
Anti-Inflammatory Actions of G-Protein-Coupled Estrogen Receptor 1 (GPER) and Brain-Derived Estrogen Following Cerebral Ischemia in Ovariectomized Rats
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
Abstract
:Simple Summary
Abstract
1. Introduction
2. Materials and Methods
2.1. Animals and Animal Model of Global Cerebral Ischemia (GCI)
2.2. Administration of Drugs
2.3. Hippocampal Tissue Preparation and Immunofluorescence Staining
2.4. Hippocampal Sample Preparation and Western Blot Analysis
2.5. Aromatase Activity
2.6. Statistical Analysis
3. Results
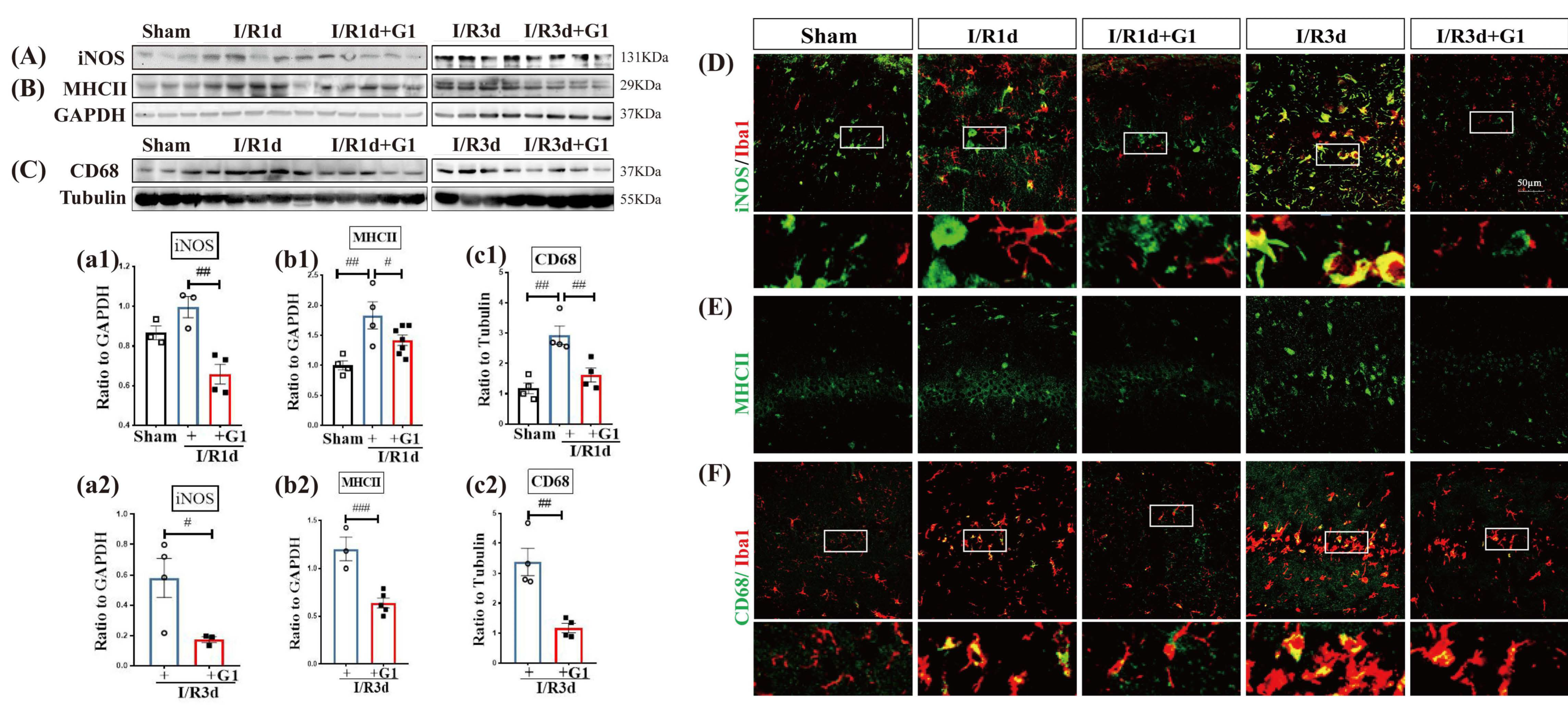
3.1. G1 Administration Suppresses M1 Phenotypic Markers of Microglia in the Hippocampal CA1 Region Following GCI
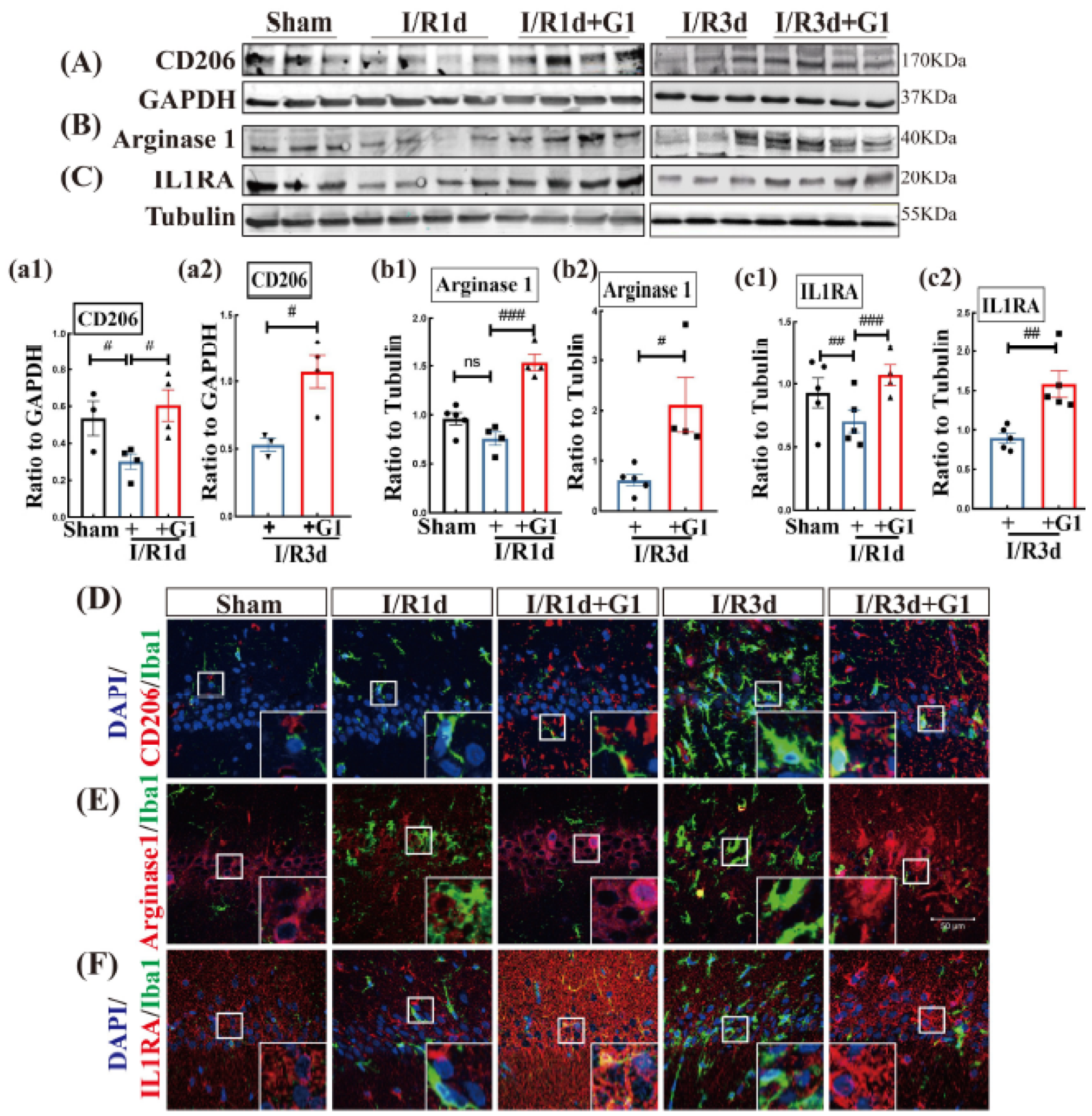
3.2. G1 Administration Enhances M2 Markers of Microglia in the Hippocampal CA1 Region Following GCI
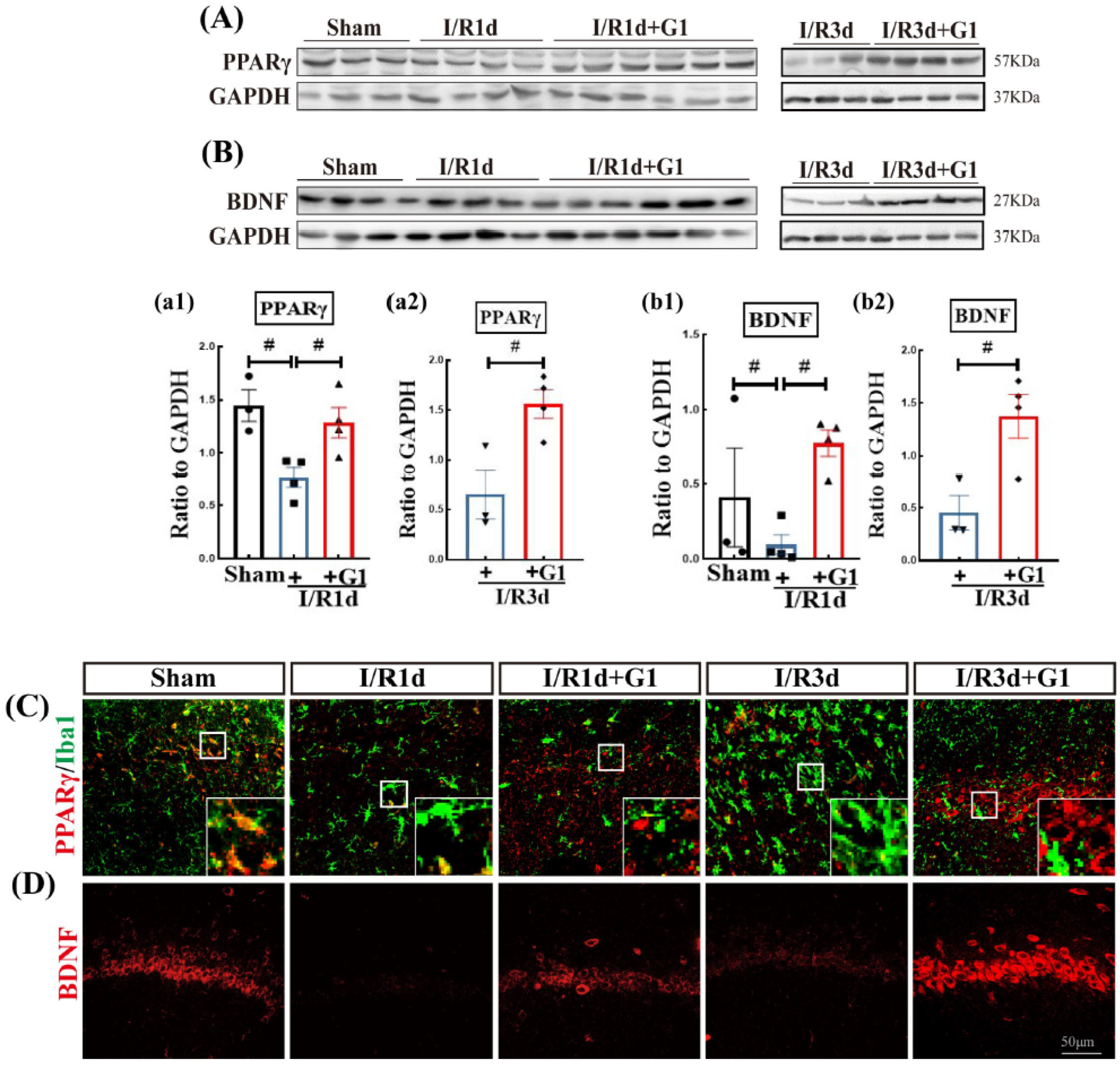
3.3. G1 Administration Increases the Levels of Brain-Derived Neurotrophic Factor (BDNF) and Pro-Survival Transcription Factor PPARγ in the Hippocampal CA1 Region after GCI
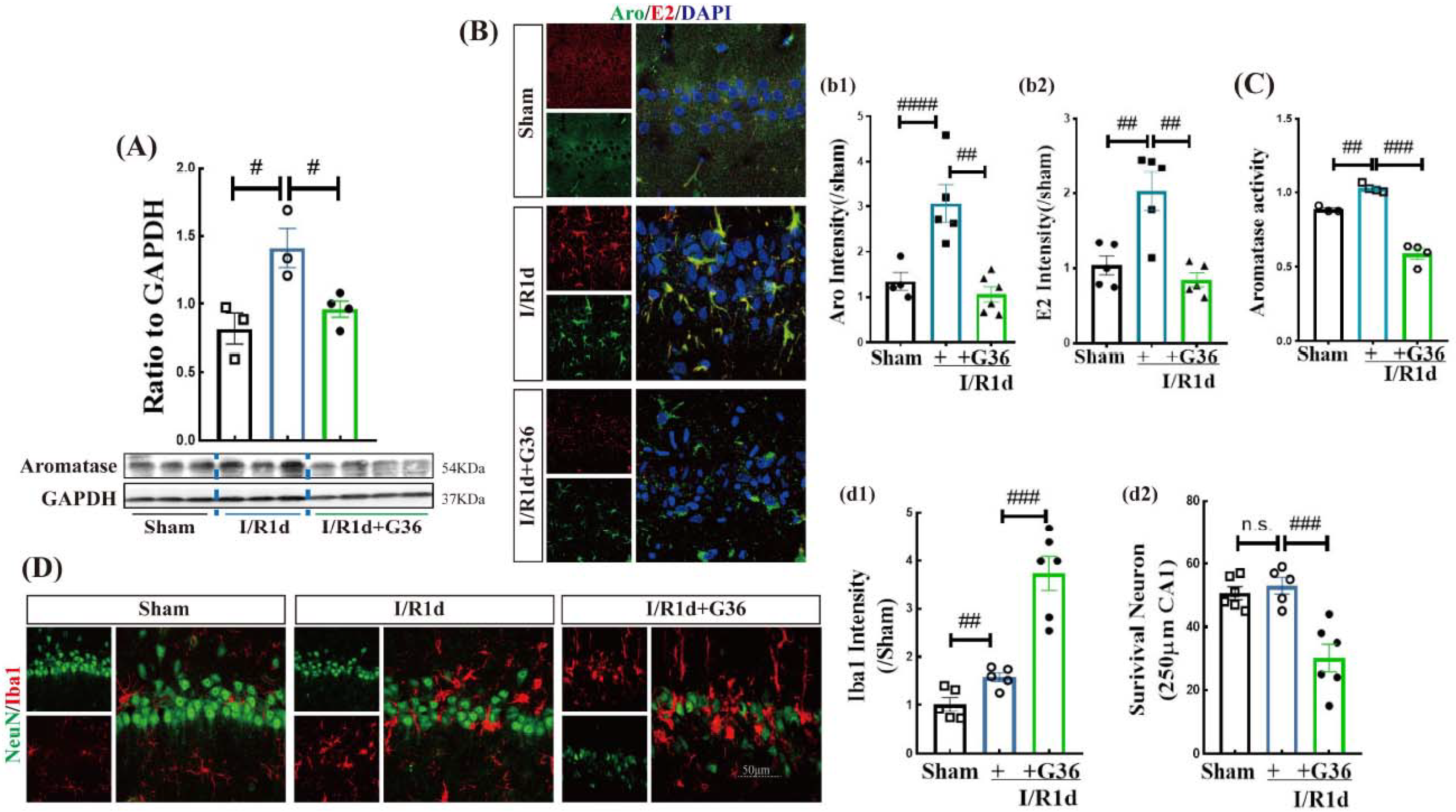
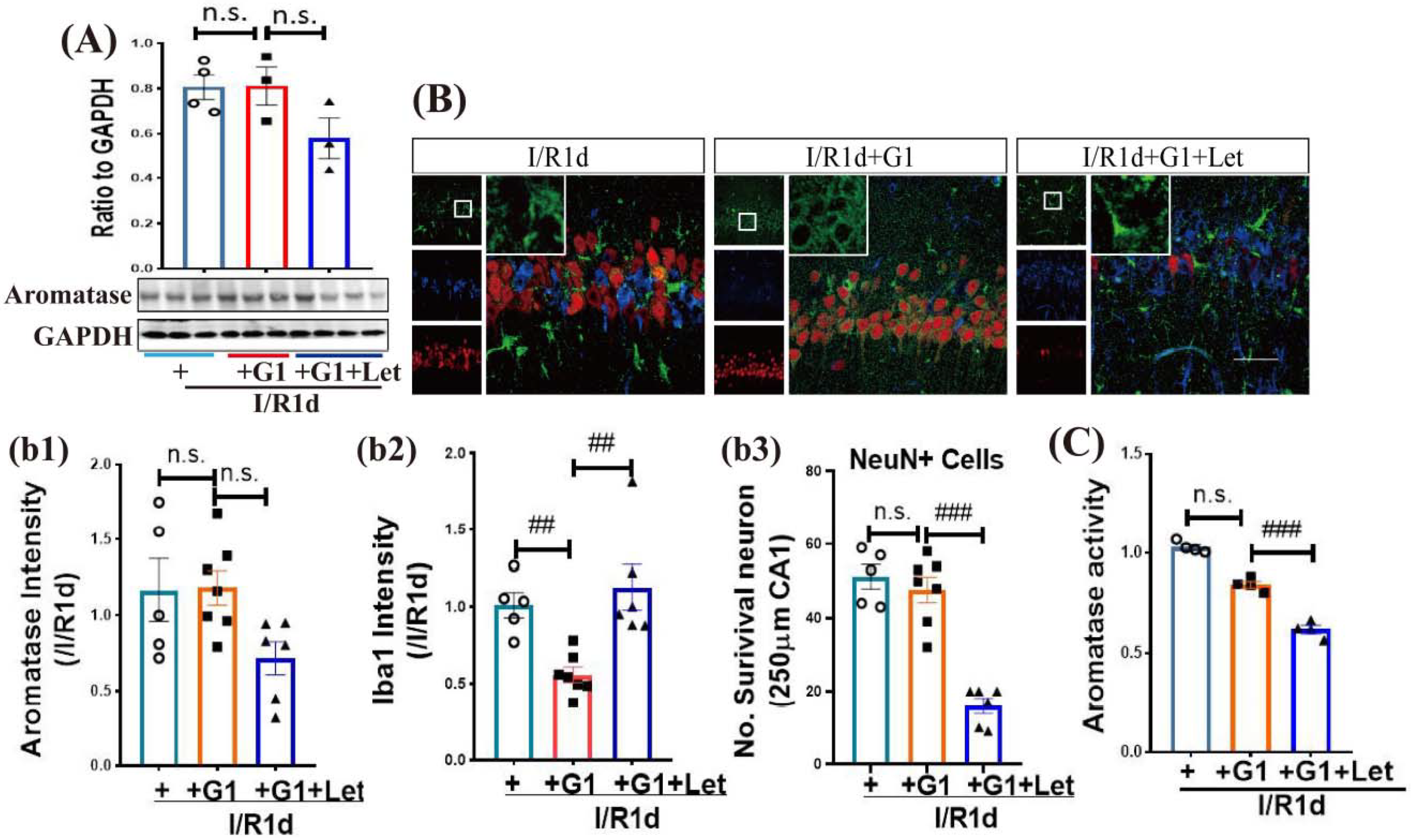
3.4. GPER Plays an Important Role in the Neuroprotective Effect of Aromatase-BDE2 Signaling on the Hippocampal CA1 Region after GCI
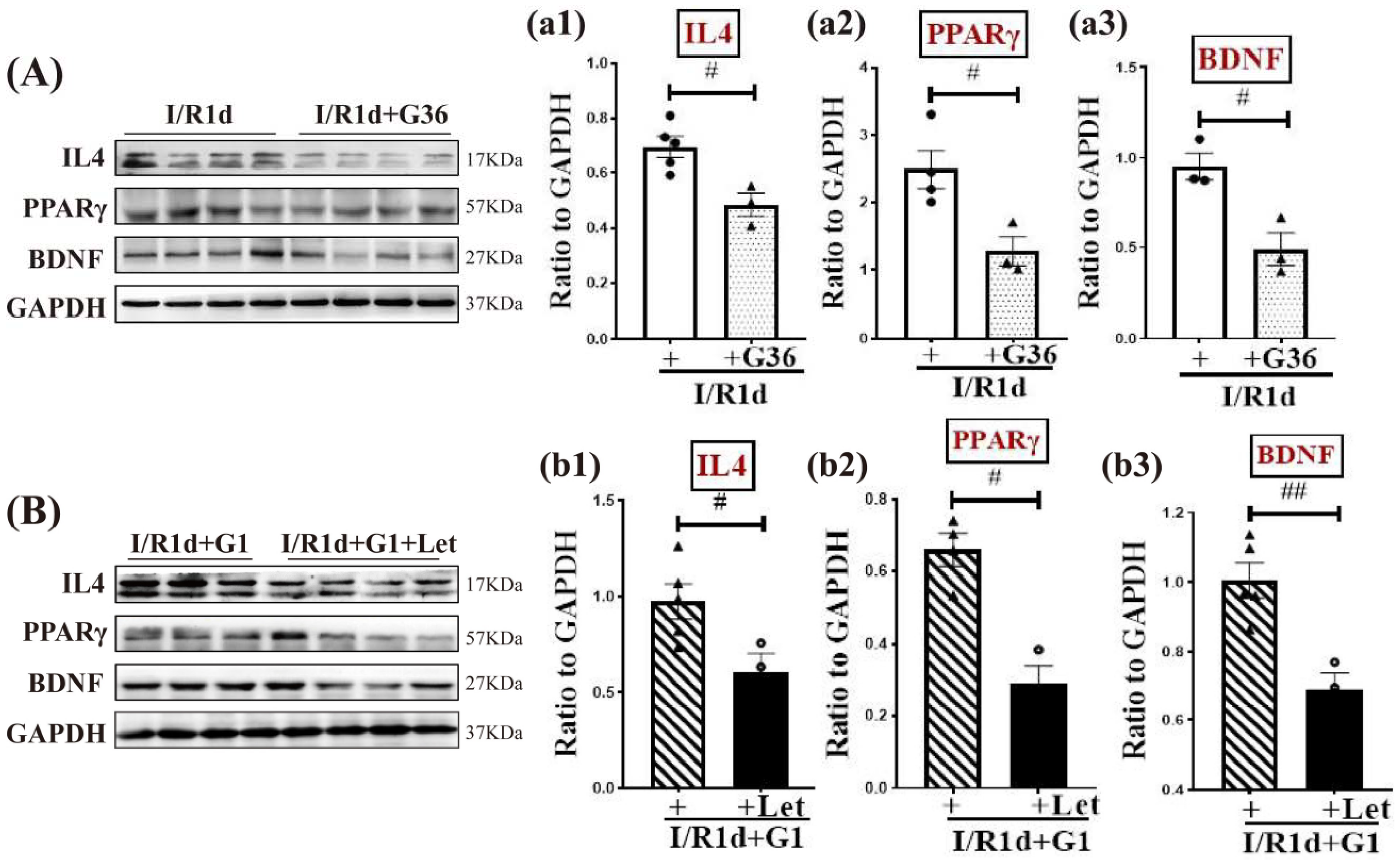
3.5. The Anti-Inflammatory Role of G1 Is Dependent upon Aro-BDE2 Signaling in the Hippocampal CA1 Region Following GCI
3.6. Synergistic Action of G1 and BDE2 Promotes Protein Expression of IL-4, PPARγ, and BDNF in the Hippocampal CA1 Region in the Early Phase of GCI
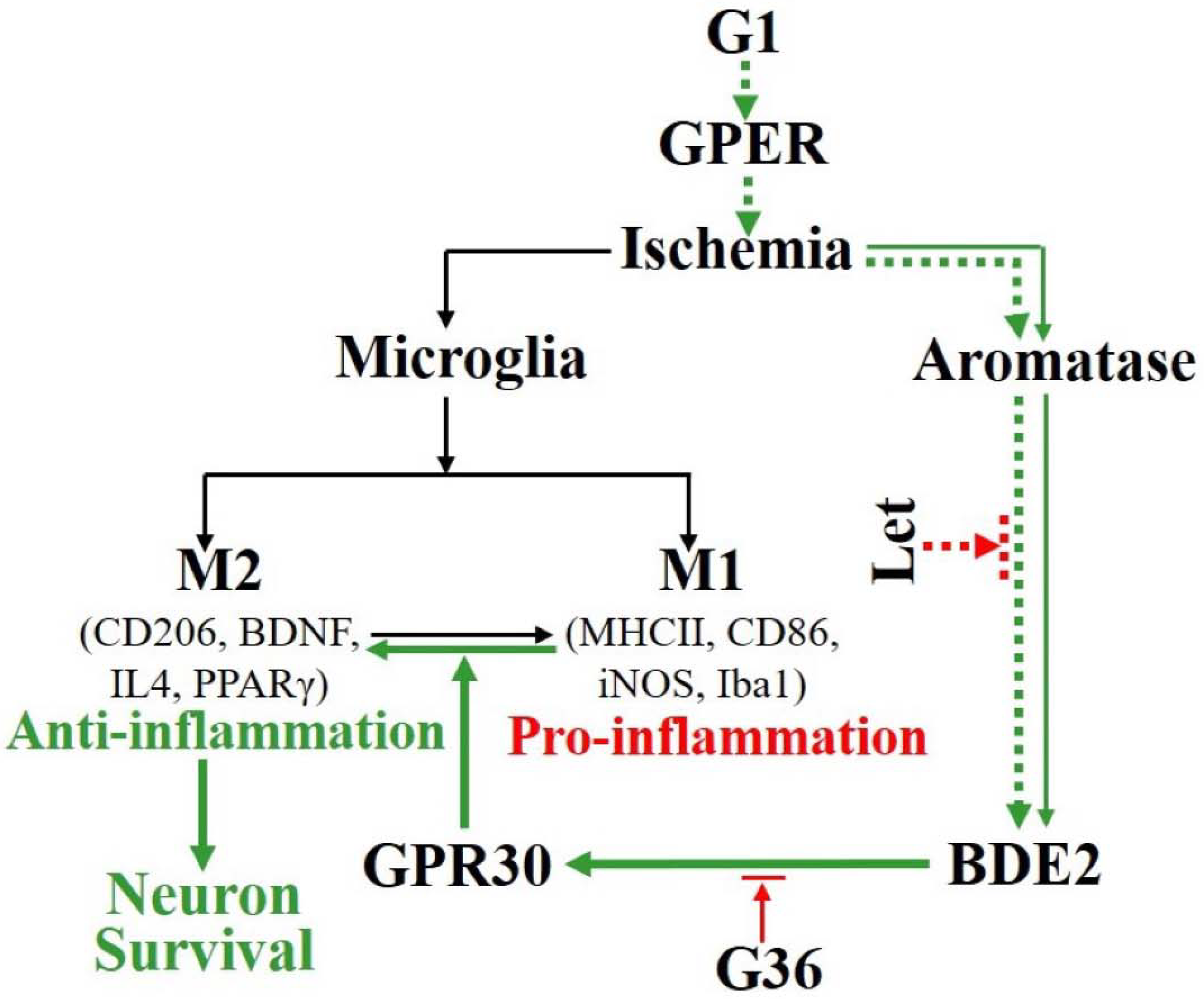
4. Discussion
5. Conclusions
Supplementary Materials
Author Contributions
Funding
Institutional Review Board Statement
Informed Consent Statement
Data Availability Statement
Acknowledgments
Conflicts of Interest
References
- Kim, M.J.; Cho, J.H.; Cho, J.H.; Park, J.H.; Ahn, J.H.; Tae, H.J.; Cho, G.S.; Yan, B.C.; Hwang, I.K.; Lee, C.H.; et al. Impact of hyperthermia before and during ischemia-reperfusion on neuronal damage and gliosis in the gerbil hippocampus induced by transient cerebral ischemia. J. Neurol. Sci. 2015, 348, 101–110. [Google Scholar] [CrossRef] [PubMed]
- Lee, J.C.; Chen, B.H.; Cho, J.H.; Kim, I.H.; Ahn, J.H.; Park, J.H.; Tae, H.J.; Cho, G.S.; Yan, B.C.; Kim, D.W.; et al. Changes in the expression of DNA-binding/differentiation protein inhibitors in neurons and glial cells of the gerbil hippocampus following transient global cerebral ischemia. Mol. Med. Rep. 2015, 11, 2477–2485. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Wood, P.L. Microglia as a unique cellular target in the treatment of stroke: Potential neurotoxic mediators produced by activated microglia. Neurol. Res. 1995, 17, 242–248. [Google Scholar] [CrossRef] [PubMed]
- Kriz, J.; Lalancette-Hébert, M. Inflammation, plasticity and real-time imaging after cerebral ischemia. Acta Neuropathol. 2009, 117, 497–509. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Cervantes, M.; Moralí, G.; Letechipía-Vallejo, G. Melatonin and ischemia-reperfusion injury of the brain. J. Pineal Res. 2008, 45, 1–7. [Google Scholar] [CrossRef]
- Thakkar, R.; Wang, R.; Wang, J.; Vadlamudi, R.K.; Brann, D.W. 17β-Estradiol Regulates Microglia Activation and Polarization in the Hippocampus Following Global Cerebral Ischemia. Oxidative Med. Cell. Longev. 2018, 2018, 4248526. [Google Scholar] [CrossRef] [Green Version]
- Mesquida-Veny, F.; Del Río, J.; Hervera, A. Macrophagic and microglial complexity after neuronal injury. Prog. Neurobiol. 2020, 101970. [Google Scholar] [CrossRef]
- Liao, B.; Zhao, W.; Beers, D.R.; Henkel, J.S.; Appel, S.H. Transformation from a neuroprotective to a neurotoxic microglial phenotype in a mouse model of ALS. Exp. Neurol. 2012, 237, 147–152. [Google Scholar] [CrossRef] [Green Version]
- Calippe, B.; Douin-Echinard, V.; Laffargue, M.; Laurell, H.; Rana-Poussine, V.; Pipy, B.; Guéry, J.C.; Bayard, F.; Arnal, J.F.; Gourdy, P. Chronic estradiol administration in vivo promotes the proinflammatory response of macrophages to TLR4 activation: Involvement of the phosphatidylinositol 3-kinase pathway. J. Immunol. 2008, 180, 7980–7988. [Google Scholar] [CrossRef] [Green Version]
- Rettew, J.A.; Huet, Y.M.; Marriott, I. Estrogens augment cell surface TLR4 expression on murine macrophages and regulate sepsis susceptibility in vivo. Endocrinology 2009, 150, 3877–3884. [Google Scholar] [CrossRef]
- Brann, D.W.; Lu, Y.; Wang, J.; Zhang, Q.; Thakkar, R.; Sareddy, G.R.; Pratap, U.P.; Tekmal, R.R.; Vadlamudi, R.K. Brain-derived estrogen and neural function. Neurosci. Biobehav. Rev. 2022, 132, 793–817. [Google Scholar] [CrossRef]
- Garcia-Ovejero, D.; Azcoitia, I.; Doncarlos, L.L.; Melcangi, R.C.; Garcia-Segura, L.M. Glia-neuron crosstalk in the neuroprotective mechanisms of sex steroid hormones. Brain Research. Brain Res. Rev. 2005, 48, 273–286. [Google Scholar] [CrossRef]
- Lu, Y.; Sareddy, G.R.; Wang, J.; Zhang, Q.; Tang, F.L.; Pratap, U.P.; Tekmal, R.R.; Vadlamudi, R.K.; Brann, D.W. Neuron-Derived Estrogen Is Critical for Astrocyte Activation and Neuroprotection of the Ischemic Brain. J. Neurosci. Off. J. Soc. Neurosci. 2020, 40, 7355–7374. [Google Scholar] [CrossRef]
- Ishihara, Y.; Itoh, K.; Tanaka, M.; Tsuji, M.; Kawamoto, T.; Kawato, S.; Vogel, C.F.A.; Yamazaki, T. Potentiation of 17β-estradiol synthesis in the brain and elongation of seizure latency through dietary supplementation with docosahexaenoic acid. Sci. Rep. 2017, 7, 6268. [Google Scholar] [CrossRef] [Green Version]
- Chu, X.L.; Zhang, T.; Kong, F.X.; Xiao, Y.Y.; Chi, X.X. The regulatory effect of genistein on P450 aromatase and follicle-stimulating hormone receptor in mouse experimental model of menopausal metabolic syndrome. J. Anim. Physiol. Anim. Nutr. 2020, 104, 371–378. [Google Scholar] [CrossRef]
- Yilmaz, M.B.; Wolfe, A.; Cheng, Y.H.; Glidewell-Kenney, C.; Jameson, J.L.; Bulun, S.E. Aromatase promoter If is regulated by estrogen receptor alpha (ESR1) in mouse hypothalamic neuronal cell lines. Biol. Reprod. 2009, 81, 956–965. [Google Scholar] [CrossRef]
- Sierra, A.; Veiga, S.; Honda, S.-I.; Harada, N.; Garcia-Segura, L.M. Brain aromatase is neuroprotective. J. Neurobiol. 2001, 47, 318–329. [Google Scholar] [CrossRef] [Green Version]
- Wang, J.; Sareddy, G.R.; Lu, Y.; Pratap, U.P.; Tang, F.; Greene, K.M.; Meyre, P.L.; Tekmal, R.R.; Vadlamudi, R.K.; Brann, D.W. Astrocyte-Derived Estrogen Regulates Reactive Astrogliosis and is Neuroprotective following Ischemic Brain Injury. J. Neurosci. Off. J. Soc. Neurosci. 2020, 40, 9751–9771. [Google Scholar] [CrossRef]
- Zhang, Q.G.; Wang, R.; Tang, H.; Dong, Y.; Chan, A.; Sareddy, G.R.; Vadlamudi, R.K.; Brann, D.W. Brain-derived estrogen exerts anti-inflammatory and neuroprotective actions in the rat hippocampus. Mol. Cell. Endocrinol. 2014, 389, 84–91. [Google Scholar] [CrossRef] [Green Version]
- Fester, L.; Prange-Kiel, J.; Jarry, H.; Rune, G.M. Estrogen synthesis in the hippocampus. Cell Tissue Res. 2011, 345, 285–294. [Google Scholar] [CrossRef]
- Lu, Y.; Sareddy, G.R.; Wang, J.; Wang, R.; Li, Y.; Dong, Y.; Zhang, Q.; Liu, J.; O’Connor, J.C.; Xu, J.; et al. Neuron-Derived Estrogen Regulates Synaptic Plasticity and Memory. J. Neurosci. Off. J. Soc. Neurosci. 2019, 39, 2792–2809. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Hutson, D.D.; Gurrala, R.; Ogola, B.O.; Zimmerman, M.A.; Mostany, R.; Satou, R.; Lindsey, S.H. Estrogen receptor profiles across tissues from male and female Rattus norvegicus. Biol. Sex Differ. 2019, 10, 4. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Zhang, Z.; Qin, P.; Deng, Y.; Ma, Z.; Guo, H.; Guo, H.; Hou, Y.; Wang, S.; Zou, W.; Sun, Y.; et al. The novel estrogenic receptor GPR30 alleviates ischemic injury by inhibiting TLR4-mediated microglial inflammation. J. Neuroinflamm. 2018, 15, 206. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Bai, N.; Zhang, Q.; Zhang, W.; Liu, B.; Yang, F.; Brann, D.; Wang, R. G-protein-coupled estrogen receptor activation upregulates interleukin-1 receptor antagonist in the hippocampus after global cerebral ischemia: Implications for neuronal self-defense. J. Neuroinflamm. 2020, 17, 45. [Google Scholar] [CrossRef] [PubMed]
- Kumar, A.; Foster, T.C. G Protein-Coupled Estrogen Receptor: Rapid Effects on Hippocampal-Dependent Spatial Memory and Synaptic Plasticity. Front. Endocrinol. 2020, 11, 385. [Google Scholar] [CrossRef]
- Yang, L.C.; Zhang, Q.G.; Zhou, C.F.; Yang, F.; Zhang, Y.D.; Wang, R.M.; Brann, D.W. Extranuclear estrogen receptors mediate the neuroprotective effects of estrogen in the rat hippocampus. PLoS ONE 2010, 5, e9851. [Google Scholar] [CrossRef]
- Chen, X.; Zhang, M.; Ahmed, M.; Surapaneni, K.M.; Veeraraghavan, V.P.; Arulselvan, P. Neuroprotective effects of ononin against the aluminium chloride-induced Alzheimer’s disease in rats. Saudi J. Biol. Sci. 2021, 28, 4232–4239. [Google Scholar] [CrossRef]
- Strosznajder, A.K.; Wojtowicz, S.; Jezyna, M.J.; Sun, G.Y.; Strosznajder, J.B. Recent Insights on the Role of PPAR-beta/delta in Neuroinflammation and Neurodegeneration, and Its Potential Target for Therapy. Neuromol. Med. 2021, 23, 86–98. [Google Scholar] [CrossRef]
- Mohamed, E.A.; Ahmed, H.I.; Zaky, H.S.; Badr, A.M. Sesame oil mitigates memory impairment, oxidative stress, and neurodegeneration in a rat model of Alzheimer’s disease. A pivotal role of NF-kappaB/p38MAPK/BDNF/PPAR-gamma pathways. J Ethnopharmacol. 2021, 267, 113468. [Google Scholar] [CrossRef]
- Scott, E.L.; Zhang, Q.G.; Vadlamudi, R.K.; Brann, D.W. Premature menopause and risk of neurological disease: Basic mechanisms and clinical implications. Mol. Cell. Endocrinol. 2014, 389, 2–6. [Google Scholar] [CrossRef]
- Chu, K.; Yin, B.; Wang, J.; Peng, G.; Liang, H.; Xu, Z.; Du, Y.; Fang, M.; Xia, Q.; Luo, B. Inhibition of P2X7 receptor ameliorates transient global cerebral ischemia/reperfusion injury via modulating inflammatory responses in the rat hippocampus. J. Neuroinflamm. 2012, 9, 69. [Google Scholar] [CrossRef] [Green Version]
- Wang, R.; Zhang, Q.; Han, D.; Xu, J.; Lü, Q.; Zhang, G. Inhibition of MLK3-MKK4/7-JNK1/2 pathway by Akt1 in exogenous estrogen-induced neuroprotection against transient global cerebral ischemia by a non-genomic mechanism in male rats. J. Neurochem. 2006, 99, 1543–1554. [Google Scholar] [CrossRef] [Green Version]
- Tang, H.; Zhang, Q.; Yang, L.; Dong, Y.; Khan, M.; Yang, F.; Brann, D.; Wang, R. Reprint of “GPR30 mediates estrogen rapid signaling and neuroprotection”. Mol. Cell. Endocrinol. 2014, 389, 92–98. [Google Scholar] [CrossRef]
- Liu, S.; Zhang, N.; Guo, Y.; Zhao, R.; Shi, T.; Feng, S.; Wang, S.; Yang, Q.; Li, X.; Wu, Y.; et al. G-protein-coupled receptor 30 mediates rapid neuroprotective effects of estrogen via depression of NR2B-containing NMDA receptors. J. Neurosci. Off. J. Soc. Neurosci. 2012, 32, 4887–4900. [Google Scholar] [CrossRef] [Green Version]
- Kubota, T.; Matsumoto, H.; Kirino, Y. Ameliorative effect of membrane-associated estrogen receptor G protein coupled receptor 30 activation on object recognition memory in mouse models of Alzheimer’s disease. J. Pharmacol. Sci. 2016, 131, 219–222. [Google Scholar] [CrossRef] [Green Version]
- Alavi, M.; Karimi, G.; Roohbakhsh, A. The role of orphan G protein-coupled receptors in the pathophysiology of multiple sclerosis: A review. Life Sci. 2019, 224, 33–40. [Google Scholar] [CrossRef]
- Tang, H.; Zhang, Q.; Yang, L.; Dong, Y.; Khan, M.; Yang, F.; Brann, D.W.; Wang, R. GPR30 mediates estrogen rapid signaling and neuroprotection. Mol. Cell. Endocrinol. 2014, 387, 52–58. [Google Scholar] [CrossRef] [Green Version]
- Roux, P.P.; Topisirovic, I. Signaling Pathways Involved in the Regulation of mRNA Translation. Mol. Cell. Biol. 2018, 38. [Google Scholar] [CrossRef] [Green Version]
- Valjent, E.; Caboche, J.; Vanhoutte, P. Mitogen-activated protein kinase/extracellular signal-regulated kinase induced gene regulation in brain: A molecular substrate for learning and memory? Mol. Neurobiol. 2001, 23, 83–99. [Google Scholar] [CrossRef]
- Roux, P.P.; Shahbazian, D.; Vu, H.; Holz, M.K.; Cohen, M.S.; Taunton, J.; Sonenberg, N.; Blenis, J. RAS/ERK signaling promotes site-specific ribosomal protein S6 phosphorylation via RSK and stimulates cap-dependent translation. J. Biol. Chem. 2007, 282, 14056–14064. [Google Scholar] [CrossRef]
- Sánchez-Alegría, K.; Flores-León, M.; Avila-Muñoz, E.; Rodríguez-Corona, N.; Arias, C. PI3K Signaling in Neurons: A Central Node for the Control of Multiple Functions. Int. J. Mol. Sci. 2018, 19, 3725. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Duncan, K.; Saldanha, C. Central aromatization: A dramatic and responsive defense against threat and trauma to the vertebrate brain. Front. Neuroendocrinol. 2020, 56, 100816. [Google Scholar] [CrossRef] [PubMed]
- Saldanha, C.J. Estrogen as a Neuroprotectant in Both Sexes: Stories from the Bird Brain. Front. Neurol. 2020, 11, 497. [Google Scholar] [CrossRef]
- Pedersen, A.L.; Brownrout, J.L.; Saldanha, C.J. Neuroinflammation and neurosteroidogenesis: Reciprocal modulation during injury to the adult zebra finch brain. Physiol. Behav. 2018, 187, 51–56. [Google Scholar] [CrossRef] [PubMed]
- Mehos, C.J.; Nelson, L.H.; Saldanha, C.J. A Quantification of the Injury-Induced Changes in Central Aromatase, Oestrogenic Milieu and Steroid Receptor Expression in the Zebra Finch. J. Neuroendocr. 2016, 28, 12348. [Google Scholar] [CrossRef]
- Azcoitia, I.; Sierra, A.; Veiga, S.; Garcia-Segura, L. Aromatase Expression by Reactive Astroglia Is Neuroprotective. Ann. N. Y. Acad. Sci. 2003, 1007, 298–305. [Google Scholar] [CrossRef]
- McCullough, L.D.; Blizzard, K.; Simpson, E.R.; Öz, O.K.; Hurn, P.D. Aromatase Cytochrome P450 and Extragonadal Estrogen Play a Role in Ischemic Neuroprotection. J. Neurosci. 2003, 23, 8701–8705. [Google Scholar] [CrossRef] [Green Version]
- Miller, N.R.; Jover, T.; Cohen, H.W.; Zukin, R.S.; Etgen, A.M. Estrogen can act via estrogen receptor alpha and beta to protect hippocampal neurons against global ischemia-induced cell death. Endocrinology 2005, 146, 3070–3079. [Google Scholar] [CrossRef] [Green Version]
- Dai, X.; Chen, L.; Sokabe, M. Neurosteroid estradiol rescues ischemia-induced deficit in the long-term potentiation of rat hippocampal CA1 neurons. Neuropharmacology 2007, 52, 1124–1138. [Google Scholar] [CrossRef]
- Zhang, Q.G.; Han, D.; Wang, R.M.; Dong, Y.; Yang, F.; Vadlamudi, R.K.; Brann, D.W. C terminus of Hsc70-interacting protein (CHIP)-mediated degradation of hippocampal estrogen receptor-alpha and the critical period hypothesis of estrogen neuroprotection. Proc. Natl. Acad. Sci. USA 2011, 108, E617–E624. [Google Scholar] [CrossRef]







Disclaimer/Publisher’s Note: The statements, opinions and data contained in all publications are solely those of the individual author(s) and contributor(s) and not of MDPI and/or the editor(s). MDPI and/or the editor(s) disclaim responsibility for any injury to people or property resulting from any ideas, methods, instructions or products referred to in the content. |
© 2023 by the authors. Licensee MDPI, Basel, Switzerland. This article is an open access article distributed under the terms and conditions of the Creative Commons Attribution (CC BY) license (https://creativecommons.org/licenses/by/4.0/).
Share and Cite
Xu, J.; Bai, J.; Gao, F.; Xu, C.; Huang, Y.; Li, D.; Wang, L.; Wang, R. Anti-Inflammatory Actions of G-Protein-Coupled Estrogen Receptor 1 (GPER) and Brain-Derived Estrogen Following Cerebral Ischemia in Ovariectomized Rats. Biology 2023, 12, 99. https://doi.org/10.3390/biology12010099
Xu J, Bai J, Gao F, Xu C, Huang Y, Li D, Wang L, Wang R. Anti-Inflammatory Actions of G-Protein-Coupled Estrogen Receptor 1 (GPER) and Brain-Derived Estrogen Following Cerebral Ischemia in Ovariectomized Rats. Biology. 2023; 12(1):99. https://doi.org/10.3390/biology12010099
Chicago/Turabian StyleXu, Jing, Jing Bai, Fujia Gao, Chao Xu, Yuanyuan Huang, Danyang Li, Lu Wang, and Ruimin Wang. 2023. "Anti-Inflammatory Actions of G-Protein-Coupled Estrogen Receptor 1 (GPER) and Brain-Derived Estrogen Following Cerebral Ischemia in Ovariectomized Rats" Biology 12, no. 1: 99. https://doi.org/10.3390/biology12010099
APA StyleXu, J., Bai, J., Gao, F., Xu, C., Huang, Y., Li, D., Wang, L., & Wang, R. (2023). Anti-Inflammatory Actions of G-Protein-Coupled Estrogen Receptor 1 (GPER) and Brain-Derived Estrogen Following Cerebral Ischemia in Ovariectomized Rats. Biology, 12(1), 99. https://doi.org/10.3390/biology12010099