
Giant Multinucleated Cells in Aging and Senescence—An Abridgement

 ,
, 
 ,
,  and
and 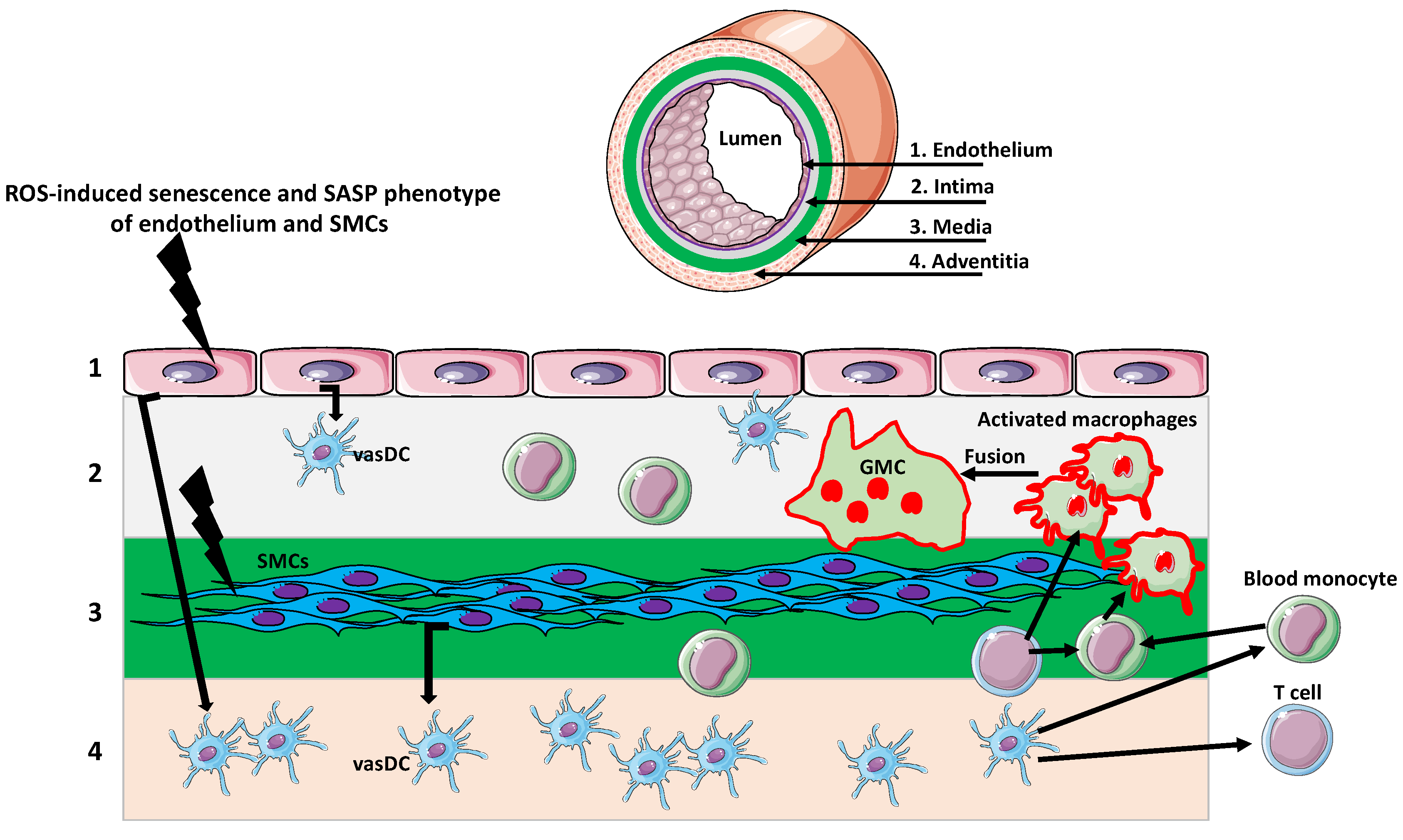{kind=link}
{kind=link}
Abstract
:Simple Summary
Abstract
1. Introduction
Aging Versus Senescence
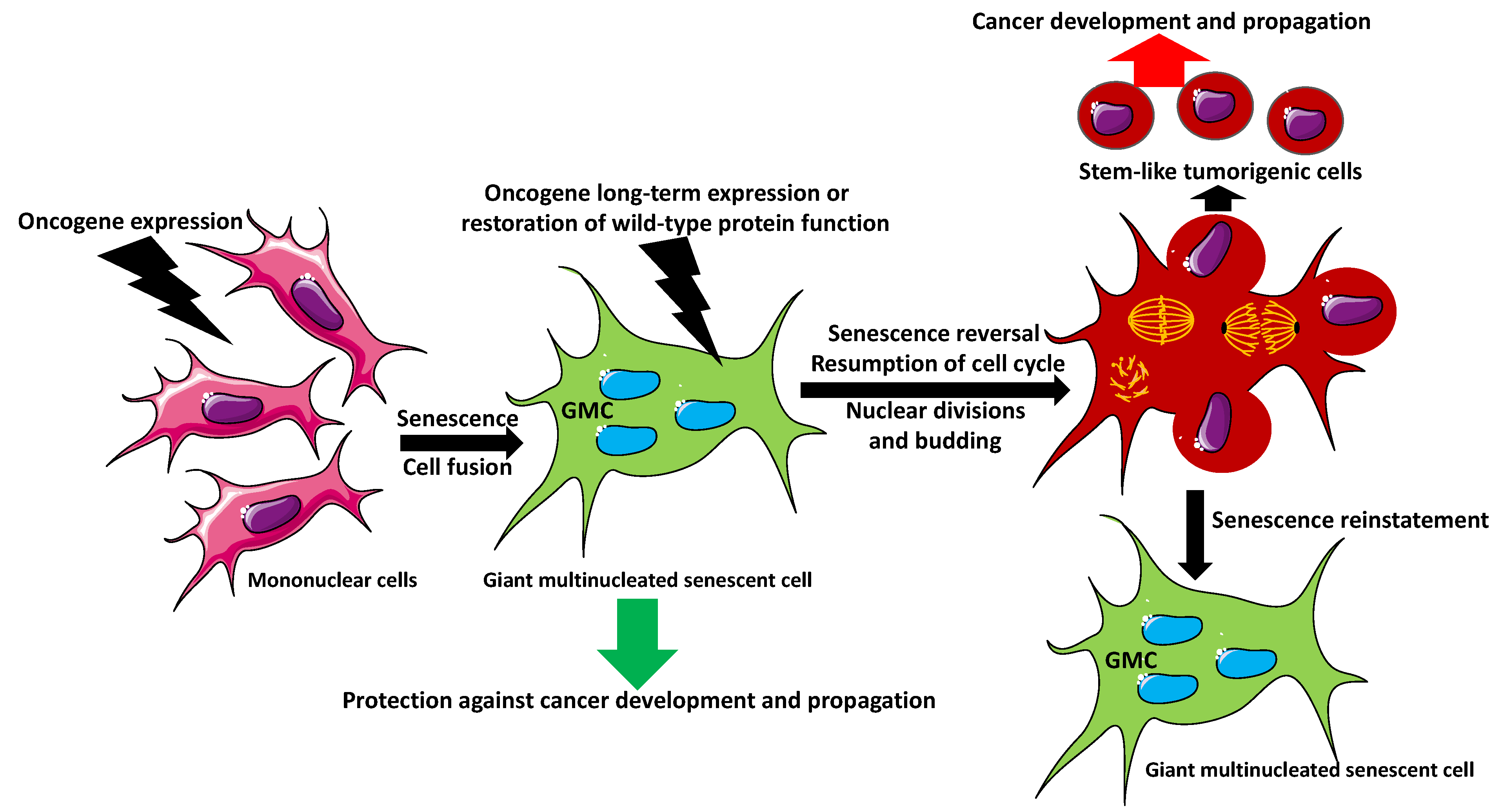
2. Giant Multinucleated Cell Formation
3. Giant Cells in Aging Arteries
4. Giant Multinucleated Cells in Aging Gonads
5. Can Senescent Multinucleated Giant Cells Initiate Cancer?
6. Multinucleation in Cancer
7. Molecular Promoters and Repressors of Aging and Growth—A Lesson from a Worm and Fly
8. Conclusions
Author Contributions
Funding
Institutional Review Board Statement
Informed Consent Statement
Data Availability Statement
Acknowledgments
Conflicts of Interest
Abbreviations
| AKT | serine/threonine-specific protein kinases family |
| Bcl-2 | B-cell lymphoma-2 protein |
| cGAS | DNA-sensing receptor cyclic GMP–AMP synthase |
| DAF-2 | the insulin-like growth factor 1 receptor |
| DAF-16/FOXO | forkhead box transcription factor O |
| DC | dendritic cell |
| DDR | DNA-damage response pathway |
| dNTP | deoxyribonucleotide triphosphate |
| E2F | E2 family transcription factor |
| ECM | extracellular matrix |
| FOXO | forkhead box transcription factor O |
| GCA | giant cell arteritis |
| GMC | giant multinucleated cell |
| GM-CSF | granulocyte-macrophage colony stimulating factor |
| GRO-a | growth-regulated protein alpha |
| Hsp70 | heat shock protein 70 |
| ICAM-1 | intercellular adhesion molecule 1 |
| IFN γ | interferon γ |
| IL-1a | interleukin 1 alpha (IL-1α) also known as hematopoietin 1 |
| IL-6 | interleukin 6 |
| IL-8 | interleukin 8, a chemoattractant cytokine |
| IL-15 | interleukin 15 |
| IGF | insulin-like growth factor |
| MIP-1a | macrophage inflammatory protein-1 alpha (MIP-1 alpha)/CCL3 |
| OXPHOS | oxidative phosphorylation pathway |
| pRB | retinoblastoma protein |
| PI 3-kinase | phosphatidylinositol 3-kinase |
| p16Ink4a/RB | cyclin-dependent kinase inhibitor/retinoblastoma tumor suppressor pathway |
| p53/p21CIP1 | tumor protein p53/cyclin-dependent kinase inhibitor 1 |
| RAS | rat sarcoma virus small GTPase |
| HRAS | Harvey rat sarcoma virus or transforming protein p21, small GTPase |
| ROS | reactive oxygen species |
| RRM2 | ribonucleotide reductase regulatory subunit M2 |
| SA-βgal | senescence-associated beta-galactosidase |
| SAHFs | senescence-associated heterochromatin foci |
| SASP | senescence-associated secretory phenotype |
| STING | stimulator of interferon genes |
| vasDC | vascular dendritic cells |
| VEGF | vascular endothelial growth factor |
References
- Terzi, M.Y.; Izmirli, M.; Gogebakan, B. The cell fate: Senescence or quiescence. Mol. Biol. Rep. 2016, 43, 1213–1220. [Google Scholar] [CrossRef] [PubMed]
- Hayflick, L.; Moorhead, P.S. The serial cultivation of human diploid cell strains. Exp. Cell Res. 1961, 25, 585–621. [Google Scholar] [CrossRef]
- Allsopp, R.C. Models of initiation of replicative senescence by loss of telomeric DNA. Exp. Gerontol. 1996, 31, 235–243. [Google Scholar] [CrossRef]
- Kuilman, T.; Michaloglou, C.; Mooi, W.J.; Peeper, D.S. The essence of senescence. Genes Dev. 2010, 24, 2463–2479. [Google Scholar] [CrossRef] [Green Version]
- Zhang, R.; Chen, W.; Adams, P.D. Molecular dissection of formation of senescence-associated heterochromatin foci. Mol. Cell Biol. 2007, 27, 2343–2358. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Mooi, W.J.; Peeper, D.S. Oncogene-induced cell senescence-halting on the road to cancer. N. Engl. J. Med. 2006, 355, 1037–1046. [Google Scholar] [CrossRef] [PubMed]
- Kohli, J.; Veenstra, I.; Demaria, M. The struggle of a good friend getting old: Cellular senescence in viral responses and therapy. EMBO Rep. 2021, 22, e52243. [Google Scholar] [CrossRef]
- Wei, W.; Ji, S. Cellular senescence: Molecular mechanisms and pathogenicity. J. Cell Physiol. 2018, 233, 9121–9135. [Google Scholar] [CrossRef]
- Muñoz-Espín, D.; Cañamero, M.; Maraver, A.; Gómez-López, G.; Contreras, J.; Murillo-Cuesta, S.; Rodríguez-Baeza, A.; Varela-Nieto, I.; Ruberte, J.; Collado, M.; et al. Programmed cell senescence during mammalian embryonic development. Cell 2013, 155, 1104–1118. [Google Scholar] [CrossRef] [Green Version]
- Storer, M.; Mas, A.; Robert-Moreno, A.; Pecoraro, M.; Ortells, M.C.; Di Giacomo, V.; Yosef, R.; Pilpel, N.; Krizhanovsky, V.; Sharpe, J.; et al. Senescence is a developmental mechanism that contributes to embryonic growth and patterning. Cell 2013, 155, 1119–1130. [Google Scholar] [CrossRef] [Green Version]
- Yang, Y.; Shi, L.; Fu, X.; Ma, G.; Yang, Z.; Li, Y.; Zhou, Y.; Yuan, L.; Xia, Y.; Zhong, X.; et al. Metabolic and epigenetic dysfunctions underlie the arrest of in vitro fertilized human embryos in a senescent-like state. PLoS Biol. 2022, 20, e3001682. [Google Scholar] [CrossRef] [PubMed]
- Calcinotto, A.; Kohli, J.; Zagato, E.; Pellegrini, L.; Demaria, M.; Alimonti, A. Cellular Senescence: Aging, Cancer, and Injury. Physiol. Rev. 2019, 99, 1047–1078. [Google Scholar] [CrossRef] [PubMed]
- Chakradeo, S.; Elmore, L.W.; Gewirtz, D.A. Is Senescence Reversible? Curr. Drug Targets 2016, 17, 460–466. [Google Scholar] [CrossRef] [PubMed]
- Beauséjour, C.M.; Krtolica, A.; Galimi, F.; Narita, M.; Lowe, S.W.; Yaswen, P.; Campisi, J. Reversal of human cellular senescence: Roles of the p53 and p16 pathways. EMBO J. 2003, 22, 4212–4222. [Google Scholar] [CrossRef]
- Kumari, R.; Jat, P. Mechanisms of Cellular Senescence: Cell Cycle Arrest and Senescence Associated Secretory Phenotype. Front Cell Dev. Biol. 2021, 9, 645593. [Google Scholar] [CrossRef]
- McHugh, D.; Gil, J. Senescence and aging: Causes, consequences, and therapeutic avenues. J. Cell Biol. 2018, 217, 65–77. [Google Scholar] [CrossRef]
- Birch, J.; Gil, J. Senescence and the SASP: Many therapeutic avenues. Genes Dev. 2020, 34, 156–1576. [Google Scholar] [CrossRef]
- Cuollo, L.; Antonangeli, F.; Santoni, A.; Soriani, A. The Senescence-Associated Secretory Phenotype (SASP) in the Challenging Future of Cancer Therapy and Age-Related Diseases. Biology 2020, 9, 485. [Google Scholar] [CrossRef]
- Lopes-Paciencia, S.; Saint-Germain, E.; Rowell, M.C.; Ruiz, A.F.; Kalegari, P.; Ferbeyre, G. The senescence-associated secretory phenotype and its regulation. Cytokine 2019, 117, 15–22. [Google Scholar] [CrossRef]
- Coppé, J.P.; Desprez, P.Y.; Krtolica, A.; Campisi, J. The senescence-associated secretory phenotype: The dark side of tumor suppression. Annu. Rev. Pathol. 2010, 5, 99–118. [Google Scholar] [CrossRef] [Green Version]
- Neurohr, G.E.; Terry, R.L.; Lengefeld, J.; Bonney, M.; Brittingham, G.P.; Moretto, F.; Miettinen, T.P.; Vaites, L.P.; Soares, L.M.; Paulo, J.A.; et al. Excessive cell growth causes cytoplasm dilution and contributes to senescence. Cell 2019, 176, 1083–1097.e18. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- González-Gualda, E.; Baker, A.G.; Fruk, L.; Muñoz-Espín, D. A guide to assessing cellular senescence in vitro and in vivo. FEBS J. 2021, 288, 56–80. [Google Scholar] [CrossRef] [PubMed]
- Dikovskaya, D.; Cole, J.J.; Mason, S.M.; Nixon, C.; Karim, S.A.; McGarry, L.; Clark, W.; Hewitt, R.N.; Sammons, M.A.; Zhu, J.; et al. Mitotic stress is an integral part of the oncogene-induced senescence program that promotes multinucleation and cell cycle arrest. Cell Rep. 2015, 12, 1483–1496. [Google Scholar] [CrossRef] [Green Version]
- Radspieler, M.M.; Schindeldecker, M.; Stenzel, P.; Försch, S.; Tagscherer, K.E.; Herpel, E.; Hohenfellner, M.; Hatiboglu, G.; Roth, W.; Macher-Goeppinger, S. Lamin-B1 is a senescence-associated biomarker in clear-cell renal cell carcinoma. Oncol. Lett. 2019, 18, 2654–2660. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Vergnes, L.; Péterfy, M.; Bergo, M.O.; Young, S.G.; Reue, K. Lamin B1 is required for mouse development and nuclear integrity. Proc. Natl. Acad. Sci. USA 2004, 101, 10428–10433. [Google Scholar] [CrossRef] [Green Version]
- Narita, M.; Nũnez, S.; Heard, E.; Narita, M.; Lin, A.W.; Hearn, S.A.; Spector, D.L.; Hannon, G.J.; Lowe, S.W. Rb-mediated heterochromatin formation and silencing of E2F target genes during cellular senescence. Cell 2003, 113, 703–716. [Google Scholar] [CrossRef] [Green Version]
- De Oliveira Mann, C.C.; Kranzusch, P.J. cGAS conducts micronuclei DNA surveillance. Trends Cell Biol. 2017, 27, 697–698. [Google Scholar] [CrossRef] [PubMed]
- Motwani, M.; Pesiridis, S.; Fitzgerald, K.A. DNA sensing by the cGAS-STING pathway in health and disease. Nat. Rev. Genet. 2019, 20, 657–674. [Google Scholar] [CrossRef]
- Vasileiou, P.V.S.; Evangelou, K.; Vlasis, K.; Fildisis, G.; Panayiotidis, M.I.; Chronopoulos, E.; Passias, P.G.; Kouloukoussa, M.; Gorgoulis, V.G.; Havaki, S. Mitochondrial Homeostasis and Cellular Senescence. Cells 2019, 8, 686. [Google Scholar] [CrossRef] [Green Version]
- Victorelli, S.; Passos, J.F. Reactive Oxygen Species Detection in Senescent Cells. Methods Mol. Biol. 2019, 1896, 21–29. [Google Scholar] [CrossRef]
- Passos, J.F.; Nelson, G.; Wang, C.; Richter, T.; Simillion, C.; Proctor, C.J.; Miwa, S.; Olijslagers, S.; Hallinan, J.; Wipat, A.; et al. Feedback between p21 and reactive oxygen production is necessary for cell senescence. Mol. Syst. Biol. 2010, 6, 347. [Google Scholar] [CrossRef]
- Cory, S.; Huang, D.C.S.; Adams, J.M. The Bcl-2 family: Roles in cell survival and oncogenesis. Oncogene 2003, 22, 8590–8607. [Google Scholar] [CrossRef] [Green Version]
- Yosef, R.; Pilpel, N.; Tokarsky-Amiel, R.; Biran, A.; Ovadya, Y.; Cohen, S.; Vadai, E.; Dassa, L.; Shahar, E.; Condiotti, R.; et al. Directed elimination of senescent cells by inhibition of BCL-W and BCL-XL. Nat. Commun. 2016, 7, 11190. [Google Scholar] [CrossRef]
- Helming, L.; Gordon, S. Molecular mediators of macrophage fusion. Trends Cell Biol. 2009, 19, 514–522. [Google Scholar] [CrossRef]
- Kloc, M.; Subuddhi, A.; Uosef, A.; Kubiak, J.Z.; Ghobrial, R.M. Monocyte-Macrophage Lineage Cell Fusion. Int. J. Mol. Sci. 2022, 23, 6553. [Google Scholar] [CrossRef]
- Franceschi, C.; Garagnani, P.; Parini, P.; Giuliani, C.; Santoro, A. Inflammaging: A new immune-metabolic viewpoint for age-related diseases. Nat. Rev. Endocrinol. 2018, 14, 576–590. [Google Scholar] [CrossRef] [PubMed]
- Watanabe, R.; Hashimoto, M. Aging-Related Vascular Inflammation: Giant Cell Arteritis and Neurological Disorders. Front Aging Neurosci. 2022, 14, 843305. [Google Scholar] [CrossRef]
- Mohan, S.V.; Liao, Y.J.; Kim, J.W.; Goronzy, J.J.; Weyand, C.M. Giant cell arteritis: Immune and vascular aging as disease risk factors. Arthritis Res. Ther. 2011, 13, 231. [Google Scholar] [CrossRef] [Green Version]
- Ungvari, Z.; Tarantini, S.; Donato, A.J.; Galvan, V.; Csiszar, A. Mechanisms of Vascular Aging. Circ. Res. 2018, 123, 849–867. [Google Scholar] [CrossRef]
- Tyrrell, D.J.; Blin, M.G.; Song, J.; Wood, S.C.; Zhang, M.; Beard, D.A.; Goldstein, D.R. Age-Associated Mitochondrial Dysfunction Accelerates Atherogenesis. Circ. Res. 2020, 126, 298–314. [Google Scholar] [CrossRef]
- Kloc, M.; Ghobrial, R.M. Chronic allograft rejection: A significant hurdle to transplant success. Burns Trauma. 2014, 2, 3–10. [Google Scholar] [CrossRef] [Green Version]
- Weyand, C.M.; Liao, Y.J.; Goronzy, J.J. The immunopathology of giant cell arteritis: Diagnostic and therapeutic implications. J. Neuroophthalmol. 2012, 32, 259–265. [Google Scholar] [CrossRef] [Green Version]
- Jiemy, W.F.; van Sleen, Y.; van der Geest, K.S.; Ten Berge, H.A.; Abdulahad, W.H.; Sandovici, M.; Boots, A.M.; Heeringa, P.; Brouwer, E. Distinct macrophage phenotypes skewed by local granulocyte macrophage colony-stimulating factor (GM-CSF) and macrophage colony-stimulating factor (M-CSF) are associated with tissue destruction and intimal hyperplasia in giant cell arteritis. Clin. Transl. Immunol. 2020, 9, e1164. [Google Scholar] [CrossRef] [PubMed]
- Briley, S.M.; Jasti, S.; McCracken, J.M.; Hornick, J.E.; Fegley, B.; Pritchard, M.T.; Duncan, F.E. Reproductive age-associated fibrosis in the stroma of the mammalian ovary. Reproduction 2016, 152, 245–260. [Google Scholar] [CrossRef] [Green Version]
- Foley, K.G.; Pritchard, M.T.; Duncan, F.E. Macrophage-derived multinucleated giant cells: Hallmarks of the aging ovary. Reproduction 2021, 161, V5–V9. [Google Scholar] [CrossRef]
- Aird, K.M.; Zhang, G.; Li, H.; Tu, Z.; Bitler, B.G.; Garipov, A.; Wu, H.; Wei, Z.; Wagner, S.N.; Herlyn, M.; et al. Suppression of nucleotide metabolism underlies the establishment and maintenance of oncogene-induced senescence. Cell Rep. 2013, 3, 1252–1265. [Google Scholar] [CrossRef] [Green Version]
- Leikam, C.; Hufnagel, A.L.; Otto, C.; Murphy, D.J.; Mühling, B.; Kneitz, S.; Nanda, I.; Schmid, M.; Wagner, T.U.; Haferkamp, S.; et al. In vitro evidence for senescent multinucleated melanocytes as a source for tumor-initiating cells. Cell Death Dis. 2015, 6, e1711. [Google Scholar] [CrossRef] [Green Version]
- Rajaraman, R.; Rajaraman, M.M.; Rajaraman, S.R.; Guernsey, D.L. Neosis—A paradigm of self-renewal in cancer. Cell Biol. Int. 2005, 29, 1084–1097. [Google Scholar] [CrossRef]
- Zhang, S.; Mercado-Uribe, I.; Xing, Z.; Sun, B.; Kuang, J.; Liu, J. Generation of cancer stem-like cells through the formation of polyploid giant cancer cells. Oncogene 2013, 33, 116–128. [Google Scholar] [CrossRef]
- Wang, H.F.; Xiang, W.; Xue, B.Z.; Wang, Y.H.; Yi, D.Y.; Jiang, X.B.; Zhao, H.Y.; Fu, P. Cell fusion in cancer hallmarks: Current research status and future indications. Oncol Lett. 2021, 22, 530. [Google Scholar] [CrossRef]
- Ariizumi, T.; Ogose, A.; Kawashima, H.; Hotta, T.; Umezu, H.; Endo, N. Multinucleation followed by an acytokinetic cell division in myxofibrosarcoma with giant cell proliferation. J. Exp. Clin. Cancer Res. 2009, 28, 44. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Herbein, G.; Nehme, Z. Polyploid Giant Cancer Cells, a Hallmark of Oncoviruses and a New Therapeutic Challenge. Front Oncol. 2020, 10, 567116. [Google Scholar] [CrossRef]
- Munzarova, M.; Lauerova, L.; Capkova, J. Are advanced malignant melanoma cells hybrids between melanocytes and macrophages? Melanoma Res. 1992, 2, 127–129. [Google Scholar] [CrossRef] [PubMed]
- Díaz-Carballo, D.; Saka, S.; Klein, J.; Rennkamp, T.; Acikelli, A.H.; Malak, S.; Jastrow, H.; Wennemuth, G.; Tempfer, C.; Schmitz, I.; et al. A Distinct Oncogenerative Multinucleated Cancer Cell Serves as a Source of Stemness and Tumor Heterogeneity. Cancer Res. 2018, 78, 2318–2331. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Wilson, J.D.; Roehrborn, C. Long-term consequences of castration in men: Lessons from the Skoptzy and the eunuchs of the Chinese and Ottoman courts. J. Clin. Endocrinol. Metab. 1999, 84, 4324–4331. [Google Scholar] [CrossRef]
- Patel, M.N.; Knight, C.G.; Karageorgi, C.; Leroi, A.M. Evolution of germ-line signals that regulate growth and aging in nematodes. Proc. Natl. Acad. Sci. USA 2002, 99, 769–774. [Google Scholar] [CrossRef] [Green Version]
- Hsin, H.; Kenyon, C. Signals from the reproductive system regulate the lifespan of C. elegans. Nature 1999, 399, 362–366. [Google Scholar] [CrossRef]
- Tatar, M.; Bartke, A.; Antebi, A. The endocrine regulation of aging by insulin-like signals. Science 2003, 299, 1346–1351. [Google Scholar] [CrossRef]
- Holzenberger, M.; Dupont, J.; Ducos, B.; Leneuve, P.; Géloën, A.; Even, P.C.; Cervera, P.; Le Bouc, Y. IGF-1 receptor regulates lifespan and resistance to oxidative stress in mice. Nature 2003, 421, 182–187. [Google Scholar] [CrossRef]
- Bluher, M.; Kahn, B.B.; Kahn, C.R. Extended longevity in mice lacking the insulin receptor in adipose tissue. Science 2003, 299, 572–574. [Google Scholar] [PubMed] [Green Version]
- Gems, D.; Partridge, L. Insulin/IGF signalling and ageing: Seeing the bigger picture. Curr. Opin. Genet. Dev. 2001, 11, 287–292. [Google Scholar] [CrossRef]
- Kirkwood, T.B. Genes that shape the course of ageing. Trends Endocrinol. Metab. 2003, 14, 345–347. [Google Scholar] [CrossRef] [PubMed]
- Lin, K.; Hsin, H.; Libina, N.; Kenyon, C. Regulation of the Caenorhabditis elegans longevity protein DAF-16 by insulin/IGF-1 and germline signaling. Nat. Genet. 2001, 28, 139–145. [Google Scholar] [CrossRef] [PubMed]
- Murphy, C.T.; McCarroll, S.A.; Bargmann, C.I.; Fraser, A.; Kamath, R.S.; Ahringer, J.; Li, H.; Kenyon, C. Genes that act downstream of DAF-16 to influence the lifespan of Caenorhabditis elegans. Nature 2003, 424, 277–284. [Google Scholar] [CrossRef]
- Clancy, D.J.; Gems, D.; Harshman, L.G.; Oldham, S.; Stocker, H.; Hafen, E.; Leevers, S.J.; Partridge, L. Extension of life-span by loss of CHICO, a Drosophila insulin receptor substrate protein. Science 2001, 292, 104–106. [Google Scholar] [CrossRef] [Green Version]
- Taormina, G.; Ferrante, F.; Vieni, S.; Grassi, N.; Russo, A.; Mirisola, M.G. Longevity: Lesson from Model Organisms. Genes 2019, 10, 518. [Google Scholar] [CrossRef] [Green Version]
- Piper, M.D.W.; Partridge, L. Drosophila as a model for ageing. Biochim. Biophys. Acta Mol. Basis. Dis. 2018, 1864 Pt A, 2707–2717. [Google Scholar] [CrossRef]
- Anisimov, V.N.; Bartke, A. The key role of growth hormone-insulin-IGF-1 signaling in aging and cancer. Crit. Rev. Oncol. Hematol. 2013, 87, 201–223. [Google Scholar] [CrossRef] [Green Version]
- Xu, H.; Barnes, G.T.; Yang, Q.; Tan, G.; Yang, D.; Chou, C.J.; Sole, J.; Nichols, A.; Ross, J.S.; Tartaglia, L.A.; et al. Chronic inflammation in fat plays a crucial role in the development of obesity-related insulin resistance. J. Clin. Investig. 2003, 112, 1821–1830. [Google Scholar] [CrossRef]
- Allen, E.A.; Baehrecke, E.H. Autophagy in animal development. Cell Death Differ. 2020, 27, 903–918. [Google Scholar] [CrossRef]
- Das, S.; Shukla, N.; Singh, S.S.; Kushwaha, S.; Shrivastava, R. Mechanism of interaction between autophagy and apoptosis in cancer. Apoptosis 2021, 26, 512–533. [Google Scholar] [CrossRef]
- Arsham, A.M.; Neufeld, T.P. A genetic screen in Drosophila reveals novel cytoprotective functions of the autophagy-lysosome pathway. PLoS ONE 2009, 4, e6068. [Google Scholar] [CrossRef] [PubMed]
- Kakanj, P.; Bhide, S.; Moussian, B.; Leptin, M. Autophagy-mediated plasma membrane removal promotes the formation of epithelial syncytia. EMBO J. 2022, 2, 401–412. [Google Scholar] [CrossRef] [PubMed]
- Redman, C.W.G.; Staff, A.C.; Roberts, J.M. Syncytiotrophoblast stress in preeclampsia: The convergence point for multiple pathways. Am. J. Obstet. Gynecol. 2020, 226, S907–S927. [Google Scholar] [CrossRef] [PubMed]
- Fortini, P.; Ferretti, C.; Iorio, E.; Cagnin, M.; Garribba, L.; Pietraforte, D.; Falchi, M.; Pascucci, B.; Baccarini, S.; Morani, F.; et al. The fine tuning of metabolism, autophagy and differentiation during in vitro myogenesis. Cell Death Dis. 2016, 7, e2168. [Google Scholar] [CrossRef]
- Tosato, M.; Zamboni, V.; Ferrini, A.; Cesari, M. The aging process and potential interventions to extend life expectancy. Clin. Interv. Aging 2007, 2, 401–412. [Google Scholar]
- Tower, J. Transgenic methods for increasing Drosophila life span. Mech. Age Dev. 2000, 118, 1–14. [Google Scholar] [CrossRef]
- Arking, R.; Burde, V.; Graves, K.; Hari, R.; Feldman, E.; Zeevi, A.; Soliman, S.; Saraiya, A.; Buck, S.; Vettraino, J.; et al. Forward and reverse selection for longevity in Drosophila is characterized by alteration of antioxidant gene expression and oxidative damage patterns. Exp. Gerontol. 2000, 35, 167–185. [Google Scholar] [CrossRef]
- Larsen, P.L. Aging and resistance to oxidative damage in Caenorhabditis elegans. Proc. Natl. Acad. Sci. USA 1993, 90, 8905–8909. [Google Scholar] [CrossRef] [Green Version]
- Melov, S.; Ravenscroft, J.; Malik, S.; Gill, M.S.; Walker, D.W.; Clayton, P.E.; Wallace, D.C.; Malfroy, B.; Doctrow, S.R.; Lithgow, G.J. Extension of lifespan with superoxide dismutase/catalase mimetics. Science 2000, 289, 1567–1569. [Google Scholar] [CrossRef] [Green Version]


Publisher’s Note: MDPI stays neutral with regard to jurisdictional claims in published maps and institutional affiliations. |
© 2022 by the authors. Licensee MDPI, Basel, Switzerland. This article is an open access article distributed under the terms and conditions of the Creative Commons Attribution (CC BY) license (https://creativecommons.org/licenses/by/4.0/).
Share and Cite
Kloc, M.; Uosef, A.; Subuddhi, A.; Kubiak, J.Z.; Piprek, R.P.; Ghobrial, R.M. Giant Multinucleated Cells in Aging and Senescence—An Abridgement. Biology 2022, 11, 1121. https://doi.org/10.3390/biology11081121
Kloc M, Uosef A, Subuddhi A, Kubiak JZ, Piprek RP, Ghobrial RM. Giant Multinucleated Cells in Aging and Senescence—An Abridgement. Biology. 2022; 11(8):1121. https://doi.org/10.3390/biology11081121
Chicago/Turabian StyleKloc, Malgorzata, Ahmed Uosef, Arijita Subuddhi, Jacek Z. Kubiak, Rafal P. Piprek, and Rafik M. Ghobrial. 2022. "Giant Multinucleated Cells in Aging and Senescence—An Abridgement" Biology 11, no. 8: 1121. https://doi.org/10.3390/biology11081121
APA StyleKloc, M., Uosef, A., Subuddhi, A., Kubiak, J. Z., Piprek, R. P., & Ghobrial, R. M. (2022). Giant Multinucleated Cells in Aging and Senescence—An Abridgement. Biology, 11(8), 1121. https://doi.org/10.3390/biology11081121