
Learning Hyperbolic Embedding for Phylogenetic Tree Placement and Updates


Abstract
:Simple Summary
Abstract
1. Introduction
2. Materials and Methods
2.1. Problem Statement
2.2. Euclidean Embedding and Its Limitation
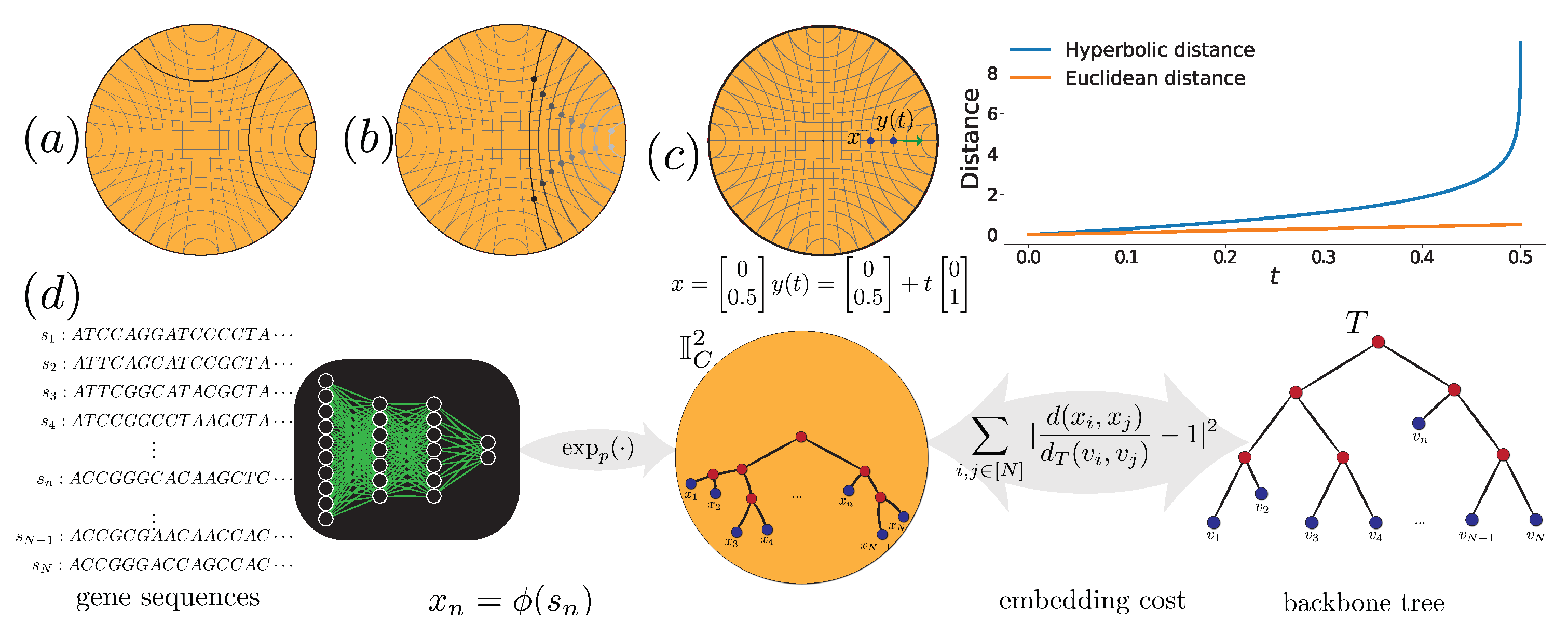
2.3. Embedding in Hyperbolic Spaces
2.3.1. Hyperbolic Spaces in Machine Learning
2.3.2. Models of Hyperbolic Spaces
2.3.3. Hyperbolic Embedding Functions
2.4. H-Depp Design
2.4.1. Loss Function
2.4.2. Neural Network Models
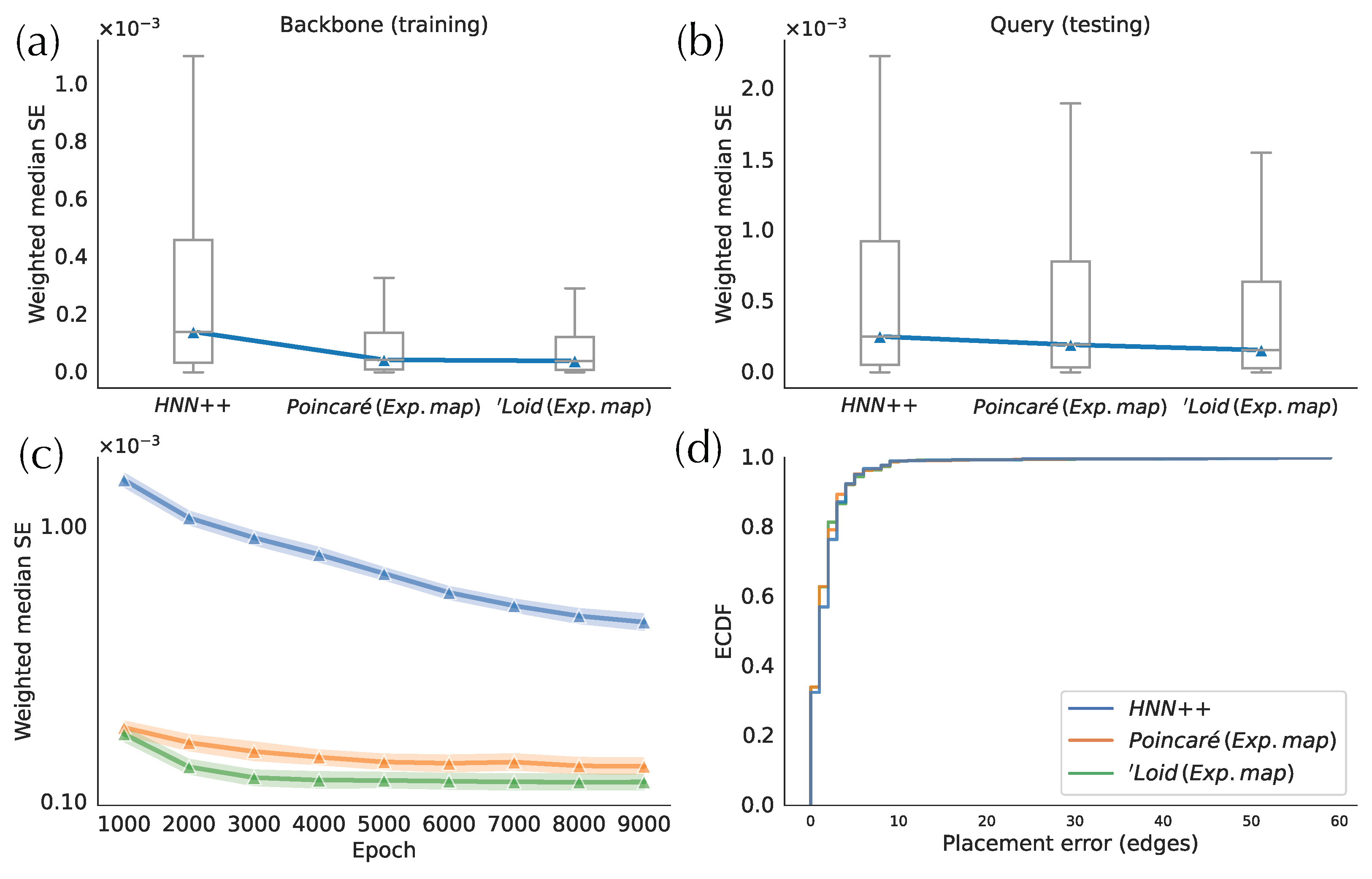
- Exponential Maps: We use a Euclidean neural network followed by the exponential map of the Poincaré ball or the ’Loid model shown in (3) and (4) and detailed in Section 2.3. We use the following cost function:where , is the Euclidean neural network, is the exponential map for the appropriate model of hyperbolic space with curvature (i.e., (3) or (4)), and s is a scale factor further discussed in Section 2.4.3.
- HNN++: We add one hyperbolic layer designed by [13] to the model used in DEPP. This all-by-all layer performs hyperbolic matrix multiplication; its inputs are the output of the previous Euclidean layer transformed by the exponential map, which outputs hyperbolic points used in the cost function (5). Finally, we perform parameter optimization using hyperbolic back-propagation [13].
2.4.3. Training: Challenges and Solutions
2.5. Experimental Evaluation Setup
2.5.1. Datasets
2.5.2. Evaluation Procedure
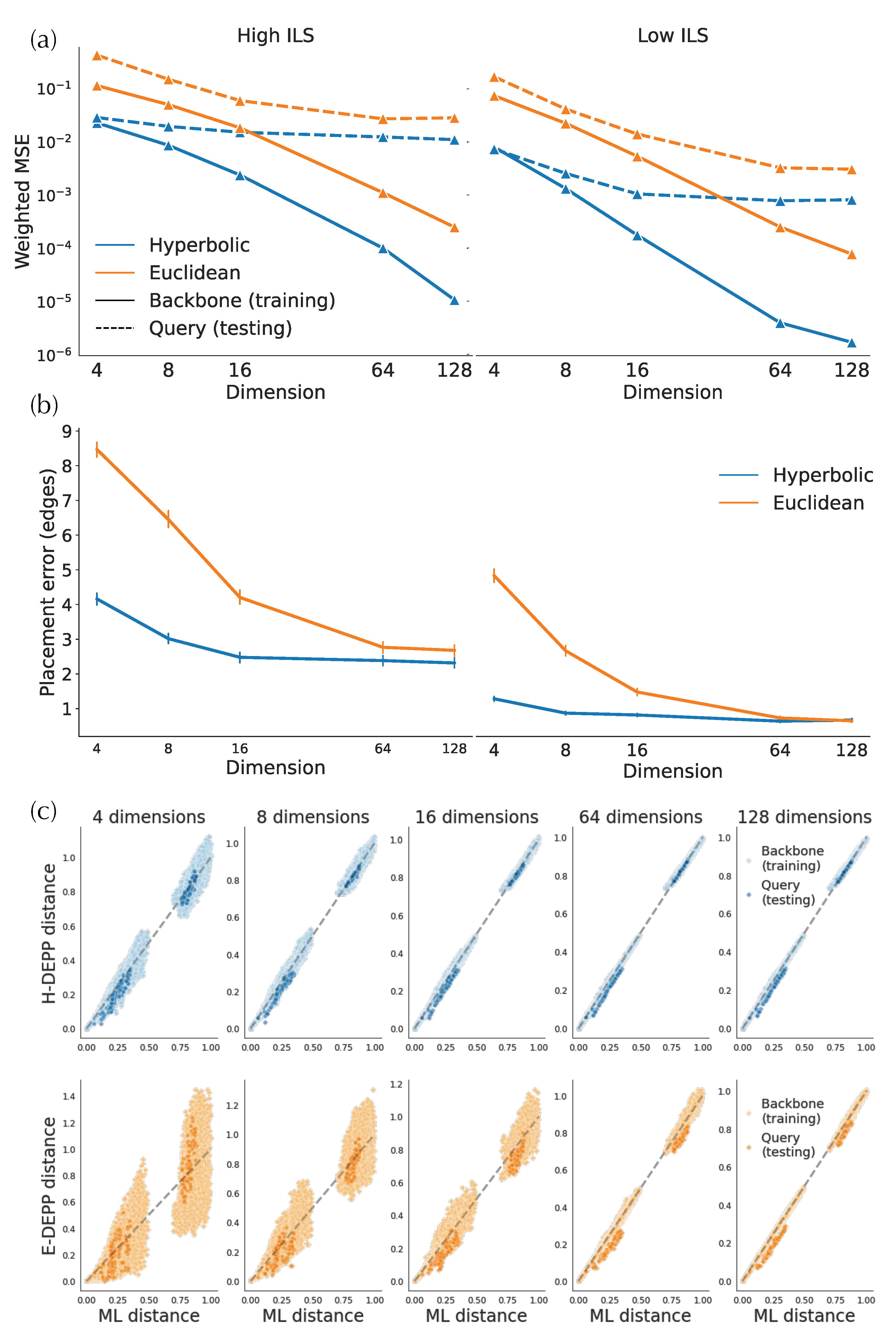
- Euclidean vs. Hyperbolic DEPP (E-DEPP vs. H-DEPP). We use H-DEPP and E-DEPP models, trained on backbone data, to compute the distances between queries and backbone species. We then use the distance matrix as the input of APPLES-2 to find the placement of queries with parameter -b 5, which limits the placement to the five smallest distances. We use 128 dimensions by default.
- APPLES2+Jukes-Cantor (JC). APPLES-2 uses the Jukes and Cantor [29] (JC) model to compute sequence distances by default because Balaban et al. [6] found no evidence of improvement in placement accuracy by using more complex models. We use RAxML-ng to re-estimate the branch lengths of the backbone trees (JC model).
- E-DEPP+FastME and H-DEPP+FastME. We use FastME [32] for tree inference with the distance matrix computed partially using H/E-DEPP. We build a distance matrix that includes the tree distance for each pair of backbone species and the embedding distance computed using the H/E-DEPP model trained on the backbone data for other pairs. We use this approach to nudge FastME to retain backbone relationships because it does not have a constrained search option.
- Jukes-Cantor (JC)+FastME. The pipeline for tree update using JC is similar to H/E-DEPP. We first compute distances between all pairs of species using the JC model and build a distance matrix similarly to the H/D-DEPP model. We then use the distance matrix as the input of FastME.
- RAxML. We run RAxML [33] setting the backbone tree as a constraint (-g) under the GTR+CAT model.
3. Results
3.1. Comparison between H-DEPP Alternatives
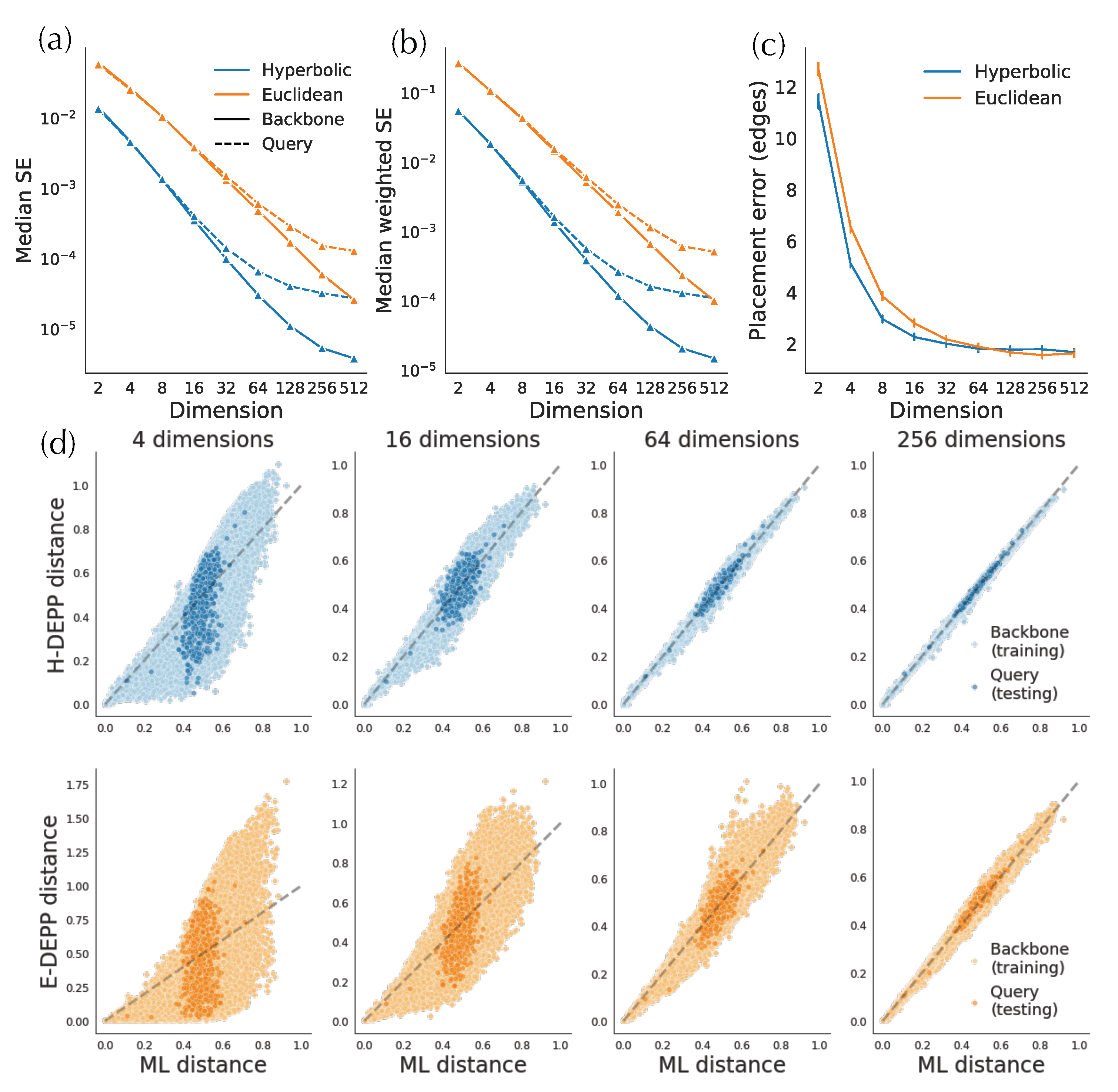
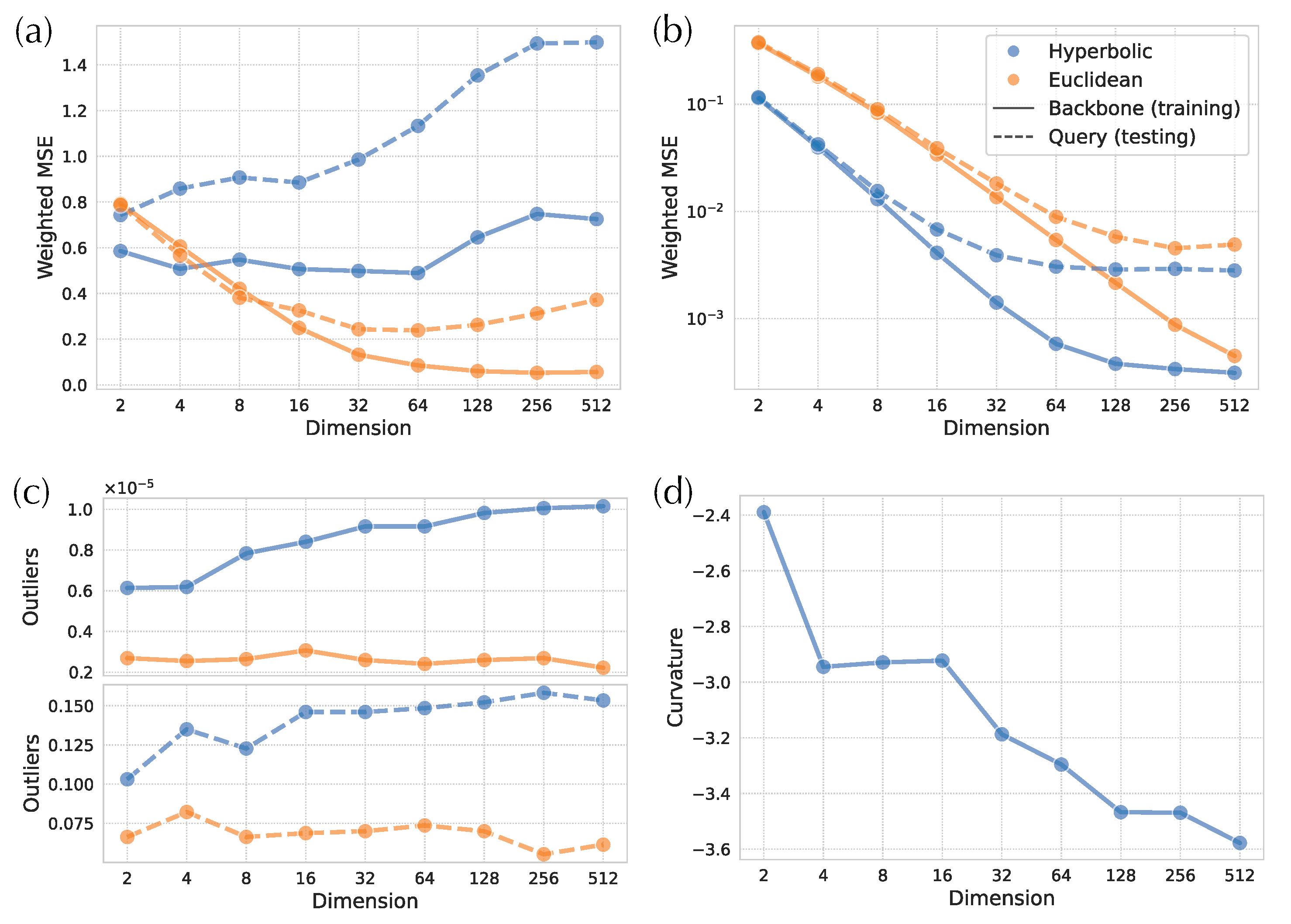
3.2. H-Depp versus E-DEPP with Varying Number of Dimensions
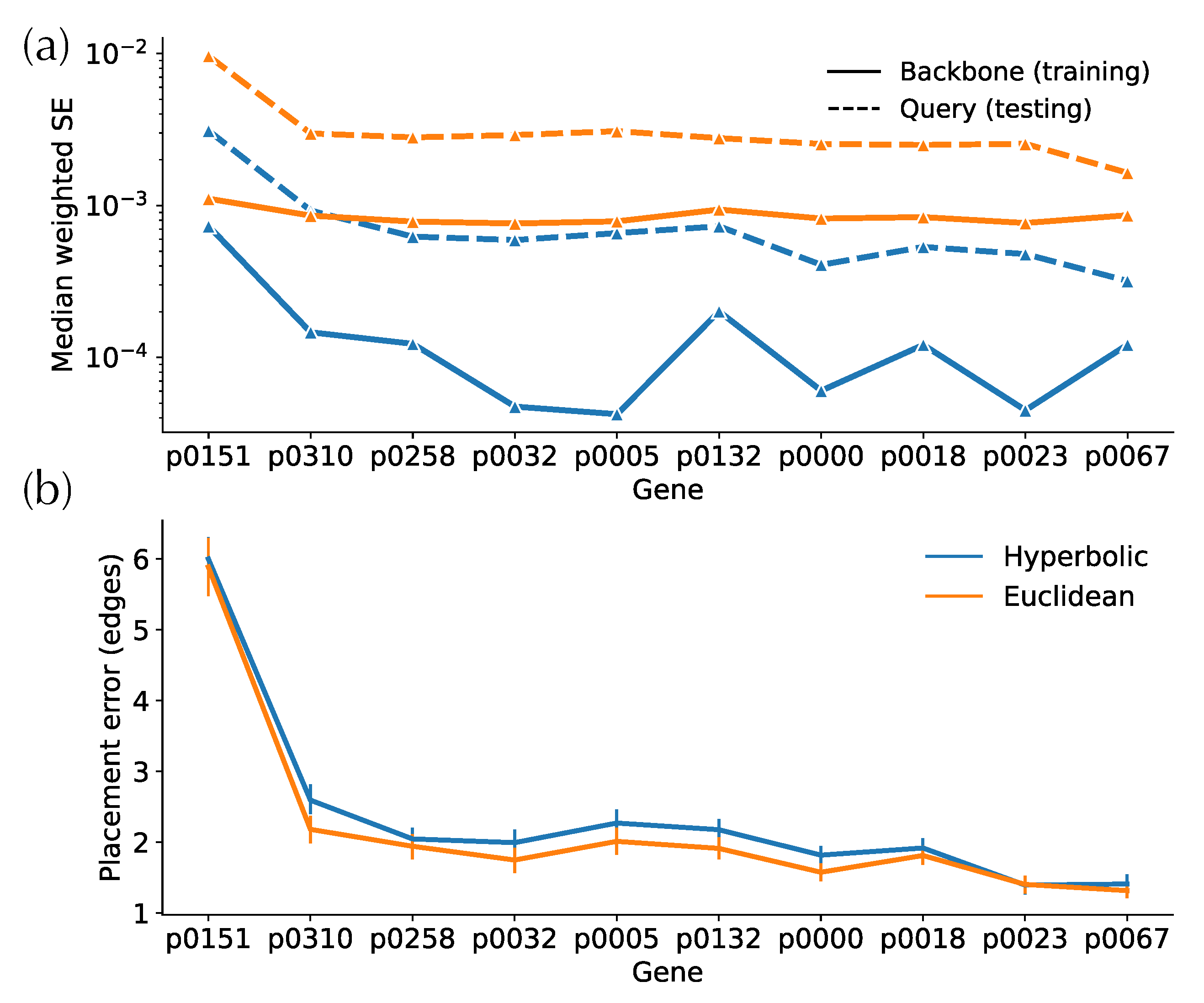
3.2.1. Distortion
3.2.2. Placement Accuracy
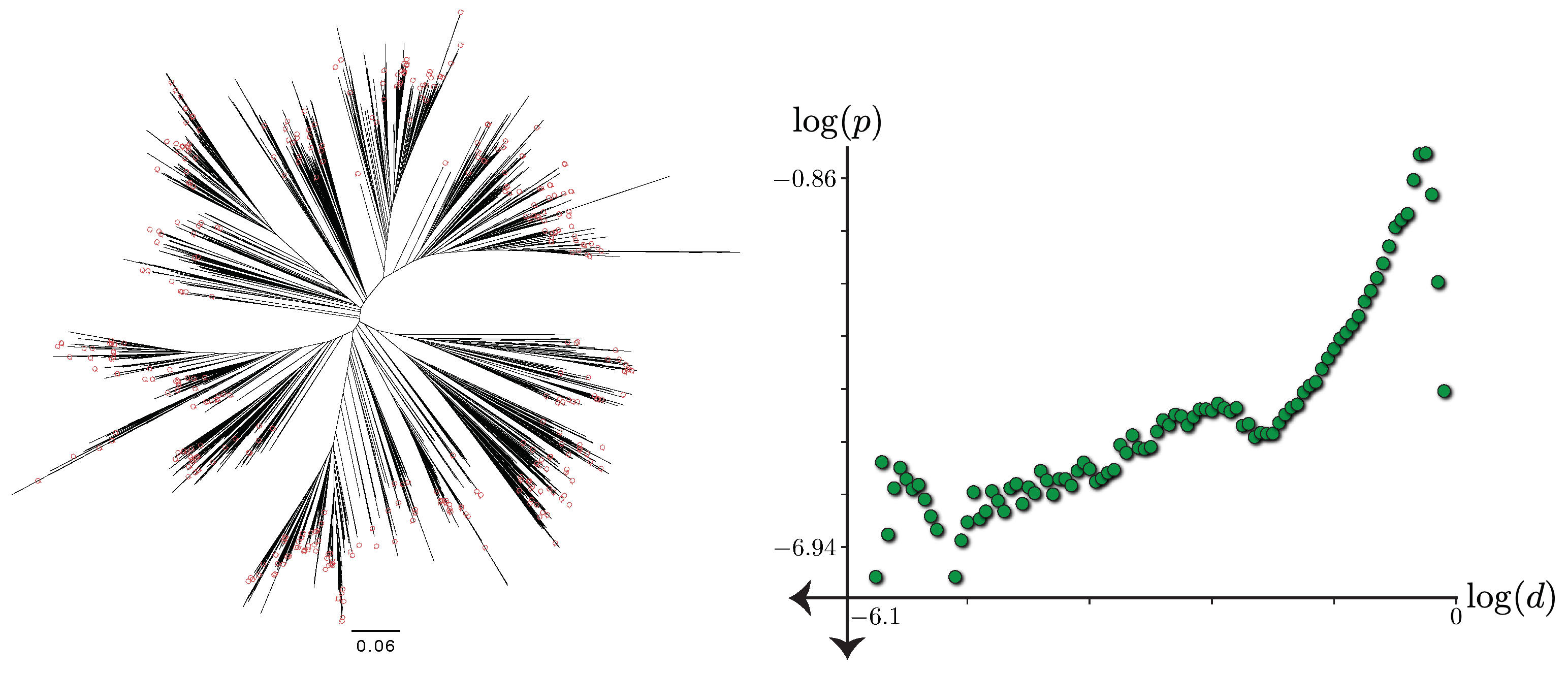
3.2.3. Small Distances and Outliers
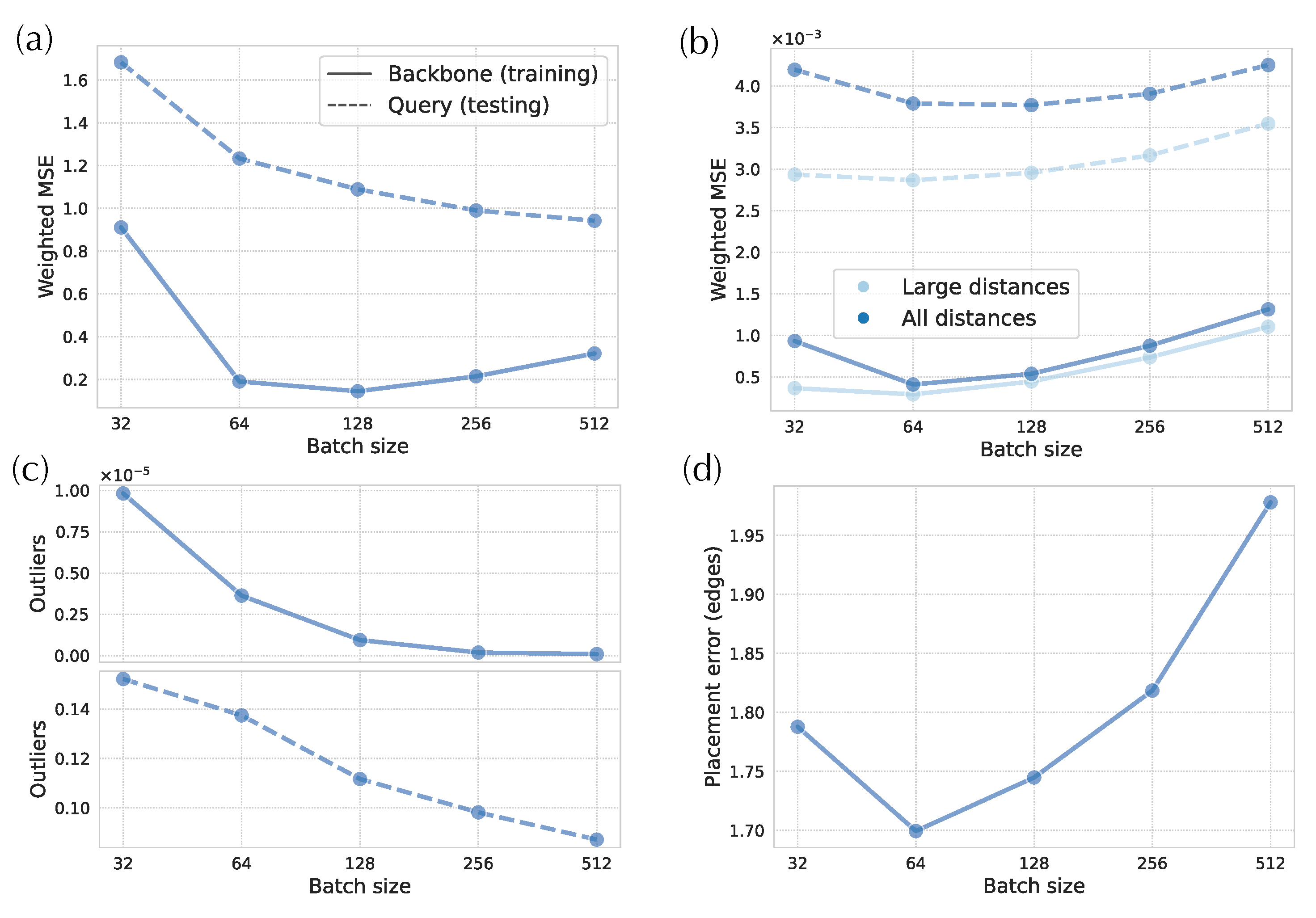
3.3. Impact of Hyper-Parameters
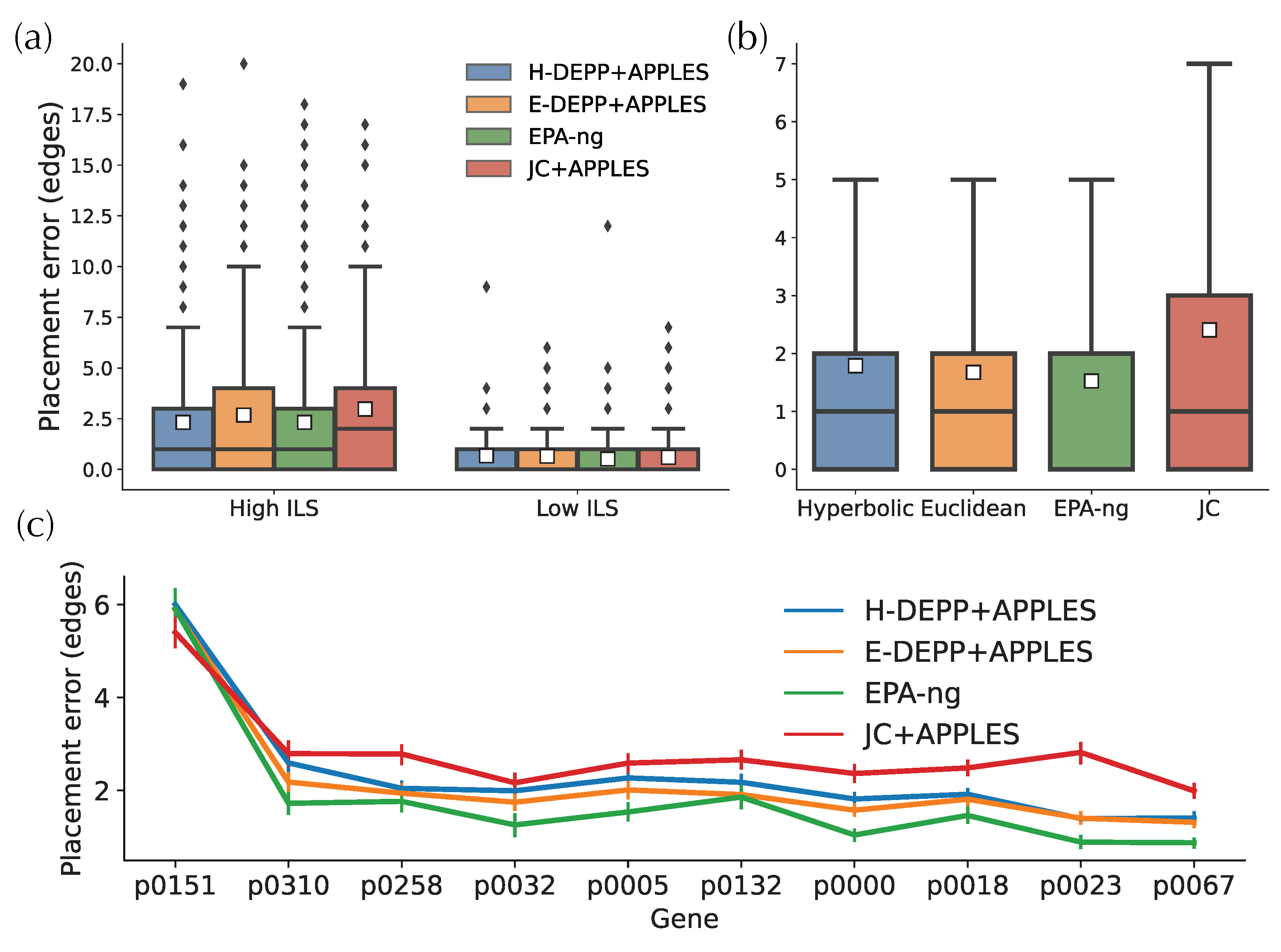
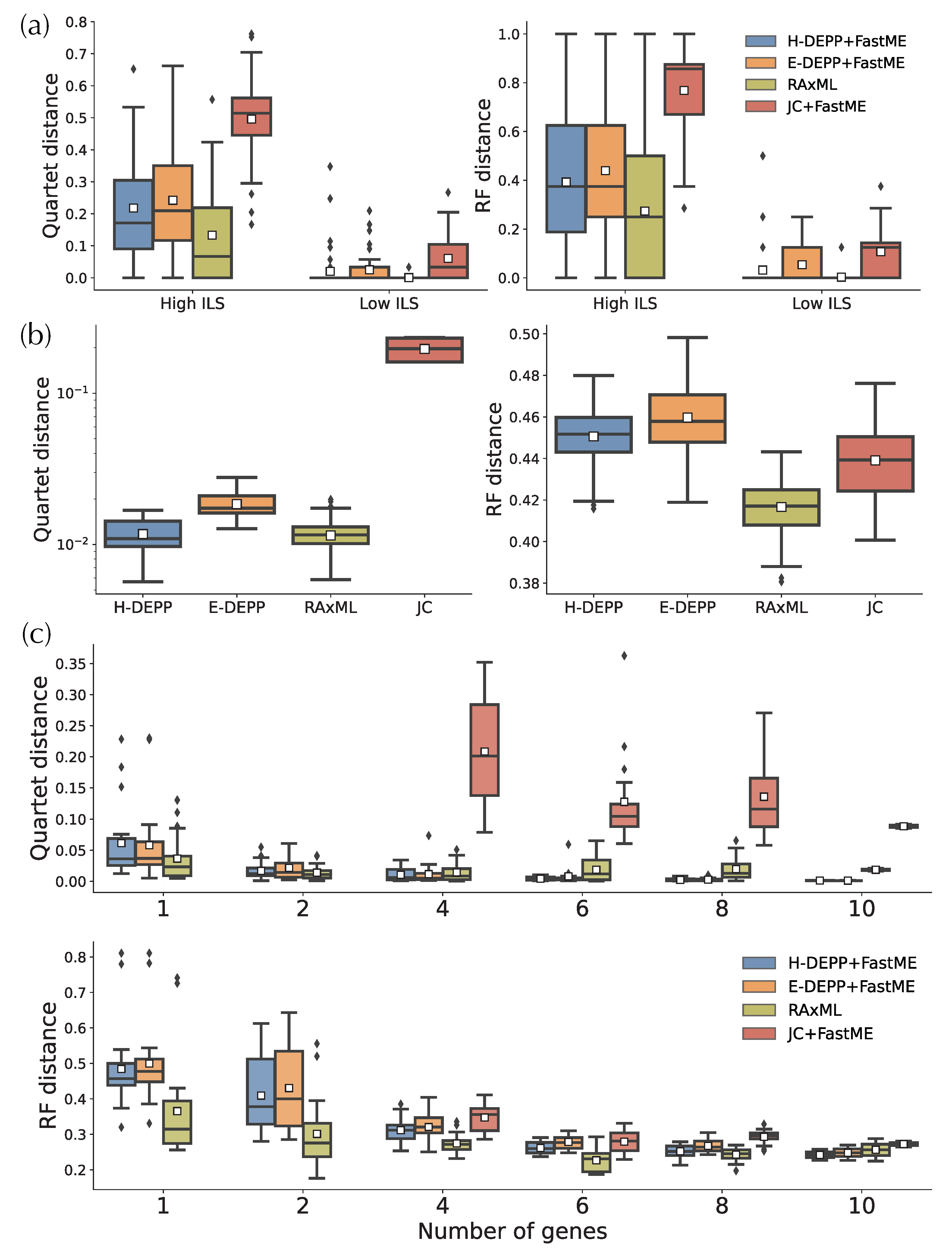
3.4. Placement Accuracy: Comparison to Alternatives
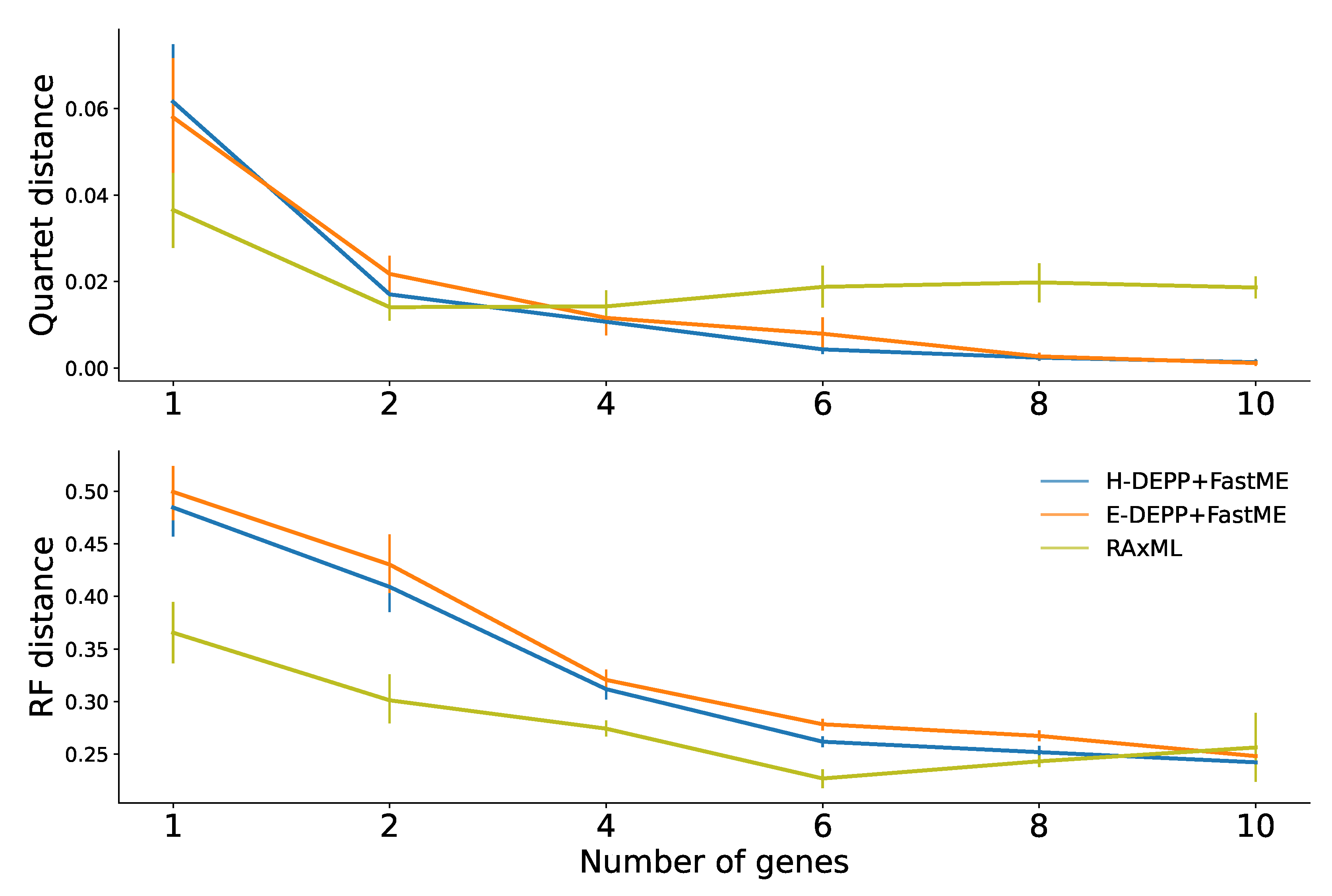
3.5. Tree Extension: Comparisons to Alternatives
4. Discussion
4.1. Tree Updates and the Impact of Discordance
4.2. Placement Accuracy and the Impact of Small Distances
4.3. Future Research
5. Conclusions
Author Contributions
Funding
Institutional Review Board Statement
Informed Consent Statement
Data Availability Statement
Conflicts of Interest
Appendix A. Extra Tables

{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
| Pseudo Count | MSE | MSE (Small) | MSE (Large) | #Outliers | |
| 0 | 0.00093 | 0.74 | 0.00036 | 416 | |
| 0.001 | 0.00066 | 0.33 | 0.00041 | 170 | |
| 0.0001 | 0.00072 | 0.52 | 0.00033 | 320 | |
| 0.00001 | 0.00145 | 1.35 | 0.00042 | 450 | |
| Pseudo Count | MSE | MSE (Small) | MSE (Large) | #Outliers | Placement Error |
| 0 | 0.0041 | 1.68 | 0.00293 | 250 | 1.78 |
| 0.001 | 0.0037 | 0.99 | 0.00296 | 146 | 1.80 |
| 0.0001 | 0.0039 | 1.39 | 0.00292 | 237 | 1.78 |
| 0.00001 | 0.0042 | 1.77 | 0.00296 | 250 | 1.75 |
Appendix B. Extra Figures


Appendix C. Commands and Versions
Appendix C.1. Software Version
- APPLES 2.0.0
- DEPP 0.2.2
- EPA-ng 0.3.8
- RAxML-ng 1.0.1
- RAxML 8.2.12
- UPP 4.3.10
- FastME 2.1.6.1
Appendix C.2. Branch Length Reestimation
- raxml-ng –evaluate –msa $sequence_file–tree $tree –model JC –threads 2–blopt nr_safe
Appendix C.3. Gene Alignment
- run_upp.py -s $input_seq -B 2000 -M -1 -T 0.33 -A 200
- run_upp.py-s $query_seq-a $backbone_seq-t $backbone_tree-A 100 -d $outdir
Appendix C.4. Placement
- APPLES+E-DEPP
- −
- Training
- ∗
- Simulated datasettrain_depp.pybackbone_tree_file=$backbone_treebackbone_seq_file=$backbone_seqpatience=5 lr=1e-4 embedding_size=128
- ∗
- WoL datasettrain_depp.pybackbone_tree_file=$backbone_treebackbone_seq_file=$backbone_seqpatience=5 lr=1e-4 embedding_size=128
- −
- Query time
- ∗
- Calculating distance matrix
- ·
- depp_distance.pyquery_seq_file=$query_seqbackbone_seq_file=$backbone_seqmodel_path=$model_pathoutdir=$out_dir
- ∗
- Placement using APPLES
- ·
- run_apples.py-d $distance_file-t $backbone_tree-o -f 0 -b 5
- APPLES+H-DEPP
- −
- Training
- ∗
- Simulated datasettrain_depp.pybackbone_tree_file=$backbone_treebackbone_seq_file=$backbone_seqweighted_method=’fm’patience=5 lr=1e-4 embedding_size=128c_epoch=6000 clr=0.005 distance_mode=’hyperbolic’
- ∗
- WoL datasettrain_depp.pybackbone_tree_file=$backbone_treebackbone_seq_file=$backbone_seqweighted_method=’fm’patience=5 lr=1e-4 embedding_size=128c_epoch=6000 clr=0.005 distance_mode=’hyperbolic’
- −
- Query time
- ∗
- Calculating distance matrix
- ·
- depp_distance.pyquery_seq_file=$query_seqbackbone_seq_file=$backbone_seqmodel_path=$model_pathoutdir=$out_dir
- ∗
- Placement using APPLES
- ·
- run_apples.py-d $distance_file-t $backbone_tree-o -f 0 -b 5
- APPLES+JCrun_apples.py-s $backbone_tree-q $query_seq-t $backbone_tree-f 0 -b 5 -D
- EPA-ng
- −
- raxml-ng–evaluate–msa $backbone_seq–tree $backbone_tree–prefix info–model GTR+G+F–threads 2 –blopt nr_safe
- −
- epa-ng–ref-msa $backbone_seq–tree $backbone_tree–query $query_seq–model $GTR_info
Appendix C.5. Tree Update
- FastME
- −
- Tree building
- ∗
- fastme -i $input_dist -o $output_tree –nni -s -m B
- −
- JC distances
- ∗
- fastme -i $input_seq -O $output_dist –dna=JC69 -c
- RAxML
- −
- raxmlHPC-PTHREADS-s $input_seq-w $output_dir-n run -p 12345-T 32 -m GTRCAT-g $backbone_tree
References
- Goyal, P.; Ferrara, E. Graph embedding techniques, applications, and performance: A survey. Knowl.-Based Syst. 2018, 151, 78–94. [Google Scholar] [CrossRef] [Green Version]
- de Vienne, D.M.; Ollier, S.; Aguileta, G. Phylo-MCOA: A Fast and Efficient Method to Detect Outlier Genes and Species in Phylogenomics Using Multiple Co-inertia Analysis. Mol. Biol. Evol. 2012, 29, 1587–1598. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Layer, M.; Rhodes, J.A. Phylogenetic trees and Euclidean embeddings. J. Math. Biol. 2017, 74, 99–111. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Jiang, Y.; Balaban, M.; Zhu, Q.; Mirarab, S. DEPP: Deep Learning Enables Extending Species Trees using Single Genes. Syst. Biol. 2022. [Google Scholar] [CrossRef]
- Balaban, M.; Sarmashghi, S.; Mirarab, S. APPLES: Scalable Distance-Based Phylogenetic Placement with or without Alignments. Syst. Biol. 2020, 69, 566–578. [Google Scholar] [CrossRef]
- Balaban, M.; Jiang, Y.; Roush, D.; Zhu, Q.; Mirarab, S. Fast and accurate distance-based phylogenetic placement using divide and conquer. Mol. Ecol. Resour. 2022, 22, 1213–1227. [Google Scholar] [CrossRef]
- Zhu, Q.; Mai, U.; Pfeiffer, W.; Janssen, S.; Asnicar, F.; Sanders, J.G.; Belda-Ferre, P.; Al-Ghalith, G.A.; Kopylova, E.; McDonald, D.; et al. Phylogenomics of 10,575 genomes reveals evolutionary proximity between domains Bacteria and Archaea. Nat. Commun. 2019, 10, 5477. [Google Scholar] [CrossRef] [Green Version]
- Parks, D.H.; Chuvochina, M.; Waite, D.W.; Rinke, C.; Skarshewski, A.; Chaumeil, P.A.; Hugenholtz, P. A standardized bacterial taxonomy based on genome phylogeny substantially revises the tree of life. Nat. Biotechnol. 2018, 36, 996–1004. [Google Scholar] [CrossRef]
- Tabaghi, P.; Dokmanić, I. Hyperbolic distance matrices. In Proceedings of the 26th ACM SIGKDD International Conference on Knowledge Discovery & Data Mining, Virtual Event, CA, USA, 6–10 July 2020; pp. 1728–1738. [Google Scholar]
- Tabaghi, P.; Peng, J.; Milenkovic, O.; Dokmanić, I. Geometry of Similarity Comparisons. arXiv 2020, arXiv:2006.09858. [Google Scholar]
- Ganea, O.; Bécigneul, G.; Hofmann, T. Hyperbolic entailment cones for learning hierarchical embeddings. In Proceedings of the International Conference on Machine Learning. PMLR, Stockholm, Sweden, 10–15 July 2018; pp. 1646–1655. [Google Scholar]
- Ganea, O.; Bécigneul, G.; Hofmann, T. Hyperbolic neural networks. Adv. Neural Inf. Process. Syst. 2018, 31, 5350–5360. [Google Scholar]
- Shimizu, R.; Mukuta, Y.; Harada, T. Hyperbolic neural networks++. arXiv 2020, arXiv:2006.08210. [Google Scholar]
- Sala, F.; De Sa, C.; Gu, A.; Ré, C. Representation tradeoffs for hyperbolic embeddings. In Proceedings of the International Conference on Machine Learning. PMLR, Stockholm, Sweden, 10–15 July 2018; pp. 4460–4469. [Google Scholar]
- Chen, W.; Han, X.; Lin, Y.; Zhao, H.; Liu, Z.; Li, P.; Sun, M.; Zhou, J. Fully hyperbolic neural networks. arXiv 2021, arXiv:2105.14686. [Google Scholar]
- Linial, N.; London, E.; Rabinovich, Y. The geometry of graphs and some of its algorithmic applications. Combinatorica 1995, 15, 215–245. [Google Scholar] [CrossRef]
- Sarkar, R. Low distortion delaunay embedding of trees in hyperbolic plane. In Proceedings of the International Symposium on Graph Drawing, Konstanz, Germany, 21–24 September 2011; pp. 355–366. [Google Scholar]
- Bachmann, G.; Bécigneul, G.; Ganea, O. Constant curvature graph convolutional networks. In Proceedings of the International Conference on Machine Learning. PMLR, Virtual, 13–18 July 2020; pp. 486–496. [Google Scholar]
- Dai, J.; Wu, Y.; Gao, Z.; Jia, Y. A hyperbolic-to-hyperbolic graph convolutional network. In Proceedings of the IEEE/CVF Conference on Computer Vision and Pattern Recognition, Virtual Event, 20–25 June 2021; pp. 154–163. [Google Scholar]
- Liu, Q.; Nickel, M.; Kiela, D. Hyperbolic graph neural networks. Adv. Neural Inf. Process. Syst. 2019, 32, 8230–8241. [Google Scholar]
- Chami, I.; Ying, Z.; Ré, C.; Leskovec, J. Hyperbolic graph convolutional neural networks. Adv. Neural Inf. Process. Syst. 2019, 32, 4868–4879. [Google Scholar]
- Skopek, O.; Ganea, O.E.; Bécigneul, G. Mixed-curvature variational autoencoders. arXiv 2020, arXiv:1911.08411. [Google Scholar]
- Gulcehre, C.; Denil, M.; Malinowski, M.; Razavi, A.; Pascanu, R.; Hermann, K.M.; Battaglia, P.; Bapst, V.; Raposo, D.; Santoro, A.; et al. Hyperbolic attention networks. arXiv 2018, arXiv:1805.09786. [Google Scholar]
- Matsumoto, H.; Mimori, T.; Fukunaga, T. Novel metric for hyperbolic phylogenetic tree embeddings. Biol. Methods Protoc. 2021, 6, bpab006. [Google Scholar] [CrossRef]
- Corso, G.; Ying, Z.; Pándy, M.; Veličković, P.; Leskovec, J.; Liò, P. Neural Distance Embeddings for Biological Sequences. Adv. Neural Inf. Process. Syst. 2021, 34, 18539–18551. [Google Scholar]
- Fitch, W.M.; Margoliash, E. Construction of Phylogenetic Trees. Science 1967, 155, 279–284. [Google Scholar] [CrossRef]
- Mirarab, S.; Warnow, T. ASTRAL-II: Coalescent-based species tree estimation with many hundreds of taxa and thousands of genes. Bioinformatics 2015, 31, i44–i52. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Robinson, D.; Foulds, L. Comparison of phylogenetic trees. Math. Biosci. 1981, 53, 131–147. [Google Scholar] [CrossRef]
- Jukes, T.H.; Cantor, C.R. Evolution of protein molecules. Mamm. Protein Metab. 1969, 3, 21–132. [Google Scholar] [CrossRef]
- Barbera, P.; Kozlov, A.M.; Czech, L.; Morel, B.; Darriba, D.; Flouri, T.; Stamatakis, A. EPA-ng: Massively Parallel Evolutionary Placement of Genetic Sequences. Syst. Biol. 2019, 68, 365–369. [Google Scholar] [CrossRef]
- Kozlov, A.M.; Darriba, D.; Flouri, T.; Morel, B.; Stamatakis, A. RAxML-NG: A fast, scalable and user-friendly tool for maximum likelihood phylogenetic inference. Bioinformatics 2019, 35, 4453–4455. [Google Scholar] [CrossRef] [Green Version]
- Lefort, V.; Desper, R.; Gascuel, O. FastME 2.0: A comprehensive, accurate, and fast distance-based phylogeny inference program. Mol. Biol. Evol. 2015, 32, 2798–2800. [Google Scholar] [CrossRef] [Green Version]
- Stamatakis, A. RAxML version 8: A tool for phylogenetic analysis and post-analysis of large phylogenies. Bioinformatics 2014, 30, 1312–1313. [Google Scholar] [CrossRef]
- Atteson, K. The Performance of Neighbor-Joining Methods of Phylogenetic Reconstruction. Algorithmica 1999, 25, 251–278. [Google Scholar] [CrossRef]
- Gascuel, O.; Steel, M. A ‘Stochastic Safety Radius’ for Distance-Based Tree Reconstruction. Algorithmica 2016, 74, 1386–1403. [Google Scholar] [CrossRef] [Green Version]
- Feurer, M.; Springenberg, J.; Hutter, F. Initializing bayesian hyperparameter optimization via meta-learning. In Proceedings of the AAAI Conference on Artificial Intelligence, Austin, TX, USA, 25–30 January 2015; Volume 29. [Google Scholar]
- Finn, C.; Abbeel, P.; Levine, S. Model-agnostic meta-learning for fast adaptation of deep networks. In Proceedings of the International Conference on Machine Learning. PMLR, Sydney, Australia, 6–11 August 2017; pp. 1126–1135. [Google Scholar]
- Ji, K.; Yang, J.; Liang, Y. Bilevel optimization: Convergence analysis and enhanced design. In Proceedings of the International Conference on Machine Learning. PMLR, Virtual, 18–24 July 2021; pp. 4882–4892. [Google Scholar]









| p | 2 | 4 | 6 | 8 | 10 | 12 | 14 | 16 | 18 | 20 | 22 | 24 |
| p-Value | p-Value | p-Value | ||||
|---|---|---|---|---|---|---|
| H-DEPP | 0.012 | 0.29 | 0.59 | 0.57 | −0.06 | |
| E-DEPP | 0.061 | 0.21 | 0.43 | 0.48 | −0.08 | |
| RAxML | 0.474 | 0.08 | 0.86 | −0.02 | 0.08 | 0.20 |
Publisher’s Note: MDPI stays neutral with regard to jurisdictional claims in published maps and institutional affiliations. |
© 2022 by the authors. Licensee MDPI, Basel, Switzerland. This article is an open access article distributed under the terms and conditions of the Creative Commons Attribution (CC BY) license (https://creativecommons.org/licenses/by/4.0/).
Share and Cite
Jiang, Y.; Tabaghi, P.; Mirarab, S. Learning Hyperbolic Embedding for Phylogenetic Tree Placement and Updates. Biology 2022, 11, 1256. https://doi.org/10.3390/biology11091256
Jiang Y, Tabaghi P, Mirarab S. Learning Hyperbolic Embedding for Phylogenetic Tree Placement and Updates. Biology. 2022; 11(9):1256. https://doi.org/10.3390/biology11091256
Chicago/Turabian StyleJiang, Yueyu, Puoya Tabaghi, and Siavash Mirarab. 2022. "Learning Hyperbolic Embedding for Phylogenetic Tree Placement and Updates" Biology 11, no. 9: 1256. https://doi.org/10.3390/biology11091256
APA StyleJiang, Y., Tabaghi, P., & Mirarab, S. (2022). Learning Hyperbolic Embedding for Phylogenetic Tree Placement and Updates. Biology, 11(9), 1256. https://doi.org/10.3390/biology11091256