. [Google Scholar] [CrossRef] [Green Version]
- Picca, A.; Lezza, A.M.S. Regulation of mitochondrial biogenesis through TFAM–mitochondrial DNA interactions: Useful insights from aging and calorie restriction studies. Mitochondrion 2015, 25, 67–75. [Google Scholar] [CrossRef] [PubMed]
- Ljubicic, V.; Joseph, A.-M.; Saleem, A.; Uguccioni, G.; Collu-Marchese, M.; Lai, R.Y.; Nguyen, L.M.-D.; Hood, D.A. Transcriptional and post-transcriptional regulation of mitochondrial biogenesis in skeletal muscle: Effects of exercise and aging. Biochim. Biophys. Acta 2009, 1800, 223–234. [Google Scholar] [CrossRef] [PubMed]
- Vaughan, R.A.; Mermier, C.M.; Bisoffi, M.; Trujillo, K.A.; Conn, C.A. Dietary stimulators of the PGC-1 superfamily and mitochondrial biosynthesis in skeletal muscle. J. Physiol. Biochem. 2014, 70, 271–284. [Google Scholar] [CrossRef]
- Canto, C.; Jiang, L.Q.; Deshmukh, A.S.; Mataki, C.; Coste, A.; Lagouge, M.; Zierath, J.R.; Auwerx, J. Interdependence of AMPK and SIRT1 for Metabolic Adaptation to Fasting and Exercise in Skeletal Muscle. Cell Metab. 2010, 11, 213–219. [Google Scholar] [CrossRef] [Green Version]
- Puigserver, P.; Rhee, J.; Lin, J.; Wu, Z.; Yoon, J.; Zhang, C.-Y.; Krauss, S.; Mootha, V.K.; Lowell, B.B.; Spiegelman, B.M. Cytokine Stimulation of Energy Expenditure through p38 MAP Kinase Activation of PPARγ Coactivator-1. Mol. Cell 2001, 8, 971–982. [Google Scholar] [CrossRef]
- Fan, M.; Rhee, J.; St-Pierre, J.; Handschin, C.; Puigserver, P.; Lin, J.; Jäeger, S.; Erdjument-Bromage, H.; Tempst, P.; Spiegelman, B.M. Suppression of mitochondrial respiration through recruitment of p160 myb binding protein to PGC-1α: Modulation by p38 MAPK. Genes Dev. 2004, 18, 278–289. [Google Scholar] [CrossRef] [Green Version]
- Theeuwes, W.F.; Gosker, H.R.; Schols, A.; Langen, R.C.J.; Remels, A.H.V. Regulation of PGC-1α expression by a GSK-3β-TFEB signaling axis in skeletal muscle. Biochim. Biophys. Acta Mol. Cell Res. 2019, 1867, 118610. [Google Scholar] [CrossRef]
- Rodgers, J.T.; Lerin, C.; Haas, W.; Gygi, S.P.; Spiegelman, B.M.; Puigserver, P. Nutrient control of glucose homeostasis through a complex of PGC-1α and SIRT1. Nature 2005, 434, 113–118. [Google Scholar] [CrossRef]
- Anderson, K.A.; Ribar, T.J.; Lin, F.; Noeldner, P.K.; Green, M.F.; Muehlbauer, M.J.; Witters, L.A.; Kemp, B.E.; Means, A.R. Hypothalamic CaMKK2 Contributes to the Regulation of Energy Balance. Cell Metab. 2008, 7, 377–388. [Google Scholar] [CrossRef] [Green Version]
- Woods, A.; Johnstone, S.R.; Dickerson, K.; Leiper, F.C.; Fryer, L.G.D.; Neumann, D.; Schlattner, U.; Wallimann, T.; Carlson, M.; Carling, D. LKB1 Is the Upstream Kinase in the AMP-Activated Protein Kinase Cascade. Curr. Biol. 2003, 13, 2004–2008. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Shackelford, D.B.; Shaw, R.J. The LKB1–AMPK pathway: Metabolism and growth control in tumour suppression. Nat. Cancer 2009, 9, 563–575. [Google Scholar] [CrossRef]
- Hawley, S.A.; Selbert, M.A.; Goldstein, E.G.; Edelman, A.M.; Carling, D.; Hardie, D.G. 5′-AMP Activates the AMP-activated Protein Kinase Cascade, and Ca21/Calmodulin Activates the Calmodulin-dependent Protein Kinase I Cascade, via Three Independent Mechanisms. J. Biol. Chem. 1995, 270, 27186–27191. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Hudson, E.R.; Pan, D.A.; James, J.; Lucocq, J.M.; Hawley, S.A.; Green, K.A.; Baba, O.; Terashima, T.; Hardie, D.G. A Novel Domain in AMP-Activated Protein Kinase Causes Glycogen Storage Bodies Similar to Those Seen in Hereditary Cardiac Arrhythmias. Curr. Biol. 2003, 13, 861. [Google Scholar] [CrossRef] [Green Version]
- Barone, E.; Di Domenico, F.; Perluigi, M.; Butterfield, D.A. The interplay among oxidative stress, brain insulin resistance and AMPK dysfunction contribute to neurodegeneration in type 2 diabetes and Alzheimer disease. Free Radic. Biol. Med. 2021, 176, 16–33. [Google Scholar] [CrossRef]
- Hardie, D.G. AMPK and Raptor: Matching Cell Growth to Energy Supply. Mol. Cell 2008, 30, 263–265. [Google Scholar] [CrossRef]
- Herzig, S.; Shaw, R.J. AMPK: Guardian of metabolism and mitochondrial homeostasis. Nat. Rev. Mol. Cell Biol. 2018, 19, 121–135. [Google Scholar] [CrossRef] [Green Version]
- Saha, A.K.; Xu, X.J.; Balon, T.; Brandon, A.; Kraegen, E.W.; Ruderman, N.B. Insulin resistance due to nutrient excess: Is it a consequence of AMPK downregulation? Cell Cycle 2011, 10, 3447. [Google Scholar] [CrossRef] [Green Version]
- Tang, B.L. Sirt1 and the Mitochondria. Mol. Cells 2016, 39, 87–95. [Google Scholar] [CrossRef] [Green Version]
- Michishita, E.; Park, J.Y.; Burneskis, J.M.; Barrett, J.C.; Horikawa, I. Evolutionarily Conserved and Nonconserved Cellular Localizations and Functions of Human SIRT Proteins. Mol. Biol. Cell 2005, 16, 4623–4635. [Google Scholar] [CrossRef] [Green Version]
- Canto, C.; Gerhart-Hines, Z.; Feige, J.N.; Lagouge, M.; Noriega, L.; Milne, J.C.; Elliott, P.J.; Puigserver, P.; Auwerx, J. AMPK regulates energy expenditure by modulating NAD+ metabolism and SIRT1 activity. Nature 2009, 458, 1056–1060. [Google Scholar] [CrossRef] [PubMed]
- Houtkooper, R.H.; Cantó, C.; Wanders, R.J.; Auwerx, J. The Secret Life of NAD+: An Old Metabolite Controlling New Metabolic Signaling Pathways. Endocr. Rev. 2010, 31, 194–223. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Ruderman, N.B.; Xu, X.J.; Nelson, L.; Cacicedo, J.M.; Saha, A.K.; Lan, F.; Ido, Y. AMPK and SIRT1: A long-standing partnership? Am. J. Physiol. Endocrinol. Metab. 2010, 298, E751–E760. [Google Scholar] [CrossRef] [PubMed]
- Price, N.L.; Gomes, A.P.; Ling, A.J.; Duarte, F.V.; Martin-Montalvo, A.; North, B.J.; Agarwal, B.; Ye, L.; Ramadori, G.; Teodoro, J.S.; et al. SIRT1 Is Required for AMPK Activation and the Beneficial Effects of Resveratrol on Mitochondrial Function. Cell Metab. 2012, 15, 675–690. [Google Scholar] [CrossRef] [Green Version]
- Ghosh, H.S.; McBurney, M.; Robbins, P.D. SIRT1 Negatively Regulates the Mammalian Target of Rapamycin. PLoS ONE 2010, 5, e9199. [Google Scholar] [CrossRef] [Green Version]
- Patti, M.E.; Butte, A.J.; Crunkhorn, S.; Cusi, K.; Berria, R.; Kashyap, S.; Miyazaki, Y.; Kohane, I.; Costello, M.; Saccone, R.; et al. Coordinated reduction of genes of oxidative metabolism in humans with insulin resistance and diabetes: Potential role of PGC1 and NRF1. Proc. Natl. Acad. Sci. USA 2003, 100, 8466–8471. [Google Scholar] [CrossRef] [Green Version]
- Quiros, P.M.; Mottis, A.; Auwerx, J. Mitonuclear communication in homeostasis and stress. Nat. Rev. Mol. Cell Biol. 2016, 17, 213–226. [Google Scholar] [CrossRef]
- Yang, C.; Sui, G.; Li, D.; Wang, L.; Zhang, S.; Lei, P.; Chen, Z.; Wang, F. Exogenous IGF-1 alleviates depression-like behavior and hippocampal mitochondrial dysfunction in high-fat diet mice. Physiol. Behav. 2021, 229, 113236. [Google Scholar] [CrossRef]
- Anderson, R.; Prolla, T. PGC-1alpha in aging and anti-aging interventions. Biochim. Biophys. Acta. 2009, 1790, 1059. [Google Scholar] [CrossRef] [Green Version]
- Qin, W.; Haroutunian, V.; Katsel, P.; Cardozo, C.P.; Ho, L.; Buxbaum, J.; Pasinetti, G.M. PGC-1α Expression Decreases in the Alzheimer Disease Brain as a Function of Dementia. Arch. Neurol. 2009, 66, 352–361. [Google Scholar] [CrossRef]
- Lagouge, M.; Argmann, C.; Gerhart-Hines, Z.; Meziane, H.; Lerin, C.; Daussin, F.; Messadeq, N.; Milne, J.; Lambert, P.; Elliott, P.; et al. Resveratrol improves mitochondrial function and protects against metabolic disease by activating SIRT1 and PGC-1α. Cell 2006, 127, 1109–1122. [Google Scholar] [CrossRef] [PubMed]
- Hara, K.; Tobe, K.; Okada, T.; Kadowaki, H.; Akanuma, Y.; Ito, C.; Kimura, S. A genetic variation in the PGC-1 gene could confer insulin resistance and susceptibility to Type II diabetes. Diabetologia 2002, 45, 740–743. [Google Scholar] [CrossRef] [PubMed]
- St-Pierre, J.; Drori, S.; Uldry, M.; Silvaggi, J.M.; Rhee, J.; Jager, S.; Handschin, C.; Zheng, K.; Lin, J.; Yang, W.; et al. Suppression of Reactive Oxygen Species and Neurodegeneration by the PGC-1 Transcriptional Coactivators. Cell 2006, 127, 397–408. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Cheng, A.; Wan, R.; Yang, J.-L.; Kamimura, N.; Son, T.G.; Ouyang, X.; Luo, Y.; Okun, E.; Mattson, M.P. Involvement of PGC-1α in the formation and maintenance of neuronal dendritic spines. Nat. Commun. 2012, 3, 1250. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Sweeney, G.; Song, J. The association between PGC-1α and Alzheimer’s disease. Anat. Cell Biol. 2016, 49, 1–6. [Google Scholar] [CrossRef] [Green Version]
- Besseiche, A.; Riveline, J.-P.; Gautier, J.-F.; Bréant, B.; Blondeau, B. Metabolic roles of PGC-1α and its implications for type 2 diabetes. Diabetes Metab. 2015, 41, 347–357. [Google Scholar] [CrossRef]
- Lamb, Y.N. Imeglimin Hydrochloride: First Approval. Drugs 2021, 81, 1683–1690. [Google Scholar] [CrossRef]
- Braakhuis, A.J.; Nagulan, R.; Somerville, V. The Effect of MitoQ on Aging-Related Biomarkers: A Systematic Review and Meta-Analysis. Oxidative Med. Cell. Longev. 2018, 2018, 8575263. [Google Scholar] [CrossRef]
- Shabalina, I.G.; Vyssokikh, M.Y.; Gibanova, N.; Csikasz, R.I.; Edgar, D.; Hallden-Waldemarson, A.; Rozhdestvenskaya, Z.; Bakeeva, L.E.; Vays, V.B.; Pustovidko, A.V.; et al. Improved health-span and lifespan in mtDNA mutator mice treated with the mitochondrially targeted antioxidant SkQ1. Aging 2017, 9, 315–339. [Google Scholar] [CrossRef] [Green Version]
- Feniouk, B.A.; Skulachev, V. Cellular and Molecular Mechanisms of Action of Mitochondria-Targeted Antioxidants. Curr. Aging Sci. 2017, 10, 41–48. [Google Scholar] [CrossRef]
- Singh, A.; Faccenda, D.; Campanella, M. Pharmacological advances in mitochondrial therapy. eBioMedicine 2021, 65, 103244. [Google Scholar] [CrossRef]
- Kamm, D.R.; Pyles, K.D.; Sharpe, M.C.; Healy, L.N.; Colca, J.R.; McCommis, K.S. Novel insulin sensitizer MSDC-0602K improves insulinemia and fatty liver disease in mice, alone and in combination with liraglutide. J. Biol. Chem. 2021, 296, 100807. [Google Scholar] [CrossRef]
- Wang, W.; Han, R.; He, H.; Wang, Z.; Luan, X.; Li, J.; Feng, L.; Chen, S.; Aman, Y.; Xie, C. Delineating the Role of Mitophagy Inducers for Alzheimer Disease Patients. Aging Dis. 2021, 12, 852–867. [Google Scholar] [CrossRef]
- Ng, L.F.; Gruber, J.; Cheah, I.K.; Goo, C.K.; Cheong, W.F.; Shui, G.; Sit, K.P.; Wenk, M.R.; Halliwell, B. The mitochondria-targeted antioxidant MitoQ extends lifespan and improves healthspan of a transgenic Caenorhabditis elegans model of Alzheimer disease. Free Radic. Biol. Med. 2014, 71, 390–401. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Cusi, K.; Alkhouri, N.; Harrison, S.A.; Fouqueray, P.; Moller, D.E.; Hallakou-Bozec, S.; Bolze, S.; Grouin, J.M.; Megnien, S.J.; Dubourg, J.; et al. Efficacy and safety of PXL770, a direct AMP kinase activator, for the treatment of non-alcoholic fatty liver disease (STAMP-NAFLD): A randomised, double-blind, placebo-controlled, phase 2a study. Lancet Gastroenterol. Hepatol. 2021, 6, 889–902. [Google Scholar] [CrossRef]
- Chang, C.-Y.; Liang, M.-Z.; Chen, L. Current progress of mitochondrial transplantation that promotes neuronal regeneration. Transl. Neurodegener. 2019, 8, 17. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Chang, J.C.; Wu, S.-L.; Liu, K.-H.; Chen, Y.-H.; Chuang, C.-S.; Cheng, F.-C.; Su, H.-L.; Wei, Y.-H.; Kuo, S.-J.; Liu, C.-S. Allogeneic/xenogeneic transplantation of peptide-labeled mitochondria in Parkinson’s disease: Restoration of mitochondria functions and attenuation of 6-hydroxydopamine–induced neurotoxicity. Transl. Res. 2015, 170, 40–56. [Google Scholar] [CrossRef]
- McCully, J.D.; Levitsky, S.; Del Nido, P.J.; Cowan, D.B. Mitochondrial transplantation for therapeutic use. Clin. Transl. Med. 2016, 5, 16. [Google Scholar] [CrossRef] [Green Version]
- Gollihue, J.L.; Patel, S.P.; Eldahan, K.C.; Cox, D.H.; Donahue, R.; Taylor, B.K.; Sullivan, P.G.; Rabchevsky, A.G. Effects of Mitochondrial Transplantation on Bioenergetics, Cellular Incorporation, and Functional Recovery after Spinal Cord Injury. J. Neurotrauma 2018, 35, 1800–1818. [Google Scholar] [CrossRef] [PubMed]
- Shi, X.; Zhao, M.; Fu, C.; Fu, A. Intravenous administration of mitochondria for treating experimental Parkinson’s disease. Mitochondrion 2017, 34, 91–100. [Google Scholar] [CrossRef]
- Chang, J.-C.; Chao, Y.-C.; Chang, H.-S.; Wu, Y.-L.; Chang, H.-J.; Lin, Y.-S.; Cheng, W.-L.; Lin, T.-T.; Liu, C.-S. Intranasal delivery of mitochondria for treatment of Parkinson’s Disease model rats lesioned with 6-hydroxydopamine. Sci. Rep. 2021, 19, 10597. [Google Scholar] [CrossRef]
- Zhang, L.; Zhang, Z.; Khan, A.; Zheng, H.; Yuan, C.; Jiang, H. Advances in drug therapy for mitochondrial diseases. Ann. Transl. Med. 2020, 8, 17. [Google Scholar] [CrossRef] [PubMed]
- Chang, J.C.; Hoel, F.; Liu, K.-H.; Wei, Y.-H.; Cheng, F.-C.; Kuo, S.-J.; Tronstad, K.J.; Liu, C.-S. Peptide-mediated delivery of donor mitochondria improves mitochondrial function and cell viability in human cybrid cells with the MELAS A3243G mutation. Sci. Rep. 2017, 7, 10710. [Google Scholar] [CrossRef] [Green Version]
- Wu, S.; Zhang, A.; Li, S.; Chatterjee, S.; Qi, R.; Segura-Ibarra, V.; Ferrari, M.; Gupte, A.; Blanco, E.; Hamilton, D.J. Polymer Functionalization of Isolated Mitochondria for Cellular Transplantation and Metabolic Phenotype Alteration. Adv. Sci. 2018, 5, 1700530. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Picone, P.; Porcelli, G.; Bavisotto, C.C.; Nuzzo, D.; Galizzi, G.; Biagio, P.L.S.; Bulone, D.; Di Carlo, M. Synaptosomes: New vesicles for neuronal mitochondrial transplantation. J. Nanobiotechnol. 2021, 19, 6. [Google Scholar] [CrossRef] [PubMed]


Publisher’s Note: MDPI stays neutral with regard to jurisdictional claims in published maps and institutional affiliations. |
© 2022 by the authors. Licensee MDPI, Basel, Switzerland. This article is an open access article distributed under the terms and conditions of the Creative Commons Attribution (CC BY) license (https://creativecommons.org/licenses/by/4.0/).
Share and Cite
Galizzi, G.; Di Carlo, M. Insulin and Its Key Role for Mitochondrial Function/Dysfunction and Quality Control: A Shared Link between Dysmetabolism and Neurodegeneration. Biology 2022, 11, 943. https://doi.org/10.3390/biology11060943
Galizzi G, Di Carlo M. Insulin and Its Key Role for Mitochondrial Function/Dysfunction and Quality Control: A Shared Link between Dysmetabolism and Neurodegeneration. Biology. 2022; 11(6):943. https://doi.org/10.3390/biology11060943
Chicago/Turabian StyleGalizzi, Giacoma, and Marta Di Carlo. 2022. "Insulin and Its Key Role for Mitochondrial Function/Dysfunction and Quality Control: A Shared Link between Dysmetabolism and Neurodegeneration" Biology 11, no. 6: 943. https://doi.org/10.3390/biology11060943
APA StyleGalizzi, G., & Di Carlo, M. (2022). Insulin and Its Key Role for Mitochondrial Function/Dysfunction and Quality Control: A Shared Link between Dysmetabolism and Neurodegeneration. Biology, 11(6), 943. https://doi.org/10.3390/biology11060943