
Rapid Scan Electron Paramagnetic Resonance Spectroscopy Is a Suitable Tool to Study Intermolecular Interactions of Intrinsically Disordered Protein

Abstract
:Simple Summary
Abstract
1. Introduction
2. Materials and Methods
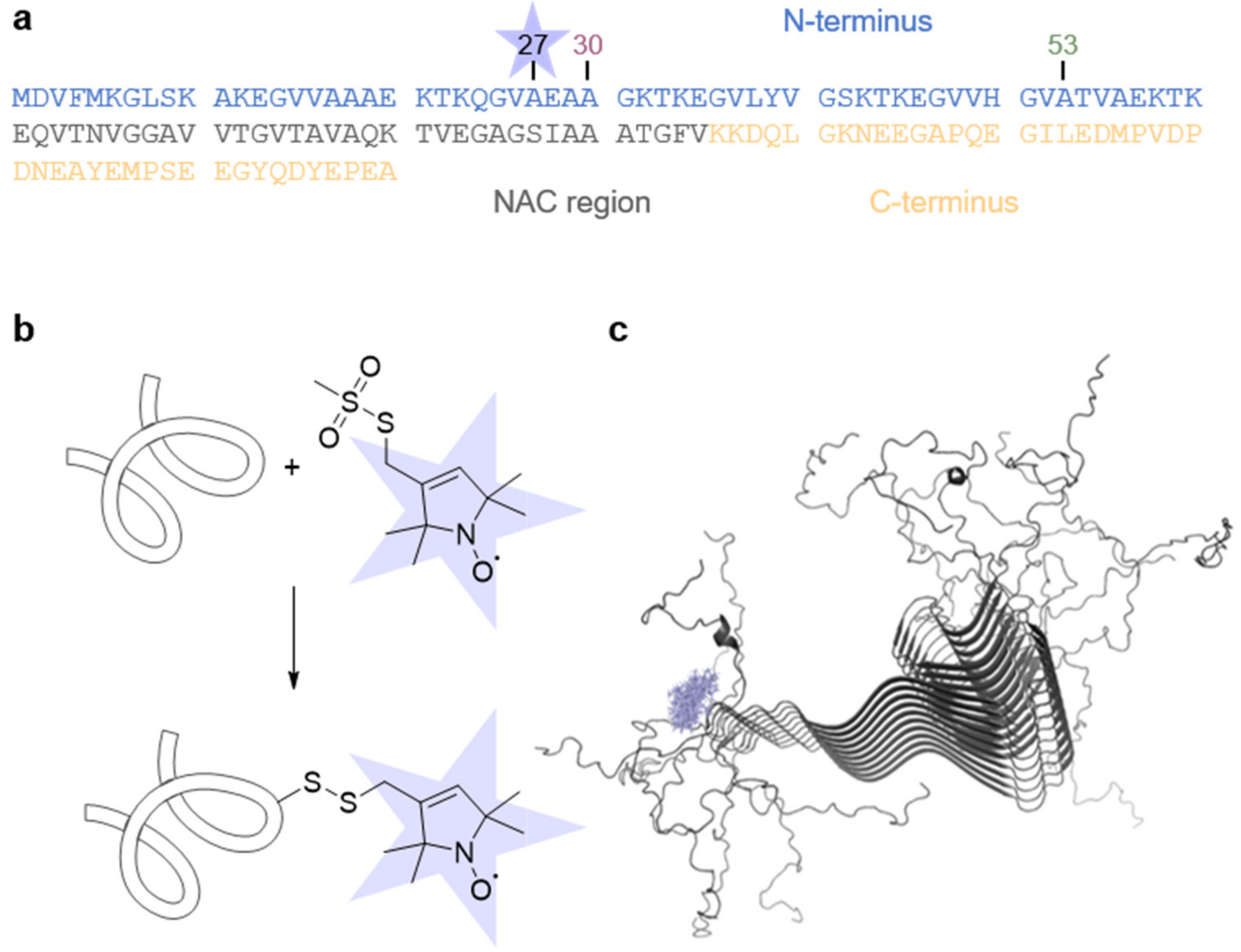
2.1. Protein Production and Spin Labeling
2.2. Aggregation Assay
2.3. Rapid Scan EPR Spectroscopy
3. Results
3.1. Spin-Labeled α-Synuclein Variants Aggregate in the Presence of Ethanol
3.2. Spin-Dilution Is Not a Prerequisite in Our Experimental Setup
3.3. Circular Dichroism Reveals the Global Aggregation Process of α-Synuclein
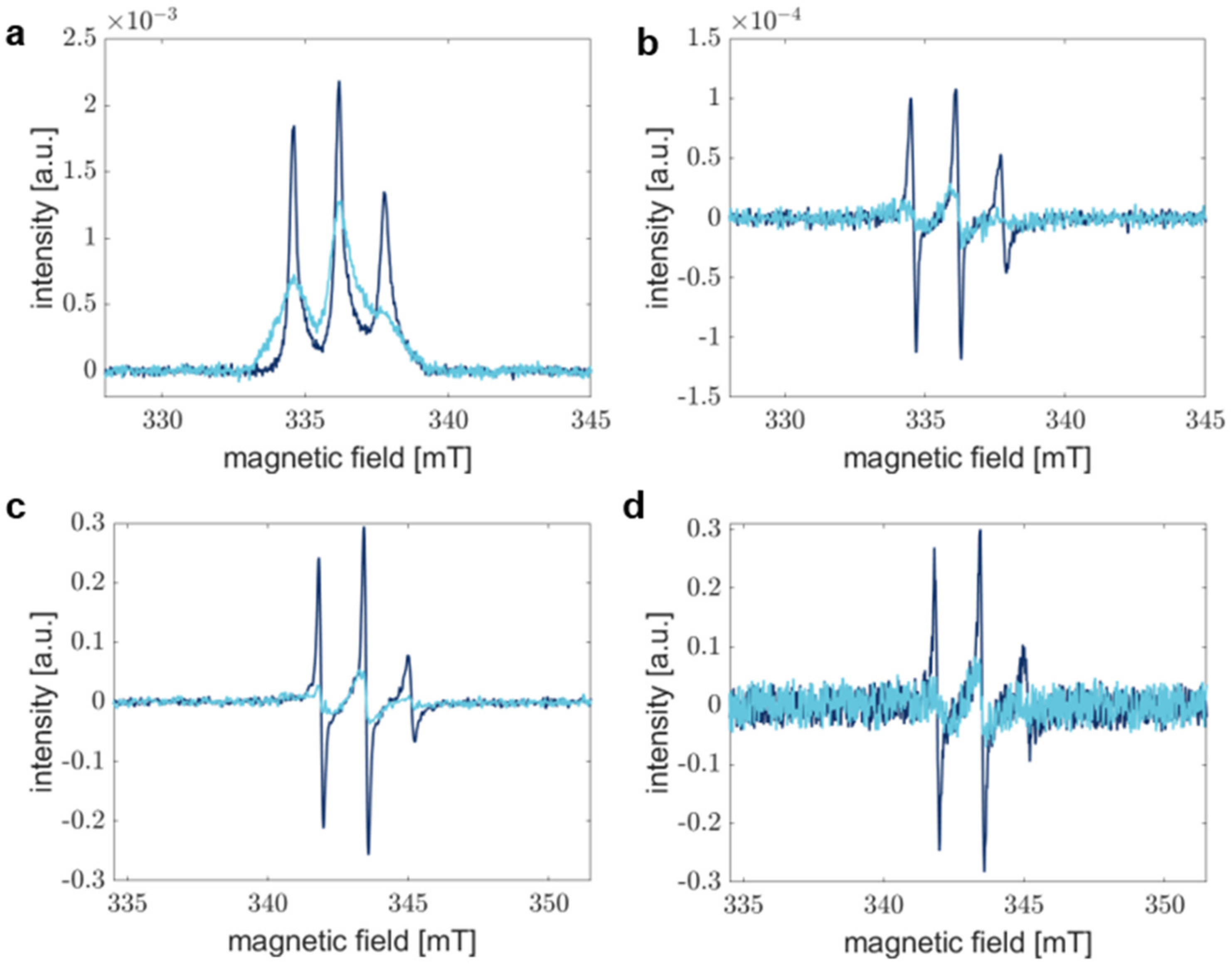
3.4. The Signal-to-Noise Ratio of Rapid Scan EPR Spectroscopy Outperforms Conventional Continuous Wave EPR Spectroscopy
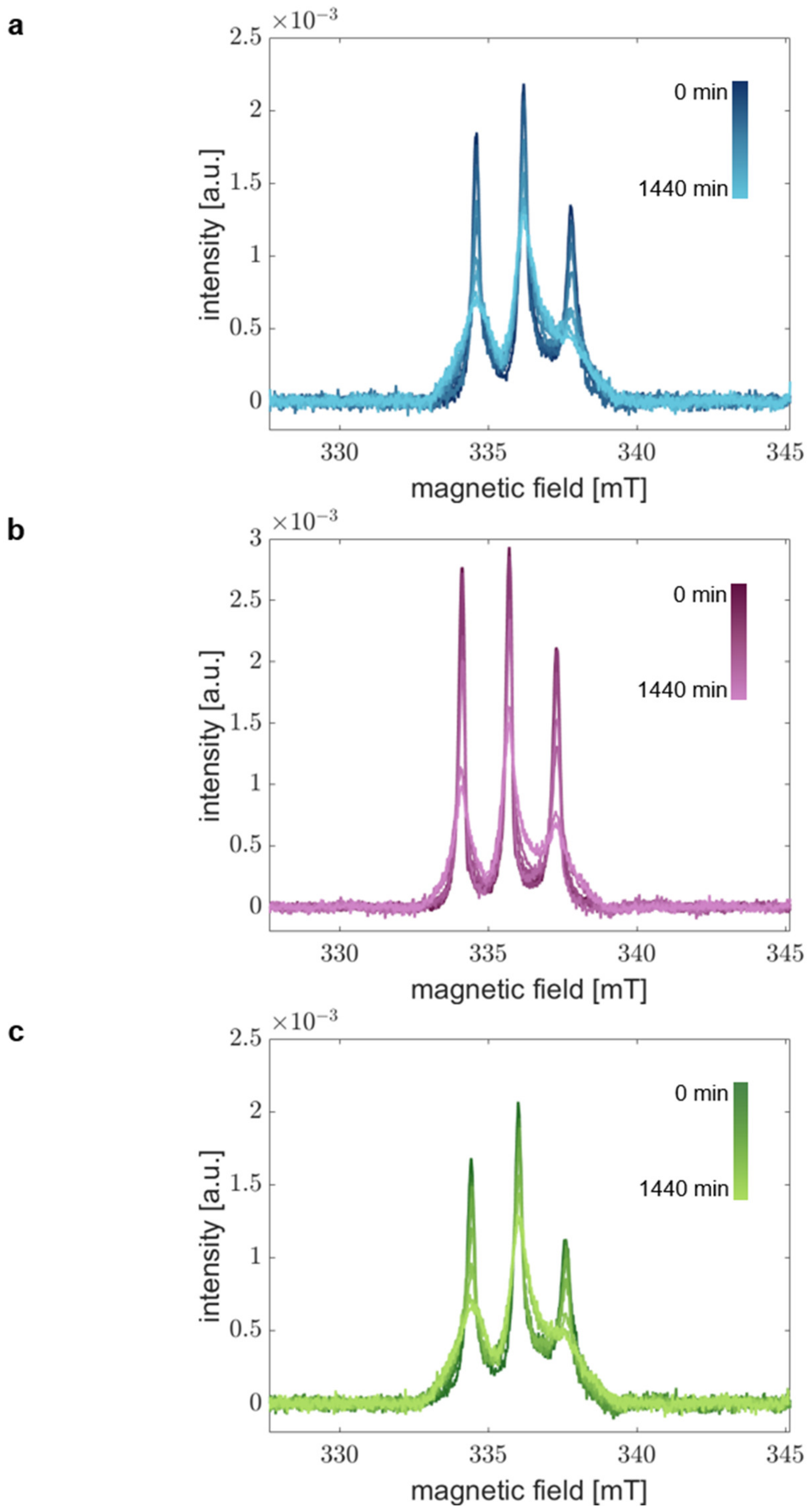
3.5. Rapid Scan Experiments Capture the Local Kinetics of α-Synuclein Aggregation
4. Discussion
5. Conclusions
Supplementary Materials
Author Contributions
Funding
Institutional Review Board Statement
Informed Consent Statement
Data Availability Statement
Acknowledgments
Conflicts of Interest
References
- Braun, P.; Gingras, A.C. History of Protein-Protein Interactions: From Egg-White to Complex Networks. Proteomics 2012, 12, 1478–1498. [Google Scholar] [CrossRef]
- Uversky, V.N. Intrinsically Disordered Proteins from A to Z. Int. J. Biochem. Cell Biol. 2011, 43, 1090–1103. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Wright, P.E.; Dyson, H.J. Intrinsically Disordered Proteins in Cellular Signaling and Regulation. Nat. Rev. Mol. Cell Biol. 2015, 16, 18–29. [Google Scholar] [CrossRef] [PubMed]
- Dyson, H.J.; Wright, P.E. Coupling of Folding and Binding for Unstructured Proteins. Curr. Opin. Struct. Biol. 2002, 12, 54–60. [Google Scholar] [CrossRef] [PubMed]
- Davidson, W.S.; Jonas, A.; Clayton, D.F.; George, J.M. Stabilization of Alpha-Synuclein Secondary Structure upon Binding to Synthetic Membranes. J. Biol. Chem. 1998, 273, 9443–9449. [Google Scholar] [CrossRef] [Green Version]
- Ulmer, T.S.; Bax, A.; Cole, N.B.; Nussbaum, R.L. Structure and Dynamics of Micelle-Bound Human α-Synuclein. J. Biol. Chem. 2005, 280, 9595–9603. [Google Scholar] [CrossRef] [Green Version]
- Spillantini, M.G.; Crowther, R.A.; Jakes, R.; Hasegawa, M.; Goedert, M. Alpha-Synuclein in Filamentous Inclusions of Lewy Bodies from Parkinson’s Disease and Dementia with Lewy Bodies. Proc. Natl. Acad. Sci. USA 1998, 95, 6469–6473. [Google Scholar] [CrossRef] [Green Version]
- Atik, A.; Stewart, T.; Zhang, J. Alpha-Synuclein as a Biomarker for Parkinson’s Disease. Brain Pathol. 2016, 26, 410–418. [Google Scholar] [CrossRef]
- Li, X.; Dong, C.; Hoffmann, M.; Garen, C.R.; Cortez, L.M.; Petersen, N.O.; Woodside, M.T. Early Stages of Aggregation of Engineered α-Synuclein Monomers and Oligomers in Solution. Sci. Rep. 2019, 9, 1734. [Google Scholar] [CrossRef] [Green Version]
- Villar-Piqué, A.; Lopes da Fonseca, T.; Outeiro, T.F. Structure, Function and Toxicity of Alpha-Synuclein: The Bermuda Triangle in Synucleinopathies. J. Neurochem. 2016, 139, 240–255. [Google Scholar] [CrossRef]
- Cremades, N.; Cohen, S.I.A.; Deas, E.; Abramov, A.Y.; Chen, A.Y.; Orte, A.; Sandal, M.; Clarke, R.W.; Dunne, P.; Aprile, F.A.; et al. Direct Observation of the Interconversion of Normal and Toxic Forms of α-Synuclein. Cell 2012, 149, 1048–1059. [Google Scholar] [CrossRef] [Green Version]
- Chen, S.W.; Drakulic, S.; Deas, E.; Ouberai, M.; Aprile, F.A.; Arranz, R.; Ness, S.; Roodveldt, C.; Guilliams, T.; De-Genst, E.J.; et al. Structural Characterization of Toxic Oligomers That Are Kinetically Trapped during α-Synuclein Fibril Formation. Proc. Natl. Acad. Sci. USA 2015, 112, E1994–E2003. [Google Scholar] [CrossRef] [Green Version]
- Krüger, R.; Kuhn, W.; Müller, T.; Woitalla, D.; Graeber, M.; Kösel, S.; Przuntek, H.; Epplen, J.T.; Schols, L.; Riess, O. Ala30Pro Mutation in the Gene Encoding α-Synuclein in Parkinson’s Disease. Nat. Genet. 1998, 18, 106–108. [Google Scholar] [CrossRef]
- Polymeropoulos, M.H.; Lavedan, C.; Leroy, E.; Ide, S.E.; Dehejia, A.; Dutra, A.; Pike, B.; Root, H.; Rubenstein, J.; Boyer, R.; et al. Mutation in the Alpha-Synuclein Gene Identified in Families with Parkinson’s Disease. Science 1997, 276, 2045–2047. [Google Scholar] [CrossRef] [Green Version]
- Conway, K.A.; Harper, J.D.; Lansbury, P.T. Accelerated in Vitro Fibril Formation by a Mutant α-Synuclein Linked to Early-Onset Parkinson Disease. Nat. Med. 1998, 4, 1318–1320. [Google Scholar] [CrossRef]
- Conway, K.A.; Lee, S.J.; Rochet, J.C.; Ding, T.T.; Williamson, R.E.; Lansbury, P.T. Acceleration of Oligomerization, Not Fibrillization, Is a Shared Property of Both α-Synuclein Mutations Linked to Early-Onset Parkinson’s Disease: Implications for Pathogenesis and Therapy. Proc. Natl. Acad. Sci. USA 2000, 97, 571–576. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Lemkau, L.R.; Comellas, G.; Kloepper, K.D.; Woods, W.S.; George, J.M.; Rienstra, C.M. Mutant Protein A30P α-Synuclein Adopts Wild-Type Fibril Structure, despite Slower Fibrillation Kinetics. J. Biol. Chem. 2012, 287, P11526–P11532. [Google Scholar] [CrossRef] [Green Version]
- Narhi, L.; Wood, S.J.; Steavenson, S.; Jiang, Y.; Wu, G.M.; Anafi, D.; Kaufman, S.A.; Martin, F.; Sitney, K.; Denis, P.; et al. Both Familial Parkinson’s Disease Mutations Accelerate Alpha-Synuclein Aggregation. J. Biol. Chem. 1999, 274, 9843–9846. [Google Scholar] [CrossRef] [Green Version]
- Li, J.; Uversky, V.N.; Fink, A.L. Effect of Familial Parkinson’s Disease Point Mutations A30P and A53T on the Structural Properties, Aggregation, and Fibrillation of Human α-Synuclein. Biochemistry 2001, 40, 11604–11613. [Google Scholar] [CrossRef]
- Hoyer, W.; Antony, T.; Cherny, D.; Heim, G.; Jovin, T.M.; Subramaniam, V. Dependence of A-Synuclein Aggregate Morphology on Solution Conditions. J. Mol. Biol. 2002, 322, 383–393. [Google Scholar] [CrossRef]
- Flagmeier, P.; Meisl, G.; Vendruscolo, M.; Knowles, T.P.J.; Dobson, C.M.; Buell, A.K.; Galvagnion, C. Mutations Associated with Familial Parkinson’s Disease Alter the Initiation and Amplification Steps of α-Synuclein Aggregation. Proc. Natl. Acad. Sci. USA 2016, 113, 10328–10333. [Google Scholar] [CrossRef] [Green Version]
- Fallah, M.A.; Gerding, H.R.; Scheibe, C.; Drescher, M.; Karreman, C.; Schildknecht, S.; Leist, M.; Hauser, K. Simultaneous IR-Spectroscopic Observation of α-Synuclein, Lipids, and Solvent Reveals an Alternative Membrane-Induced Oligomerization Pathway. ChemBioChem 2017, 18, 2312–2316. [Google Scholar] [CrossRef] [PubMed]
- Gautam, S.; Karmakar, S.; Bose, A.; Chowdhury, P.K. β-Cyclodextrin and Curcumin, a Potent Cocktail for Disaggregating and/or Inhibiting Amyloids: A Case Study with α-Synuclein. Biochemistry 2014, 53, 4081–4083. [Google Scholar] [CrossRef] [PubMed]
- Gautam, S.; Karmakar, S.; Batra, R.; Sharma, P.; Pradhan, P.; Singh, J.; Kundu, B.; Chowdhury, P.K. Polyphenols in Combination with β-Cyclodextrin Can Inhibit and Disaggregate α-Synuclein Amyloids under Cell Mimicking Conditions: A Promising Therapeutic Alternative. Biochim. Biophys. Acta Proteins Proteom. 2017, 1865, 589–603. [Google Scholar] [CrossRef] [PubMed]
- Munishkina, L.A.; Phelan, C.; Uversky, V.N.; Fink, A.L. Conformational Behavior and Aggregation of α-Synuclein in Organic Solvents: Modeling the Effects of Membranes. Biochemistry 2003, 42, 2720–2730. [Google Scholar] [CrossRef] [PubMed]
- Altenbach, C.; Flitsch, S.L.; Khorana, H.G.; Hubbell, W.L. Structural Studies on Transmembrane Proteins. 2. Spin Labeling of Bacteriorhodopsin Mutants at Unique Cysteines. Biochemistry 1989, 28, 7806–7812. [Google Scholar] [CrossRef]
- Hubbell, W.L.; Altenbach, C. Investigation of Structure and Dynamics in Membrane Proteins Using Site-Directed Spin Labeling. Curr. Opin. Struct. Biol. 1994, 4, 566–573. [Google Scholar] [CrossRef]
- Chen, M.; Margittai, M.; Chen, J.; Langen, R. Investigation of Alpha-Synuclein Fibril Structure by Site-Directed Spin Labeling. J. Biol. Chem. 2007, 282, 24970–24979. [Google Scholar] [CrossRef] [Green Version]
- Der-Sarkissian, A.; Jao, C.C.; Chen, J.; Langen, R. Structural Organization of Alpha-Synuclein Fibrils Studied by Site-Directed Spin Labeling. J. Biol. Chem. 2003, 278, 37530–37535. [Google Scholar] [CrossRef] [Green Version]
- Karyagina, I.; Becker, S.; Giller, K.; Riedel, D.; Jovin, T.M.; Griesinger, C.; Bennati, M. Electron Paramagnetic Resonance Spectroscopy Measures the Distance between the External β-Strands of Folded α-Synuclein in Amyloid Fibrils. Biophys. J. 2011, 101, L1–L3. [Google Scholar] [CrossRef]
- Pornsuwan, S.; Giller, K.; Riedel, D.; Becker, S.; Griesinger, C.; Bennati, M. Long-Range Distances in Amyloid Fibrils of α-Synuclein from PELDOR Spectroscopy. Angew. Chem. Int. Ed. 2013, 52, 10290–10294. [Google Scholar] [CrossRef] [Green Version]
- Strohäker, T.; Jung, B.C.; Liou, S.-H.; Fernandez, C.O.; Riedel, D.; Becker, S.; Halliday, G.M.; Bennati, M.; Kim, W.S.; Lee, S.-J.; et al. Structural Heterogeneity of α-Synuclein Fibrils Amplified from Patient Brain Extracts. Nat. Commun. 2019, 10, 5535. [Google Scholar] [CrossRef] [Green Version]
- Zurlo, E.; Kumar, P.; Meisl, G.; Dear, A.J.; Mondal, D.; Claessens, M.M.A.E.; Knowles, T.P.J.; Huber, M. In Situ Kinetic Measurements of α-Synuclein Aggregation Reveal Large Population of Short-Lived Oligomers. PLoS ONE 2021, 16, e0245548. [Google Scholar] [CrossRef]
- Zurlo, E.; Passerini, L.; Kumar, P.; Huber, M. In Situ Continuous Wave Electron Paramagnetic Resonance Investigation of the Amyloid Aggregation of Parkinson’s Protein Alpha-Synuclein—The Second Spin-Label Position. Appl. Magn. Reson. 2021, 53, 1133–1150. [Google Scholar] [CrossRef]
- Eaton, S.S.; Eaton, G.R. Rapid-Scan EPR of Nitroxide Spin Labels and Semiquinones. Methods Enzymol. 2015, 563, 3–21. [Google Scholar] [CrossRef] [Green Version]
- Eaton, G.R.; Eaton, S.S. Advances in Rapid Scan EPR Spectroscopy. Methods Enzymol. 2022, 666, 1–24. [Google Scholar] [CrossRef]
- Braun, T.S.; Stehle, J.; Kacprzak, S.; Carl, P.; Höfer, P.; Subramaniam, V.; Drescher, M. Intracellular Protein-Lipid Interactions Studied by Rapid-Scan Electron Paramagnetic Resonance Spectroscopy. J. Phys. Chem. Lett. 2021, 12, 2471–2475. [Google Scholar] [CrossRef]
- Tseitlin, M.; Eaton, S.S.; Eaton, G.R. Uncertainty Analysis for Absorption and First-Derivative EPR Spectra. Concepts Magn. Reson. Part A 2012, 40, 295–305. [Google Scholar] [CrossRef]
- Cattani, J.; Braun, T.; Drescher, M. Probing Alpha-Synuclein Conformations by Electron Paramagnetic Resonance (EPR) Spectroscopy. In Methods in Molecular Biology; Holtzbrinck Springer Nature Publishing Group: New York, NY, USA, 2019; Volume 1948, pp. 247–260. [Google Scholar]
- Eichhoff, U.; Höfer, P. 75 Years of EPR. EPR Milestones in 60 Years Bruker History. Appl. Magn. Reson. 2020, 51, 1723–1737. [Google Scholar] [CrossRef]
- Stoll, S.; Schweiger, A. EasySpin, a Comprehensive Software Package for Spectral Simulation and Analysis in EPR. J. Magn. Reson. 2006, 178, 42–55. [Google Scholar] [CrossRef]
- Jakes, R.; Spillantini, M.G.; Goedert, M. Identification of Two Distinct Synucleins from Human Brain. FEBS Lett. 1994, 345, 27–32. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Burré, J. The Synaptic Function of-Synuclein. J. Parkinsons. Dis. 2015, 5, 699–713. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Fusco, G.; De Simone, A.; Gopinath, T.; Vostrikov, V.; Vendruscolo, M.; Dobson, C.M.; Veglia, G. Direct Observation of the Three Regions in α-Synuclein That Determine Its Membrane-Bound Behaviour. Nat. Commun. 2014, 5, 3827. [Google Scholar] [CrossRef] [Green Version]
- Tuttle, M.D.; Comellas, G.; Nieuwkoop, A.J.; Covell, D.J.; Berthold, D.A.; Kloepper, K.D.; Courtney, J.M.; Kim, J.K.; Barclay, A.M.; Kendall, A.; et al. Solid-State NMR Structure of a Pathogenic Fibril of Full-Length Human α-Synuclein. Nat. Struct. Mol. Biol. 2016, 23, 409–415. [Google Scholar] [CrossRef] [PubMed]
- Chakraborty, R.; Chattopadhyay, K. Cryo-Electron Microscopy Uncovers Key Residues within the Core of Alpha-Synuclein Fibrils. ACS Chem. Neurosci. 2019, 10, 1135–1136. [Google Scholar] [CrossRef] [Green Version]
- Guerrero-Ferreira, R.; Taylor, N.M.; Mona, D.; Ringler, P.; Lauer, M.E.; Riek, R.; Britschgi, M.; Stahlberg, H. Cryo-EM Structure of Alpha-Synuclein Fibrils. Elife 2018, 7, e36402. [Google Scholar] [CrossRef]
- Rodriguez, J.A.; Ivanova, M.I.; Sawaya, M.R.; Cascio, D.; Reyes, F.E.; Shi, D.; Sangwan, S.; Guenther, E.L.; Johnson, L.M.; Zhang, M.; et al. Structure of the Toxic Core of α-Synuclein from Invisible Crystals. Nature 2015, 525, 486–490. [Google Scholar] [CrossRef] [Green Version]
- Mitchell, D.G.; Quine, R.W.; Tseitlin, M.; Eaton, S.S.; Eaton, G.R. X-Band Rapid-Scan EPR of Nitroxyl Radicals. J. Magn. Reson. 2012, 214, 221–226. [Google Scholar] [CrossRef]
- Uversky, V.N.; Oldfield, C.J.; Dunker, A.K. Intrinsically Disordered Proteins in Human Diseases: Introducing the D2 Concept. Annu. Rev. Biophys. 2008, 37, 215–246. [Google Scholar] [CrossRef]
- Snead, D.; Eliezer, D. Alpha-Synuclein Function and Dysfunction on Cellular Membranes. Exp. Neurobiol. 2014, 23, 292–313. [Google Scholar] [CrossRef]
- Nemani, V.M.; Lu, W.; Berge, V.; Nakamura, K.; Onoa, B.; Lee, M.K.; Chaudhry, F.A.; Nicoll, R.A.; Edwards, R.H. Increased Expression of α-Synuclein Reduces Neurotransmitter Release by Inhibiting Synaptic Vesicle Reclustering after Endocytosis. Neuron 2010, 65, 66–79. [Google Scholar] [CrossRef] [Green Version]
- Murphy, D.D.; Rueter, S.M.; Trojanowski, J.Q.; Lee, V.M.Y. Synucleins Are Developmentally Expressed, and α-Synuclein Regulates the Size of the Presynaptic Vesicular Pool in Primary Hippocampal Neurons. J. Neurosci. 2000, 20, 3214–3220. [Google Scholar] [CrossRef]




{kind=link}
{kind=link}
{kind=link}
| Accumulation Time | RS | CW |
|---|---|---|
| monomer | 121 | 26 |
| fibril | 54 | 4 |
Disclaimer/Publisher’s Note: The statements, opinions and data contained in all publications are solely those of the individual author(s) and contributor(s) and not of MDPI and/or the editor(s). MDPI and/or the editor(s) disclaim responsibility for any injury to people or property resulting from any ideas, methods, instructions or products referred to in the content. |
© 2023 by the authors. Licensee MDPI, Basel, Switzerland. This article is an open access article distributed under the terms and conditions of the Creative Commons Attribution (CC BY) license (https://creativecommons.org/licenses/by/4.0/).
Share and Cite
Dröden, J.; Drescher, M. Rapid Scan Electron Paramagnetic Resonance Spectroscopy Is a Suitable Tool to Study Intermolecular Interactions of Intrinsically Disordered Protein. Biology 2023, 12, 79. https://doi.org/10.3390/biology12010079
Dröden J, Drescher M. Rapid Scan Electron Paramagnetic Resonance Spectroscopy Is a Suitable Tool to Study Intermolecular Interactions of Intrinsically Disordered Protein. Biology. 2023; 12(1):79. https://doi.org/10.3390/biology12010079
Chicago/Turabian StyleDröden, Jessica, and Malte Drescher. 2023. "Rapid Scan Electron Paramagnetic Resonance Spectroscopy Is a Suitable Tool to Study Intermolecular Interactions of Intrinsically Disordered Protein" Biology 12, no. 1: 79. https://doi.org/10.3390/biology12010079
APA StyleDröden, J., & Drescher, M. (2023). Rapid Scan Electron Paramagnetic Resonance Spectroscopy Is a Suitable Tool to Study Intermolecular Interactions of Intrinsically Disordered Protein. Biology, 12(1), 79. https://doi.org/10.3390/biology12010079