
Machine Learning Algorithms Highlight tRNA Information Content and Chargaff’s Second Parity Rule Score as Important Features in Discriminating Probiotics from Non-Probiotics


 ,
,  , ,
, ,  , and
, and
Abstract
:Simple Summary
Abstract
1. Introduction
2. Materials and Methods
2.1. Dataset
2.2. Features Encoding
2.3. Recursive Feature Selection and Models Training/Testing
2.4. Algorithm Comparison and Evaluation of Predictive Performance on Validation and Test Sets
3. Results
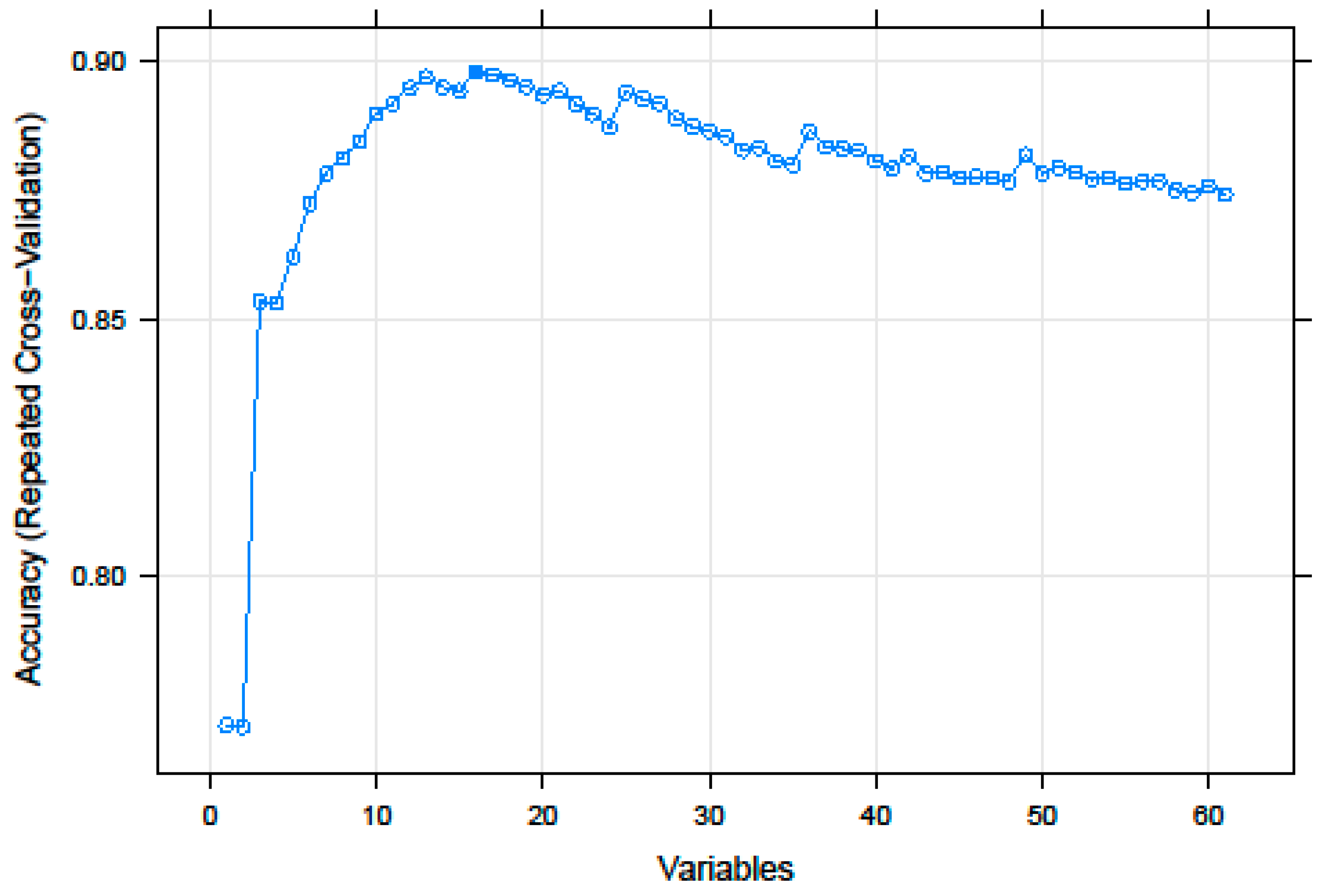
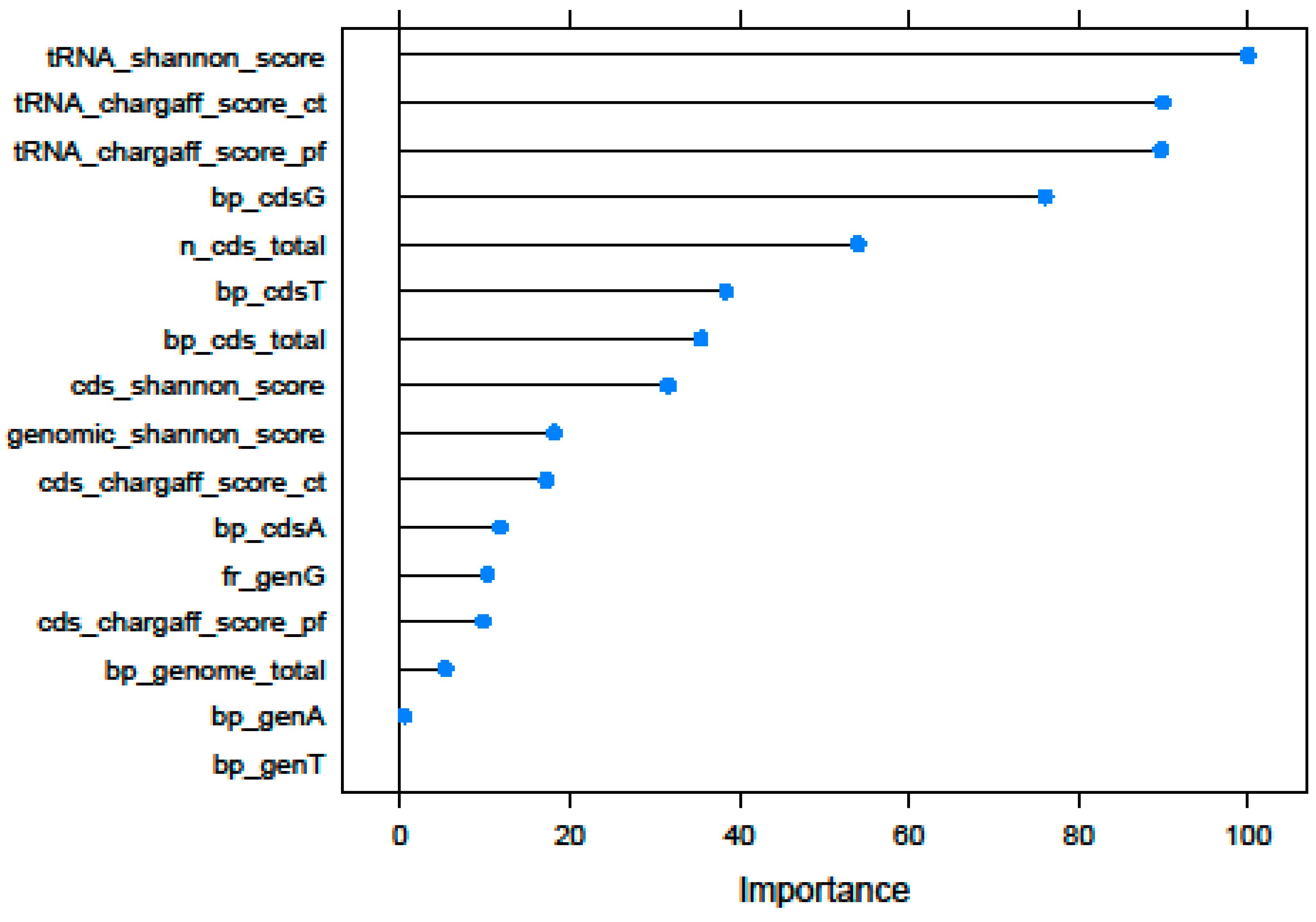
3.1. Recursive Feature Selection
3.2. Algorithm Comparison and Evaluation of Predictive Performance on Validation Set
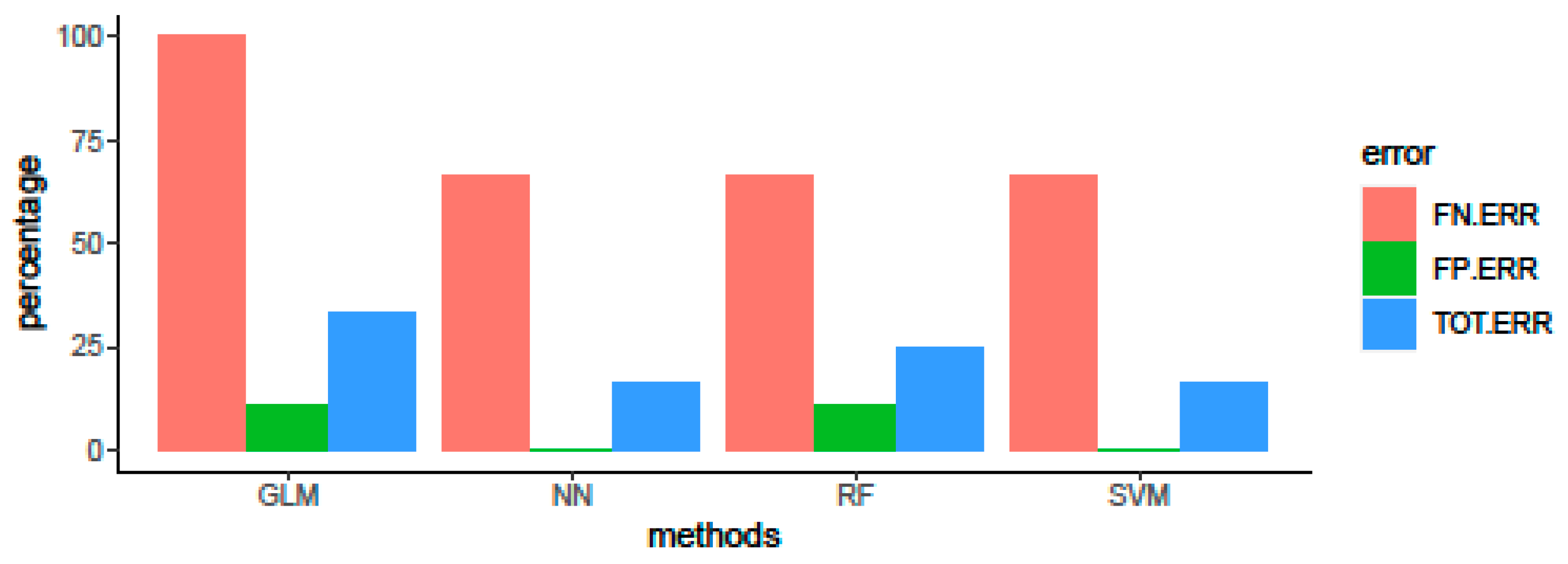
3.3. Algorithm Comparison and Evaluation of Predictive Performance on Test Set
4. Discussion
5. Conclusions
Supplementary Materials
Author Contributions
Funding
Institutional Review Board Statement
Informed Consent Statement
Data Availability Statement
Conflicts of Interest
References
- Mackowiak, P.A. Recycling Metchnikoff: Probiotics, the Intestinal Microbiome and the Quest for Long Life. Front. Public Health 2014, 1, 52–55. [Google Scholar] [CrossRef] [PubMed]
- Ohkusa TKoido, S.; Nishikawa, Y.; Sato, N. Gut Microbiota and Chronic Constipation: A Review and Update. Front. Med. 2019, 6, 19–28. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Chmielewska, A.; Szajewska, H. Systematic review of randomised controlled trials: Probiotics for functional constipation. World J. Gastroenterol. 2010, 16, 69–75. [Google Scholar] [PubMed]
- Guarino, A.; Lo Vecchio, A.; Canani, R.B. Probiotics as prevention and treatment for diarrhea. Curr. Opin. Gastroenterol. 2009, 25, 18–23. [Google Scholar] [CrossRef] [PubMed]
- Aragon, G.; Graham, D.B.; Borum, M.; Doman, D.B. Probiotic Therapy for Irritable Bowel Syndrome. Gastroenterol. Hepatol. 2010, 6, 39–44. [Google Scholar]
- McFarland, L.V.; Dublin, S. Meta-analysis of probiotics for the treatment of irritable bowel syndrome. World J. Gastroenterol. 2008, 14, 2650–2661. [Google Scholar] [CrossRef]
- Brenner, H.; Rothenbacher, D.; Arndt, V. Epidemiology of stomach cancer. Methods Mol. Biol. 2009, 472, 467–477. [Google Scholar]
- Moayyedi, P.; Ford, A.C.; Talley, N.J.; Cremonini, F.; Foxx-Orenstein, A.E.; Brandt, L.J.; Quigley, E.M. The efficacy of probiotics in the treatment of irritable bowel syndrome: A systematic review. Gut 2010, 59, 325–332. [Google Scholar] [CrossRef]
- Jiang, X.-E.; Yang, S.-M.; Zhou, X.-J.; Zhang, Y. Effects of mesalazine combined with bifid triple viable on intestinal flora, immunoglobulin and levels of cal, MMP-9, and MPO in feces of patients with ulcerative colitis. Eur. Rev. Med. Pharmacol. Sci. 2020, 24, 935–942. [Google Scholar]
- Weichselbaum, E. Potential benefits of probiotics—Main findings of an in-depth review. Br. J. Community Nurs. 2010, 15, 110–114. [Google Scholar] [CrossRef] [Green Version]
- Sachdeva, A.; Nagpal, J. Effect of fermented milk-based probiotic preparations on Helicobacter pylori eradication: A systematic review and meta-analysis of randomized-controlled trials. Eur. J. Gastroenterol. Hepatol. 2009, 21, 45–53. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Maldonado Galdeano, C.; Cazorla, S.I.; Lemme Dumit, J.M.; Vélez, E.; Perdigón, G. Beneficial Effects of Probiotic Consumption on the Immune System. Ann. Nutr. Metab. 2019, 74, 115–124. [Google Scholar] [CrossRef] [PubMed]
- King, S.; Glanville, J.; Sanders, M.E.; Fitzgerald, A.; Varley, D. Effectiveness of probiotics on the duration of illness in healthy children and adults who develop common acute respiratory infectious conditions: A systematic review and meta-analysis. Br. J. Nutr. 2014, 112, 41–45. [Google Scholar] [CrossRef] [PubMed]
- Lomax, A.R.; Calder, P.C. Prebiotics, immune function, infection and inflammation: A review of the evidence. Br. J. Nutr. 2009, 101, 633–658. [Google Scholar] [CrossRef] [Green Version]
- Vliagoftis, H.; Kouranos, V.D.; Betsi, G.I.; Falagas, M.E. Probiotics for the treatment of allergic rhinitis and asthma: Systematic review of randomized controlled trials. Ann. Allergy Asthma Immunol. 2008, 101, 570–579. [Google Scholar] [CrossRef]
- Kalliomäki, M.; Antoine, J.M.; Herz, U.; Rijkers, G.T.; Wells, J.M.; Mercenier, A. Guidance for substantiating the evidence for beneficial effects of probiotics: Prevention and management of allergic diseases by probiotics. J. Nutr. 2010, 140, 713S–721S. [Google Scholar] [CrossRef] [Green Version]
- Deng, X.; Cao, S.; Horn, A.L. Emerging Applications of Machine Learning in Food Safety. Annu. Rev. Food Sci. Technol. 2021, 12, 513–538. [Google Scholar] [CrossRef]
- Vischioni, C.; Giaccone, V.; Catellani, P.; Alberghini, L.; Miotti Scapin, R.; Taccioli, C. GBRAP: A Tool to Retrieve, Parse and Analyze GenBank Files of Viral and Bacterial Species. Biorxive. Available online: https://www.biorxiv.org/content/10.1101/2021.09.21.461110v2 (accessed on 31 January 2022).
- Fariselli, P.; Taccioli, C.; Pagani, L.; Maritan, A. DNA sequence symmetries from randomness: The origin of the Chargaff’s second parity rule. Brief Bioinform. 2021, 22, 2172–2181. [Google Scholar] [CrossRef]
- Koslicki, D. Topological entropy of DNA sequences. Bioinformatics 2021, 27, 1061–1067. [Google Scholar] [CrossRef] [Green Version]
- Bobbo, T.; Biffani, S.; Taccioli, C.; Penasa, M.; Cassandro, M. Comparison of machine learning methods to predict udder health status based on somatic cell counts in dairy cows. Sci. Rep. 2021, 11, 13642–13652. [Google Scholar] [CrossRef]
- Kuhn, M. Caret: Classification and Regression Training. R Package Version 6.0-92. 2021. Available online: https://CRAN.R-project.org/package=caret (accessed on 31 January 2022).
- Wickham, H.; Averick, M.; Bryan, J.; Chang, W.; McGowan, L.D.; François, R.; Grolemund, G.; Hayes, A.; Henry, L.; Hester, J.; et al. Welcome to the Tidyverse. J. Open Source Softw. 2019, 4, 1686. [Google Scholar] [CrossRef]
- R Core Team. A Language and Environment for Statistical Computing; Foundation for Statistical Computing: Vienna, Austria, 2021; Available online: www.R-project.org (accessed on 31 January 2022).
- Robin, X.; Turck, N.; Hainard, A.; Tiberti, N.; Lisacek, F.; Sanchez, J.-C.; Müller, M. pROC: An open-source package for R and S+ to analyze and compare ROC curves. BMC Bioinform. 2011, 12, 77–85. [Google Scholar] [CrossRef] [PubMed]
- Mukai, T.; Amikura, K.; Fu, X.; Söll, D.; Crnković, A. Indirect Routes to Aminoacyl-tRNA: The Diversity of Prokaryotic Cysteine Encoding Systems. Front. Genet. 2022, 12, 794509–794513. [Google Scholar] [CrossRef] [PubMed]
- Hauenstein, S.I.; Perona, J.J. Redundant synthesis of cysteinyl-tRNACys in Methanosarcina mazei. J. Biol. Chem. 2008, 283, 22007–22017. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Ewann, F.; Hoffman, P.S. Cysteine metabolism in Legionella pneumophila: Characterization of an L-cystine-utilizing mutant. Appl. Environ. Microbiol. 2006, 72, 3993–4000. [Google Scholar] [CrossRef] [Green Version]
- Zengler, K.; Zaramela, L.S. The social network of microorganisms—How auxotrophies shape complex communities. Nat. Rev. Microbiol. 2018, 16, 383–390. [Google Scholar] [CrossRef]
- Krahn, N.; Fischer, J.T.; Söll, D. Naturally Occurring tRNAs With Non-canonical Structures. Front. Microbiol. 2020, 11, 596914–596932. [Google Scholar] [CrossRef]
- Ehrlich, R.; Davyt, M.; López, I.; Chalar, C.; Marín, M. On the Track of the Missing tRNA Genes: A Source of Non-Canonical Functions? Front. Mol. Biosci. 2021, 8, 643701–643716. [Google Scholar] [CrossRef]
- Giannouli, S.; Kyritsis, A.; Malissovas, N.; Becker, H.D.; Stathopoulos, C. On the role of an unusual tRNAGly isoacceptor in Staphylococcus aureus. Biochimie 2009, 91, 344–351. [Google Scholar] [CrossRef]
- Rudinger-Thirion, J.; Lescure, A.; Paulus, C.; Frugier, M. Misfolded human tRNA isodecoder binds and neutralizes a 3′ UTR-embedded Alu element. Proc. Natl. Acad Sci. USA 2011, 108, E794–E802. [Google Scholar] [CrossRef] [Green Version]
- Huang, J.; Chen, W.; Zhou, F.; Pang, Z.; Wang, L.; Pan, T.; Wang, X. Tissue-specific reprogramming of host tRNA transcriptome by the microbiome. Genome Res. 2021, 31, 947–957. [Google Scholar] [CrossRef] [PubMed]
- Advani, V.M.; Ivanov, P. Translational control under stress: Reshaping the translatome. Bioessays 2019, 41, e1900009–e1900020. [Google Scholar] [CrossRef] [PubMed]
- Pawar, K.; Shigematsu, M.; Sharbati, S.; Kirino, Y. Infection-induced 5′-half molecules of tRNAHisGUG activate Toll-like receptor 7. PLoS Biol. 2020, 18, e3000982. [Google Scholar] [CrossRef] [PubMed]
- Hamdani, O.; Dhillon, N.; Hsieh, T.-H.S.; Fujita, T.; Ocampo, J.; Kirkland, J.G.; Lawrimore, J.; Kobayashi, T.J.; Friedman, B.; Fulton, D.; et al. tRNA Genes Affect Chromosome Structure and Function via Local Effects. Mol. Cell. Biol. 2019, 39, e00432–e00458. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Jiang, T.; Wu, H.; Yang, X.; Li, Y.; Zhang, Z.; Chen, F.; Zhao, L.; Zhang, C. Lactobacillus Mucosae Strain Promoted by a High-Fiber Diet in Genetic Obese Child Alleviates Lipid Metabolism and Modifies Gut Microbiota in ApoE -/- Mice on a Western Diet. Microorganisms 2020, 8, 1225. [Google Scholar] [CrossRef] [PubMed]
- Bui, T.P.N.; Shetty, S.A.; Lagkouvardos, I.; Ritari, J.; Chamlagain, B.; Douillard, F.P.; Paulin, L.; Piironen, V.; Clavel, T.; Plugge, C.M.; et al. Comparative genomics and physiology of the butyrate-producing bacterium Intestinimonas butyriciproducens. Environ. Microbiol. Rep. 2016, 8, 1024–1037. [Google Scholar] [CrossRef]
- Tsai, C.-C.; Hsih, H.-Y.; Chiu, H.-H.; Lai, Y.-Y.; Liu, J.-H.; Bi, Y.; Tsen, H.-Y. Antagonistic activity against Salmonella infection in vitro and in vivo for two Lactobacillus strains from swine and poultry. Int. J. Food Microbiol. 2005, 102, 185–194. [Google Scholar] [CrossRef]
- Sun, Z.; Harris, H.M.B.; McCann, A.; Guo, C.; Argimón, S.; Zhang, W.; Yang, X.; Jeffery, I.B.; Cooney, J.C.; Kagawa, T.F.; et al. Expanding the biotechnology potential of lactobacilli through comparative genomics of 213 strains and associated genera. Nat. Commun. 2015, 6, 8322–8335. [Google Scholar] [CrossRef]
- Chow, J.; Lee, S.M.; Shen, Y.; Khosravi, A.; Mazmanian, S.K. Host–Bacterial Symbiosis in Health and Disease. Adv. Immunol. 2010, 107, 243–274. [Google Scholar]
- Cani, P.D.; de Vos, W.M. Next-generation beneficial microbes: The case of Akkermansia muciniphila. Front. Microbiol. 2017, 8, 1765–1773. [Google Scholar] [CrossRef]
- Elsaghir, H.; Kumar, A.; Reddivari, R. Bacteroides Fragilis. StatPearls 2020. Available online: https://www.ncbi.nlm.nih.gov/books/NBK553032/ (accessed on 5 November 2021).




{kind=link}
{kind=link}
{kind=link}
| Id | Selected Features | Description |
|---|---|---|
| Genome | bp_genome_total | Genome size |
| bp_genA | Total number of Adenines (within the genome) | |
| bp_genT | Total number of Thymines (within the genome) | |
| fr_genG | Frequency of Guanines (number of Guanines divided by DNA total length) within the genome | |
| genomic_shannon_score | Shannon’s Entropy of total genome sequence | |
| CDS | n_cds_total | Total number of CDS elements (Coding DNA Sequences) |
| bp_cds_total | Total number of CDS nucleotides | |
| bp_cdsA | Total number of CDS Adenines | |
| bp_cdsG | Total number of CDS Cytosines | |
| bp_cdsT | Total number of CDS Thymines | |
| cds_chargaff_score_ct | Chargaff’s Second Parity rule score of total CDS sequence (ct method) | |
| cds_chargaff_score_pf | Chargaff’s Second Parity rule score of total CDS sequence (pf method) | |
| cds_shannon_score | Shannon Entropy value of total CDS sequence | |
| tRNA | tRNA_chargaff_score_ct | Chargaff’s Second Parity rule score of total tRNA sequence (ct method) |
| tRNA_chargaff_score_pf | Chargaff’s Second Parity rule score of total tRNA sequence (pf method) | |
| tRNA_shannon_score | Shannon’s Entropy value of total tRNA sequence |
| Method | Accuracy | Kappa Value |
|---|---|---|
| GLM | 0.936 | 0.869 |
| RF | 0.941 | 0.880 |
| SVM | 0.948 | 0.895 |
| NN | 0.951 | 0.900 |
| Method | Accuracy | 95% CI | Se | Sp | Precision | Kappa | F1 Score | MCC | AUC |
|---|---|---|---|---|---|---|---|---|---|
| GLM | 0.667 | 0.349–0.901 | 0.667 | 0.667 | 0.857 | 0.273 | 0.750 | 0.293 | 0.630 |
| RF | 0.750 | 0.423–0.945 | 0.778 | 0.667 | 0.875 | 0.400 | 0.823 | 0.408 | 0.704 |
| SVM | 0.833 | 0.516–0.979 | 0.778 | 1.000 | 1.000 | 0.636 | 0.875 | 0.683 | 0.815 |
| NN | 0.833 | 0.516–0.979 | 0.778 | 1.000 | 1.000 | 0.636 | 0.875 | 0.683 | 0.815 |
| Species | Order | NN Classification |
|---|---|---|
| Rickettsia prowazekii | Rickettsiales | Non-probiotic (referred as a pathogen in literature) |
| Yersinia pseudotuberculosis | Enterobacterales | Non-probiotic (referred as a pathogen in literature) |
| Vibrio cholerae | Vibrionales | Non-probiotic (referred as a pathogen in literature) |
| Bacteroides thetaiotaomicron | Bacteroidales | Non-probiotic (referred as possible probiotics in literature) |
| Bacteroides fragilis | Bacteroidales | Non-probiotic (referred as possible probiotics in literature) |
| Paucilactobacillus hokkaidonensis | Lactobacillales | Probiotic (referred as possible probiotics in literature) |
| Akkermansia muciniphila | Verrucomicrobiales | Probiotic (referred as possible probiotics in literature) |
| Levilactobacillus koreensis | Lactobacillales | Probiotic (referred as possible probiotics in literature) |
| Companilactobacillus ginsenosidimutans | Lactobacillales | Probiotic (referred as possible probiotics in literature) |
| Lactobacillus acetotolerans | Lactobacillales | Probiotic (referred as possible probiotics in literature) |
| Limosilactobacillus mucosae | Lactobacillales | Probiotic (referred as possible probiotics in literature) |
| Intestinimonas butyriciproducens | Eubacteriales | Probiotic (referred as possible probiotics in literature) |
Publisher’s Note: MDPI stays neutral with regard to jurisdictional claims in published maps and institutional affiliations. |
© 2022 by the authors. Licensee MDPI, Basel, Switzerland. This article is an open access article distributed under the terms and conditions of the Creative Commons Attribution (CC BY) license (https://creativecommons.org/licenses/by/4.0/).
Share and Cite
Bergamini, C.M.; Bianchi, N.; Giaccone, V.; Catellani, P.; Alberghini, L.; Stella, A.; Biffani, S.; Yaddehige, S.K.; Bobbo, T.; Taccioli, C. Machine Learning Algorithms Highlight tRNA Information Content and Chargaff’s Second Parity Rule Score as Important Features in Discriminating Probiotics from Non-Probiotics. Biology 2022, 11, 1024. https://doi.org/10.3390/biology11071024
Bergamini CM, Bianchi N, Giaccone V, Catellani P, Alberghini L, Stella A, Biffani S, Yaddehige SK, Bobbo T, Taccioli C. Machine Learning Algorithms Highlight tRNA Information Content and Chargaff’s Second Parity Rule Score as Important Features in Discriminating Probiotics from Non-Probiotics. Biology. 2022; 11(7):1024. https://doi.org/10.3390/biology11071024
Chicago/Turabian StyleBergamini, Carlo M., Nicoletta Bianchi, Valerio Giaccone, Paolo Catellani, Leonardo Alberghini, Alessandra Stella, Stefano Biffani, Sachithra Kalhari Yaddehige, Tania Bobbo, and Cristian Taccioli. 2022. "Machine Learning Algorithms Highlight tRNA Information Content and Chargaff’s Second Parity Rule Score as Important Features in Discriminating Probiotics from Non-Probiotics" Biology 11, no. 7: 1024. https://doi.org/10.3390/biology11071024
APA StyleBergamini, C. M., Bianchi, N., Giaccone, V., Catellani, P., Alberghini, L., Stella, A., Biffani, S., Yaddehige, S. K., Bobbo, T., & Taccioli, C. (2022). Machine Learning Algorithms Highlight tRNA Information Content and Chargaff’s Second Parity Rule Score as Important Features in Discriminating Probiotics from Non-Probiotics. Biology, 11(7), 1024. https://doi.org/10.3390/biology11071024