
Transcriptome Analysis and HPLC Profiling of Flavonoid Biosynthesis in Citrus aurantium L. during Its Key Developmental Stages


Abstract
:Simple Summary
Abstract
1. Introduction
2. Materials and Methods
2.1. Plant Sample Collection
2.2. Flavonoid Content Analysis
2.3. cDNA Library Construction and Sequence Analysis and Alignment
2.4. Differentially Expressed Genes (DEGs)
2.5. Weighted Correlation Network Analysis (WGCNA) and Coexpression Network Construction
2.6. RT-qPCR Analysis
3. Results
3.1. Quantitation of Flavonoid Contents in Citrus aurantium L.
3.2. Phylogenetic Analysis and the Expression Landscape between the Three Accessions
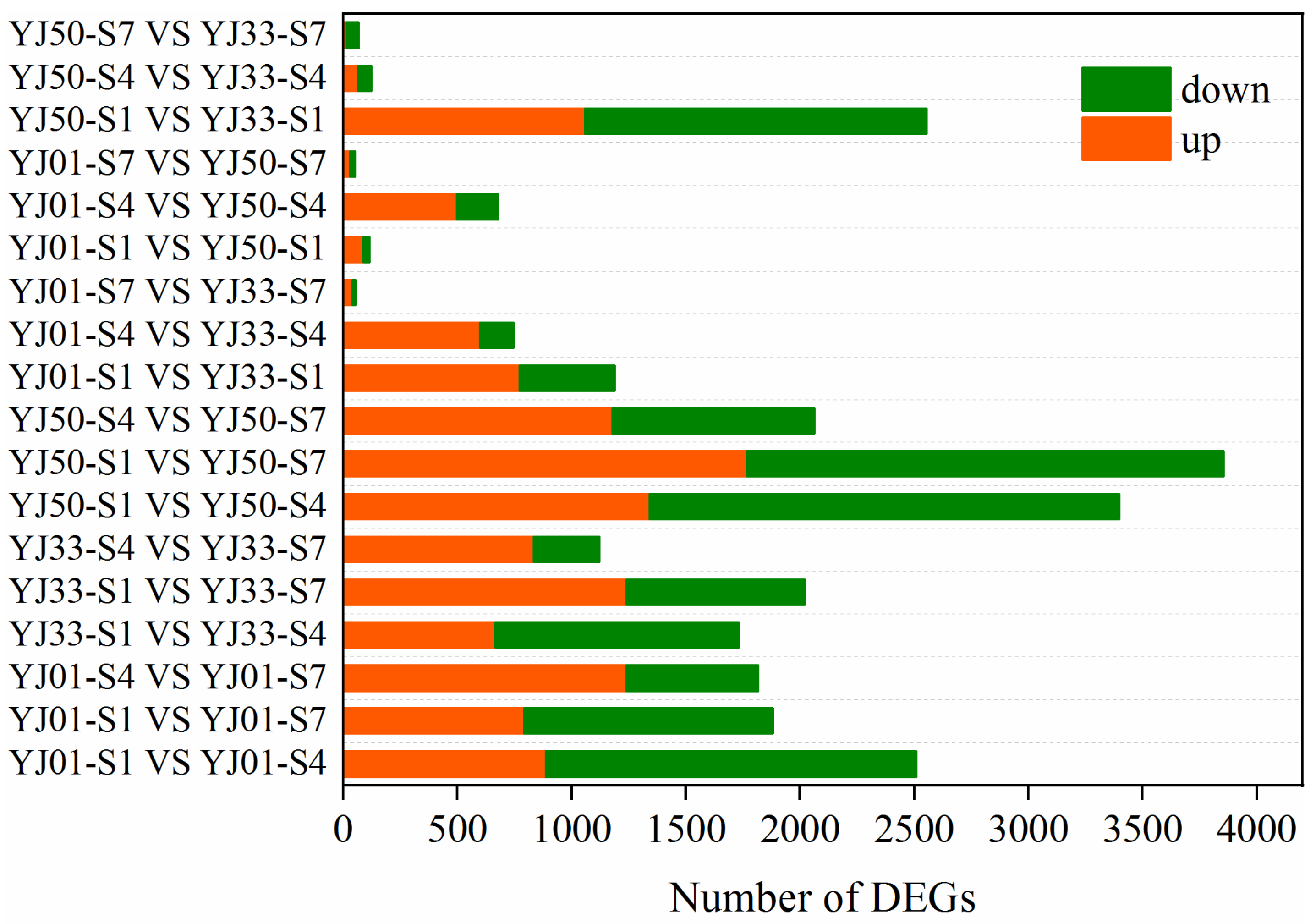
3.3. Identification and Functional Analysis of DEGs during Different Fruit Developmental Stages
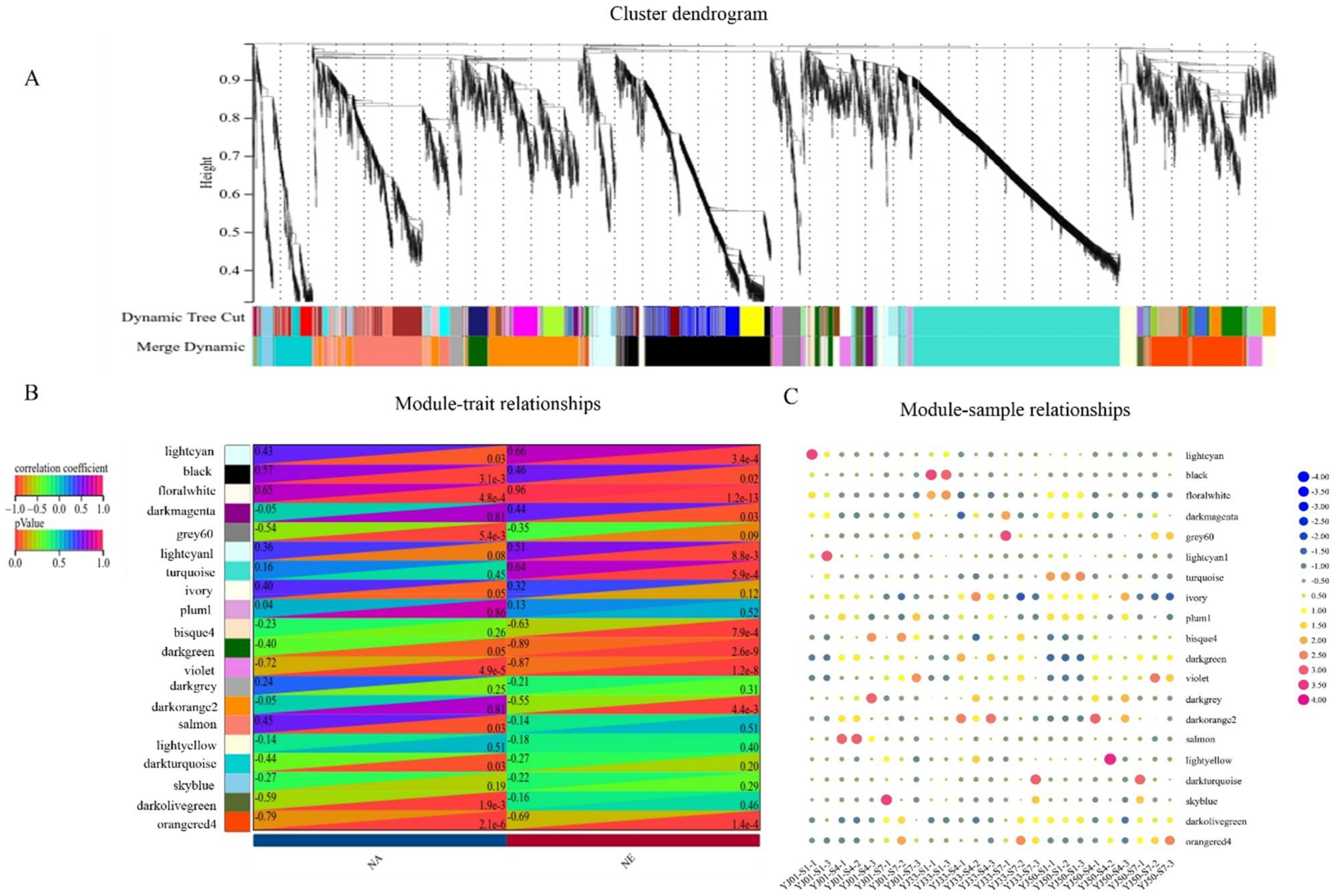
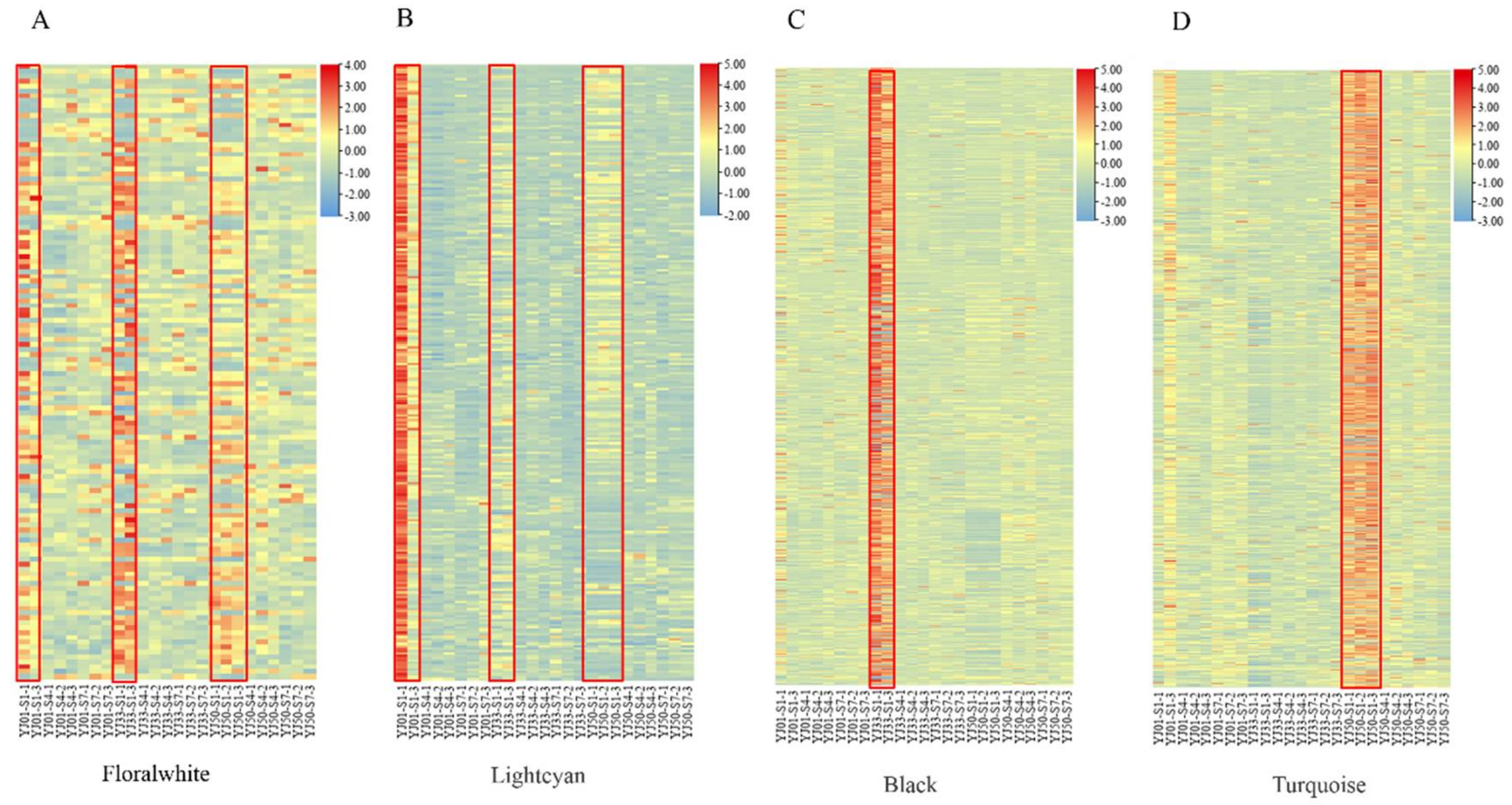
3.4. Weighted Gene Co-Expression Network Construction and Analysis
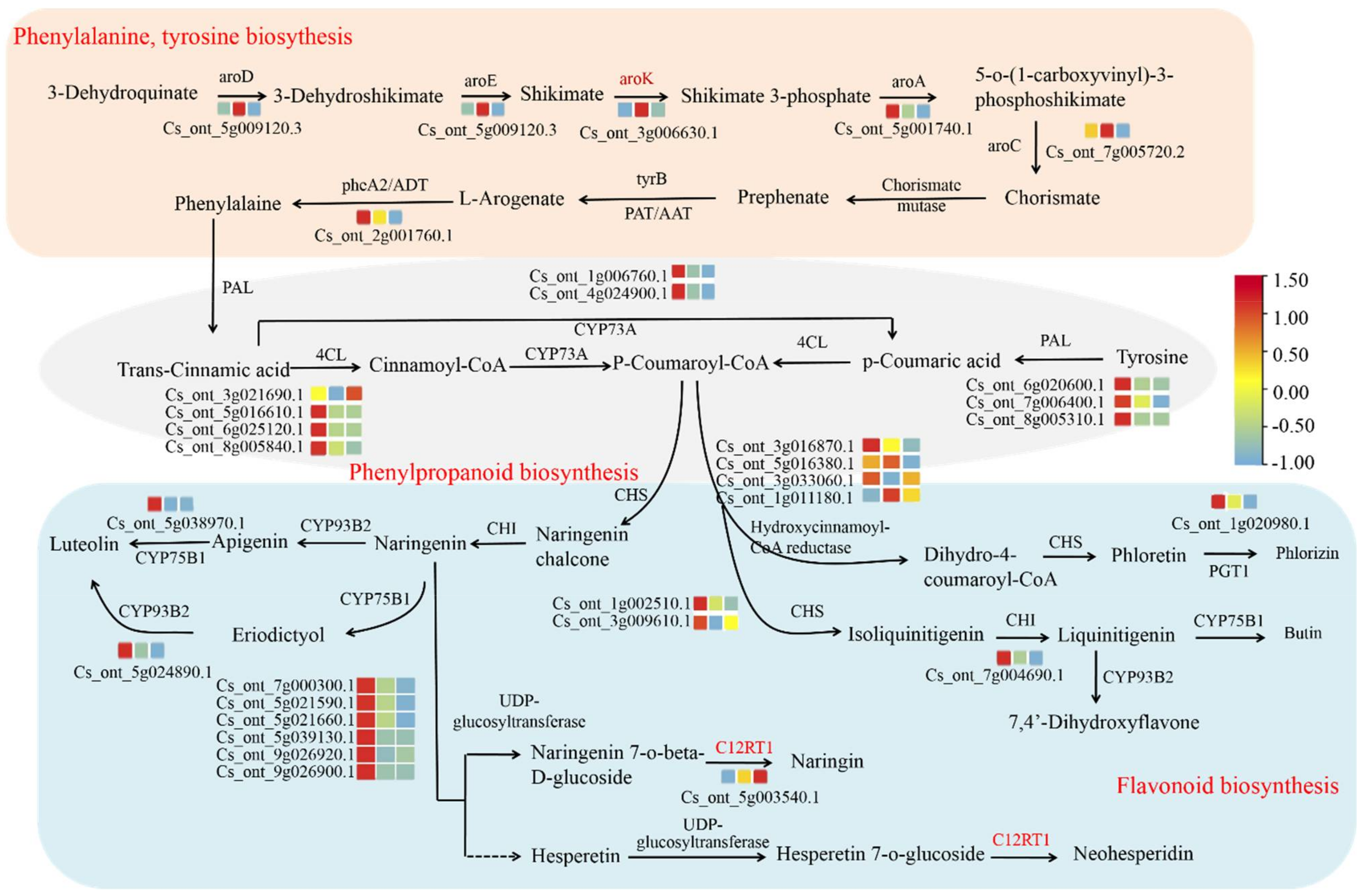
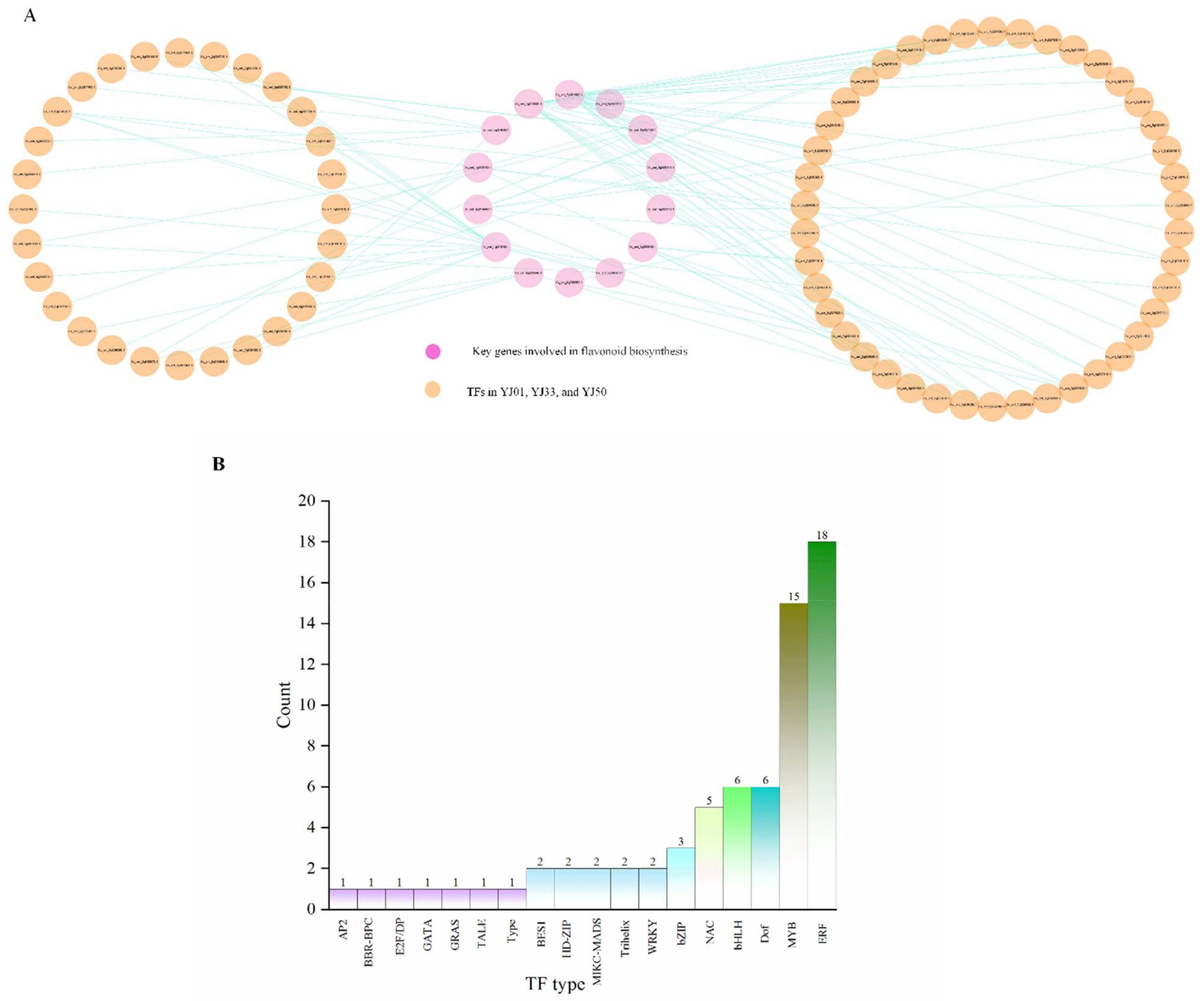
3.5. Identification of Transcriptional Regulator Networks
3.6. Validation of RNA-Seq
4. Discussion
5. Conclusions
Supplementary Materials
Author Contributions
Funding
Institutional Review Board Statement
Informed Consent Statement
Data Availability Statement
Acknowledgments
Conflicts of Interest
References
- Rao, M.J.; Zuo, H.; Xu, Q. Genomic insights into citrus domestication and its important agronomic traits. Plant Commun. 2021, 2, 100138. [Google Scholar] [CrossRef] [PubMed]
- Marya; Khan, H.; Nabavi, S.M.; Habtemariam, S. Anti-diabetic potential of peptides: Future prospects as therapeutic agents. Life Sci. 2018, 193, 153–158. [Google Scholar] [CrossRef] [PubMed]
- Suntar, I.; Khan, H.; Patel, S.; Celano, R.; Rastrelli, L. An Overview on Citrus aurantium L.: Its Functions as Food Ingredient and Therapeutic Agent. Oxidative Med. Cell. Longev. 2018, 2018, 7864269. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Li, L.; Chen, J.; Lin, L.; Pan, G.; Zhang, S.; Chen, H.; Zhang, M.; Xuan, Y.; Wang, Y.; You, Z. Quzhou Fructus Aurantii Extract suppresses inflammation via regulation of MAPK, NF-κB, and AMPK signaling pathway. Sci. Rep. 2020, 10, 1593. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Wu, J.; Huang, G.; Li, Y.; Li, X. Flavonoids from Aurantii Fructus Immaturus and Aurantii Fructus: Promising phytomedicines for the treatment of liver diseases. Chin. Med. 2020, 15, 89. [Google Scholar] [CrossRef] [PubMed]
- Paniagua, M.; Crespo, F.J.; Arís, A.; Devant, M. Effects of Flavonoids Extracted from Citrus aurantium on Performance, Behavior, and Rumen Gene Expression in Holstein Bulls Fed with High-Concentrate Diets in Pellet Form. Anim. Open Access J. MDPI 2021, 11, 1387. [Google Scholar] [CrossRef]
- Liu, W.; Feng, Y.; Yu, S.; Fan, Z.; Li, X.; Li, J.; Yin, H. The Flavonoid Biosynthesis Network in Plants. Int. J. Mol. Sci. 2021, 22, 2824. [Google Scholar] [CrossRef]
- Zhang, Y.; Huang, P.; He, W.; Sakah, K.J.; Ruan, J.; Li, Z.; Wang, T. Bioactive constituents obtained from the fruits of Citrus aurantium. J. Nat. Med. 2019, 73, 146–153. [Google Scholar] [CrossRef]
- Pellati, F.; Benvenuti, S.; Melegari, M. High-performance liquid chromatography methods for the analysis of adrenergic amines and flavanones in Citrus aurantium L. var. amara. Phytochem. Anal. PCA 2004, 15, 220–225. [Google Scholar] [CrossRef]
- Shirley, B.W. Flavonoids in seeds and grains: Physiological function, agronomic importance and the genetics of biosynthesis. Seed Sci. Res. 1998, 8, 415–422. [Google Scholar] [CrossRef]
- Petrussa, E.; Braidot, E.; Zancani, M.; Peresson, C.; Bertolini, A.; Patui, S.; Vianello, A. Plant flavonoids--biosynthesis, transport and involvement in stress responses. Int. J. Mol. Sci. 2013, 14, 14950–14973. [Google Scholar] [CrossRef] [PubMed]
- Nakabayashi, R.; Saito, K. Integrated metabolomics for abiotic stress responses in plants. Curr. Opin. Plant Biol. 2015, 24, 10–16. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Zahran, E.M.; Abdelmohsen, U.R.; Hussein, A.S.; Salem, M.A.; Khalil, H.E.; Yehia Desoukey, S.; Fouad, M.A.; Kamel, M.S. Antiulcer potential and molecular docking of flavonoids from Ocimum forskolei Benth., family Lamiaceae. Nat. Prod. Res. 2021, 35, 1933–1937. [Google Scholar] [CrossRef]
- Maleki, S.J.; Crespo, J.F.; Cabanillas, B. Anti-inflammatory effects of flavonoids. Food Chem. 2019, 299, 125124. [Google Scholar] [CrossRef]
- Kandaswami, C.; Lee, L.T.; Lee, P.P.; Hwang, J.J.; Ke, F.C.; Huang, Y.T.; Lee, M.T. The antitumor activities of flavonoids. In Vivo 2005, 19, 895–909. [Google Scholar]
- Badshah, S.L.; Faisal, S.; Muhammad, A.; Poulson, B.G.; Emwas, A.H.; Jaremko, M. Antiviral activities of flavonoids. Biomed. Pharmacother. Biomed. Pharmacother. 2021, 140, 111596. [Google Scholar] [CrossRef] [PubMed]
- Pietta, P.G. Flavonoids as antioxidants. J. Nat. Prod. 2000, 63, 1035–1042. [Google Scholar] [CrossRef]
- Vogt, T. Phenylpropanoid biosynthesis. Mol. Plant 2010, 3, 2–20. [Google Scholar] [CrossRef] [Green Version]
- Zhang, X.; Abrahan, C.; Colquhoun, T.A.; Liu, C.J. A Proteolytic Regulator Controlling Chalcone Synthase Stability and Flavonoid Biosynthesis in Arabidopsis. Plant Cell 2017, 29, 1157–1174. [Google Scholar] [CrossRef] [Green Version]
- Ito, T.; Fujimoto, S.; Suito, F.; Shimosaka, M.; Taguchi, G. C-Glycosyltransferases catalyzing the formation of di-C-glucosyl flavonoids in citrus plants. Plant J. Cell Mol. Biol. 2017, 91, 187–198. [Google Scholar] [CrossRef] [Green Version]
- Murayama, K.; Kato-Murayama, M.; Sato, T.; Hosaka, T.; Ishiguro, K.; Mizuno, T.; Kitao, K.; Honma, T.; Yokoyama, S.; Tanaka, Y.; et al. Anthocyanin 5,3’-aromatic acyltransferase from Gentiana triflora, a structural insight into biosynthesis of a blue anthocyanin. Phytochemistry 2021, 186, 112727. [Google Scholar] [CrossRef] [PubMed]
- Do, C.T.; Pollet, B.; Thévenin, J.; Sibout, R.; Denoue, D.; Barrière, Y.; Lapierre, C.; Jouanin, L. Both caffeoyl Coenzyme A 3-O-methyltransferase 1 and caffeic acid O-methyltransferase 1 are involved in redundant functions for lignin, flavonoids and sinapoyl malate biosynthesis in Arabidopsis. Planta 2007, 226, 1117–1129. [Google Scholar] [CrossRef] [PubMed]
- Chen, K.; Hu, Z.M.; Song, W.; Wang, Z.L.; He, J.B.; Shi, X.M.; Cui, Q.H.; Qiao, X.; Ye, M. Diversity of O-Glycosyltransferases Contributes to the Biosynthesis of Flavonoid and Triterpenoid Glycosides in Glycyrrhiza uralensis. ACS Synth. Biol. 2019, 8, 1858–1866. [Google Scholar] [CrossRef] [PubMed]
- Uchiyama, N.; Kim, I.H.; Kikura-Hanajiri, R.; Kawahara, N.; Konishi, T.; Goda, Y. HPLC separation of naringin, neohesperidin and their C-2 epimers in commercial samples and herbal medicines. J. Pharm. Biomed. Anal. 2008, 46, 864–869. [Google Scholar] [CrossRef] [PubMed]
- Chen, Y.; Chen, Y.; Shi, C.; Huang, Z.; Zhang, Y.; Li, S.; Li, Y.; Ye, J.; Yu, C.; Li, Z.; et al. SOAPnuke: A MapReduce acceleration-supported software for integrated quality control and preprocessing of high-throughput sequencing data. GigaScience 2018, 7, gix120. [Google Scholar] [CrossRef] [Green Version]
- Kim, D.; Paggi, J.M.; Park, C.; Bennett, C.; Salzberg, S.L. Graph-based genome alignment and genotyping with HISAT2 and HISAT-genotype. Nat. Biotechnol. 2019, 37, 907–915. [Google Scholar] [CrossRef]
- Friis, S.L.; Buchard, A.; Rockenbauer, E.; Børsting, C.; Morling, N. Introduction of the Python script STRinNGS for analysis of STR regions in FASTQ or BAM files and expansion of the Danish STR sequence database to 11 STRs. Forensic Sci. Int. Genet. 2016, 21, 68–75. [Google Scholar] [CrossRef]
- Shahriyari, L. Effect of normalization methods on the performance of supervised learning algorithms applied to HTSeq-FPKM-UQ data sets: 7SK RNA expression as a predictor of survival in patients with colon adenocarcinoma. Brief. Bioinform. 2019, 20, 985–994. [Google Scholar] [CrossRef]
- Love, M.I.; Huber, W.; Anders, S. Moderated estimation of fold change and dispersion for RNA-seq data with DESeq2. Genome Biol. 2014, 15, 550. [Google Scholar] [CrossRef] [Green Version]
- Wang, X.; Yang, H.; Zhang, L.; Han, L.; Di, S.; Wei, X.; Wu, H.; Zhang, H.; Zhao, L.; Tong, X. Network Pharmacology-Based Prediction of Mechanism of Shenzhuo Formula for Application to DKD. Evid. Based Complementary Altern. Med. 2021, 2021, 6623010. [Google Scholar] [CrossRef]
- Liang, W.; Sun, F.; Zhao, Y.; Shan, L.; Lou, H. Identification of Susceptibility Modules and Genes for Cardiovascular Disease in Diabetic Patients Using WGCNA Analysis. J. Diabetes Res. 2020, 2020, 4178639. [Google Scholar] [CrossRef] [PubMed]
- Shannon, P.; Markiel, A.; Ozier, O.; Baliga, N.S.; Wang, J.T.; Ramage, D.; Amin, N.; Schwikowski, B.; Ideker, T. Cytoscape: A software environment for integrated models of biomolecular interaction networks. Genome Res. 2003, 13, 2498–2504. [Google Scholar] [CrossRef]
- Sangya, P.; Maria, M.; Claire, D. UniProt: The universal protein knowledgebase. Nucleic Acids Res. 2017, 45, D158–D169. [Google Scholar] [CrossRef] [Green Version]
- Bakir, A.; Hosbul, T.; Cuce, F.; Artuk, C.; Taskin, G.; Caglayan, M.; Guney, M.; Kurkcu, M.F.; Yildiz, F.; Erdal, H.; et al. Investigation of Viral Load Cycle Threshold Values in Patients with SARS-CoV-2 Associated Pneumonia with Real-Time PCR Method. J. Infect. Dev. Ctries. 2021, 15, 1408–1414. [Google Scholar] [CrossRef] [PubMed]
- Wu, G.A.; Terol, J.; Ibanez, V.; López-García, A.; Pérez-Román, E.; Borredá, C.; Domingo, C.; Tadeo, F.R.; Carbonell-Caballero, J.; Alonso, R.; et al. Genomics of the origin and evolution of Citrus. Nature 2018, 554, 311–316. [Google Scholar] [CrossRef] [Green Version]
- Wang, L.; He, F.; Huang, Y.; He, J.; Yang, S.; Zeng, J.; Deng, C.; Jiang, X.; Fang, Y.; Wen, S.; et al. Genome of Wild Mandarin and Domestication History of Mandarin. Mol. Plant 2018, 11, 1024–1037. [Google Scholar] [CrossRef] [Green Version]
- Velasco, R.; Licciardello, C. A genealogy of the citrus family. Nat. Biotechnol. 2014, 32, 640–642. [Google Scholar] [CrossRef]
- Salse, J. In silico archeogenomics unveils modern plant genome organisation, regulation and evolution. Curr. Opin. Plant Biol. 2012, 15, 122–130. [Google Scholar] [CrossRef]
- Xu, Q.; Chen, L.L.; Ruan, X.; Chen, D.; Zhu, A.; Chen, C.; Bertrand, D.; Jiao, W.B.; Hao, B.H.; Lyon, M.P.; et al. The draft genome of sweet orange (Citrus sinensis). Nat. Genet. 2013, 45, 59–66. [Google Scholar] [CrossRef]
- Wu, G.A.; Prochnik, S.; Jenkins, J.; Salse, J.; Hellsten, U.; Murat, F.; Perrier, X.; Ruiz, M.; Scalabrin, S.; Terol, J.; et al. Sequencing of diverse mandarin, pummelo and orange genomes reveals complex history of admixture during citrus domestication. Nat. Biotechnol. 2014, 32, 656–662. [Google Scholar] [CrossRef]
- Reyes Jara, A.M.; Gomez Lobato, M.E.; Civello, P.M.; Martinez, G.A. Phenylalanine ammonia lyase is more relevant than Chalcone synthase and Chalcone isomerase in the biosynthesis of flavonoids during postharvest senescence of broccoli. J. Food Biochem. 2022, 46, e14054. [Google Scholar] [CrossRef] [PubMed]
- Dai, X.; Shi, X.; Yang, C.; Zhao, X.; Zhuang, J.; Liu, Y.; Gao, L.; Xia, T. Two UDP-Glycosyltransferases Catalyze the Biosynthesis of Bitter Flavonoid 7-O-Neohesperidoside through Sequential Glycosylation in Tea Plants. J. Agric. Food Chem. 2022, 70, 2354–2365. [Google Scholar] [CrossRef] [PubMed]
- Caputi, L.; Malnoy, M.; Goremykin, V.; Nikiforova, S.; Martens, S. A genome-wide phylogenetic reconstruction of family 1 UDP-glycosyltransferases revealed the expansion of the family during the adaptation of plants to life on land. Plant J. Cell Mol. Biol. 2012, 69, 1030–1042. [Google Scholar] [CrossRef] [PubMed]
- Masada, S.; Terasaka, K.; Mizukami, H. A single amino acid in the PSPG-box plays an important role in the catalytic function of CaUGT2 (Curcumin glucosyltransferase), a Group D Family 1 glucosyltransferase from Catharanthus roseus. FEBS Lett. 2007, 581, 2605–2610. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Brazier-Hicks, M.; Gershater, M.; Dixon, D.; Edwards, R. Substrate specificity and safener inducibility of the plant UDP-glucose-dependent family 1 glycosyltransferase super-family. Plant Biotechnol. J. 2018, 16, 337–348. [Google Scholar] [CrossRef]
- Bowles, D.; Lim, E.K.; Poppenberger, B.; Vaistij, F.E. Glycosyltransferases of lipophilic small molecules. Annu. Rev. Plant Biol. 2006, 57, 567–597. [Google Scholar] [CrossRef]
- Zhao, C.; Liu, X.; Gong, Q.; Cao, J.; Shen, W.; Yin, X.; Grierson, D.; Zhang, B.; Xu, C.; Li, X.; et al. Three AP2/ERF family members modulate flavonoid synthesis by regulating type IV chalcone isomerase in citrus. Plant Biotechnol. J. 2021, 19, 671–688. [Google Scholar] [CrossRef]
- Chen, J.; Yuan, Z.; Zhang, H.; Li, W.; Shi, M.; Peng, Z.; Li, M.; Tian, J.; Deng, X.; Cheng, Y.; et al. Cit1,2RhaT and two novel CitdGlcTs participate in flavor-related flavonoid metabolism during citrus fruit development. J. Exp. Bot. 2019, 70, 2759–2771. [Google Scholar] [CrossRef]
- Wan, Q.; Bai, T.; Liu, M.; Liu, Y.; Xie, Y.; Zhang, T.; Huang, M.; Zhang, J. Comparative Analysis of the Chalcone-Flavanone Isomerase Genes in Six Citrus Species and Their Expression Analysis in Sweet Orange (Citrus sinensis). Front. Genet. 2022, 13, 848141. [Google Scholar] [CrossRef]
- Shui, L.; Huo, K.; Chen, Y.; Zhang, Z.; Li, Y.; Niu, J. Integrated metabolome and transcriptome revealed the flavonoid biosynthetic pathway in developing Vernonia amygdalina leaves. PeerJ 2021, 9, e11239. [Google Scholar] [CrossRef]
- Zou, L.Q.; Wang, C.X.; Kuang, X.J.; Li, Y.; Sun, C. Advance in flavonoids biosynthetic pathway and synthetic biology. Zhongguo Zhong Yao Za Zhi Zhongguo Zhongyao Zazhi China J. Chin. Mater. Med. 2016, 41, 4124–4128. [Google Scholar] [CrossRef]
- Wu, B.; Liu, X.; Xu, K.; Zhang, B. Genome-wide characterization, evolution and expression profiling of UDP-glycosyltransferase family in pomelo (Citrus grandis) fruit. BMC Plant Biol. 2020, 20, 459. [Google Scholar] [CrossRef] [PubMed]
- Xu, W.; Dubos, C.; Lepiniec, L. Transcriptional control of flavonoid biosynthesis by MYB-bHLH-WDR complexes. Trends Plant Sci. 2015, 20, 176–185. [Google Scholar] [CrossRef]
- Yang, Y.; He, Z.; Bing, Q.; Duan, X.; Chen, S.; Zeng, M.; Liu, X. Two Dof transcription factors promote flavonoid synthesis in kumquat fruit by activating C-glucosyltransferase. Plant Sci. 2022, 318, 111234. [Google Scholar] [CrossRef]
- Wang, W.L.; Wang, Y.X.; Li, H.; Liu, Z.W.; Cui, X.; Zhuang, J. Two MYB transcription factors (CsMYB2 and CsMYB26) are involved in flavonoid biosynthesis in tea plant [Camellia sinensis (L.) O. kuntze]. BMC Plant Biol. 2018, 18, 288. [Google Scholar] [CrossRef] [PubMed]
- Rahim, M.A.; Busatto, N.; Trainotti, L. Regulation of anthocyanin biosynthesis in peach fruits. Planta 2014, 240, 913–929. [Google Scholar] [CrossRef]
- Zhai, R.; Wang, Z.; Zhang, S.; Meng, G.; Song, L.; Wang, Z.; Li, P.; Ma, F.; Xu, L. Two MYB transcription factors regulate flavonoid biosynthesis in pear fruit (Pyrus bretschneideri Rehd.). J. Exp. Bot. 2016, 67, 1275–1284. [Google Scholar] [CrossRef] [Green Version]
- Arlotta, C.; Puglia, G.D.; Genovese, C.; Toscano, V.; Karlova, R.; Beekwilder, J.; De Vos, R.C.H.; Raccuia, S.A. MYB5-like and bHLH influence flavonoid composition in pomegranate. Plant Sci. 2020, 298, 110563. [Google Scholar] [CrossRef]
- Montefiori, M.; Brendolise, C.; Dare, A.P.; Lin-Wang, K.; Davies, K.M.; Hellens, R.P.; Allan, A.C. In the Solanaceae, a hierarchy of bHLHs confer distinct target specificity to the anthocyanin regulatory complex. J. Exp. Bot. 2015, 66, 1427–1436. [Google Scholar] [CrossRef] [Green Version]










{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
| Flavonoid Type | Varieties | S1 | S2 | S3 | S4 | S5 | S6 | S7 | S8 |
|---|---|---|---|---|---|---|---|---|---|
| Naringin | YJ01 | 116.432 ± 1.08 | 183.78533 ± 0.92 | 164.96033 ± 0.08 | 106.728 ± 0.9 | 127.26367 ± 0.65 | 78.34 ± 0.11 | 77.739 ± 1.35 | 48.221 ± 0.58 |
| YJ33 | 123.87 ± 0.12 | 178.125 ± 1.81 | 130.525 ± 0.36 | 71.98 ± 2.9 | 95.73 ± 0.68 | 57.575 ± 0.37 | 41.81 ± 0.48 | 38.89 ± 0.69 | |
| YJ50 | 95.9926 ± 1.09 | 161.3924 ± 0.44 | 140.2121 ± 0.23 | 76.74742 ± 0.86 | 99.62167 ± 1.43 | 60.0242 ± 0.91 | 50.6822 ± 0.08 | 44.6606 ± 1.33 | |
| Neohesperidin | YJ01 | 178.6218 ± 1.31 | 119.7779 ± 0.24 | 57.6393 ± 0.4 | 31.41540 ± 0.25 | 39.7634 ± 0.14 | 14.4204 ± 0.15 | 13.2093 ± 0.12 | 2.9119 ± 0.04 |
| YJ33 | 143.17175 ± 0.53 | 102.65441 ± 1.21 | 26.00266 ± 0.08 | 7.57086 ± 1.04 | 15.70094 ± 0.25 | 3.97631 ± 0.05 | 2.92 ± 1.05 | 2.009 ± 1.55 | |
| YJ50 | 168.1867 ± 1.97 | 88.6930 ± 1.32 | 33.6265 ± 0.08 | 12.5768 ± 0.34 | 22.8530 ± 0.72 | 5.6430 ± 0.28 | 2.2437 ± 0.21 | 2.20816 ± 0.67 |
| Genes | Function | YJ01 | YJ33 | YJ50 | ||||||
|---|---|---|---|---|---|---|---|---|---|---|
| S1 vs. S4 | S1 vs. S7 | S4 vs. S7 | S1 vs. S4 | S1 vs. S7 | S4 vs. S7 | S1 vs. S4 | S1 vs. S7 | S4 vs. S7 | ||
| Cs_ont_3g008580.1 | CHS | + | + | + | ||||||
| Cs_ont_7g004690.1 | CHI | − | − | − | ||||||
| Cs_ont_7g019870.1 | LAR | + | ||||||||
| Cs_ont_5g040910.1 | ANS | + | + | + | + | + | + | + | ||
| Cs_ont_3g009610.1 | CHS | + | + | |||||||
| Cs_ont_6g025170.1 | HHT1 | + | + | + | + | + | + | + | ||
| Cs_ont_2g006480.1 | ANR | − | + | |||||||
| Cs_ont_6g005600.1 | SAT | + | ||||||||
| Cs_ont_1g002510.1 | CHS | − | − | − | − | − | − | |||
| Cs_ont_1g014200.1 | N/A | + | + | + | + | + | + | |||
| Cs_ont_5g038970.1 | CYP75B1 | − | − | − | − | |||||
| Cs_ont_5g024640.1 | HST | + | + | + | + | − | + | + | ||
| Cs_ont_8g011010.1 | SAT | + | − | |||||||
| Cs_ont_6g019320.1 | N/A | − | − | − | ||||||
| Cs_ont_1g006760.1 | CYP73A | − | − | − | − | |||||
| Cs_ont_1g014190.1 | CCOMT | + | + | |||||||
| Cs_ont_1g020980.1 | PGT1 | − | − | − | ||||||
| Cs_ont_5g024870.1 | CYP93B16 | + | + | |||||||
| Cs_ont_4g024900.1 | CYP73A | − | − | − | − | − | ||||
| Cs_ont_9g012610.1 | CHS1 | + | + | |||||||
| Cs_ont_8g017240.5 | CHS | + | ||||||||
| Cs_ont_1g002480.1 | ACS2 | − | ||||||||
| Cs_ont_5g024890.1 | CYP93B2 | − | − | |||||||
| Cs_ont_1g024230.1 | CYP81Q32 | − | − | − | + | + | − | |||
| Cs_ont_1g024260.1 | CYP81Q32 | − | − | − | − | |||||
| Cs_ont_9g014840.1 | UDPGT | − | − | − | ||||||
Publisher’s Note: MDPI stays neutral with regard to jurisdictional claims in published maps and institutional affiliations. |
© 2022 by the authors. Licensee MDPI, Basel, Switzerland. This article is an open access article distributed under the terms and conditions of the Creative Commons Attribution (CC BY) license (https://creativecommons.org/licenses/by/4.0/).
Share and Cite
Chen, J.; Shi, Y.; Zhong, Y.; Sun, Z.; Niu, J.; Wang, Y.; Chen, T.; Chen, J.; Luan, M. Transcriptome Analysis and HPLC Profiling of Flavonoid Biosynthesis in Citrus aurantium L. during Its Key Developmental Stages. Biology 2022, 11, 1078. https://doi.org/10.3390/biology11071078
Chen J, Shi Y, Zhong Y, Sun Z, Niu J, Wang Y, Chen T, Chen J, Luan M. Transcriptome Analysis and HPLC Profiling of Flavonoid Biosynthesis in Citrus aurantium L. during Its Key Developmental Stages. Biology. 2022; 11(7):1078. https://doi.org/10.3390/biology11071078
Chicago/Turabian StyleChen, Jing, Yaliang Shi, Yicheng Zhong, Zhimin Sun, Juan Niu, Yue Wang, Tianxin Chen, Jianhua Chen, and Mingbao Luan. 2022. "Transcriptome Analysis and HPLC Profiling of Flavonoid Biosynthesis in Citrus aurantium L. during Its Key Developmental Stages" Biology 11, no. 7: 1078. https://doi.org/10.3390/biology11071078
APA StyleChen, J., Shi, Y., Zhong, Y., Sun, Z., Niu, J., Wang, Y., Chen, T., Chen, J., & Luan, M. (2022). Transcriptome Analysis and HPLC Profiling of Flavonoid Biosynthesis in Citrus aurantium L. during Its Key Developmental Stages. Biology, 11(7), 1078. https://doi.org/10.3390/biology11071078