
Identification of Critical Genes for Ovine Horn Development Based on Transcriptome during the Embryonic Period
 ,
,  and
and
Abstract
:Simple Summary
Abstract
1. Introduction
2. Materials and Methods
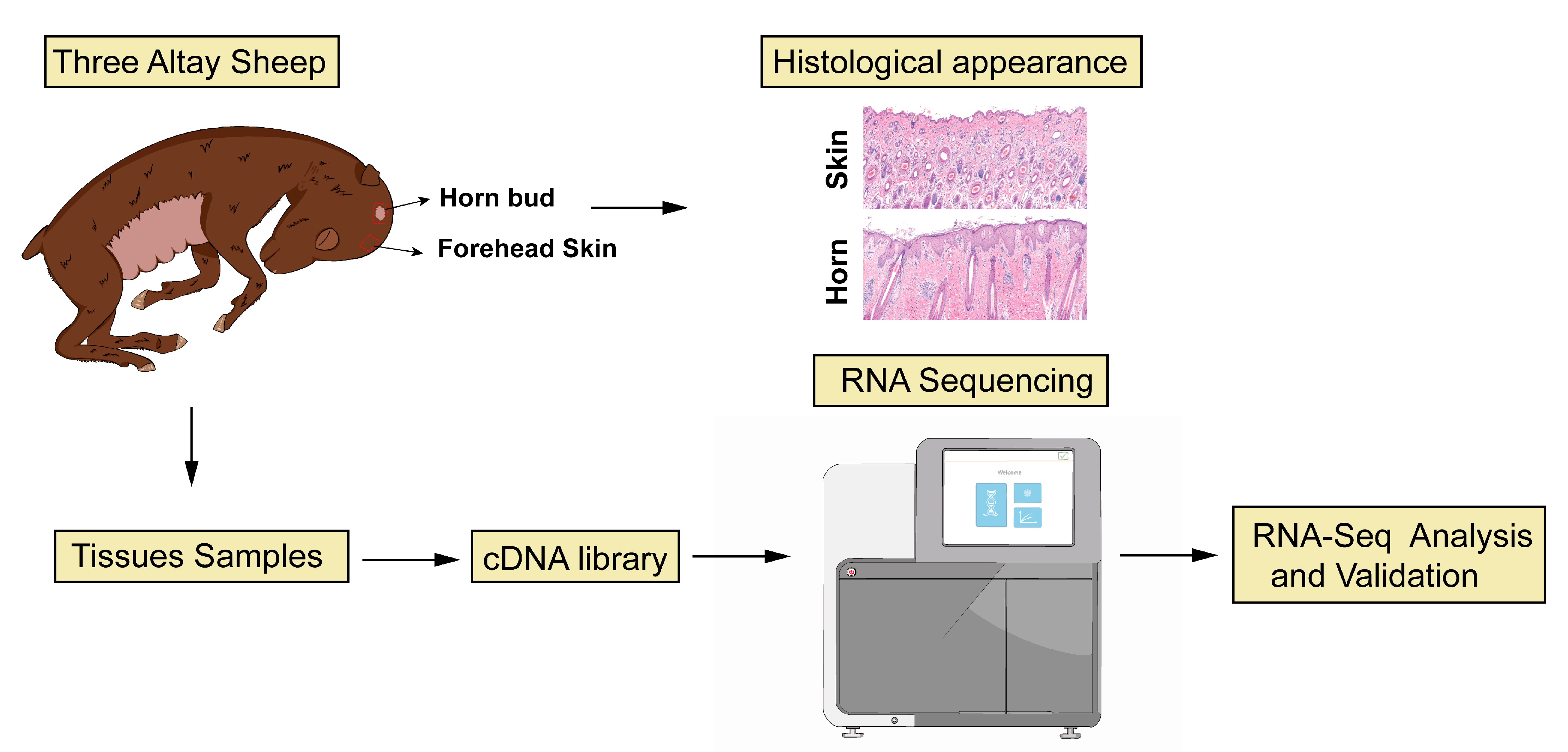
2.1. Sample Collection
2.2. Preparation of Histological Samples
2.3. RNA Extraction, cDNA Library Preparation, and Sequencing
2.4. Quality Control and Mapping of Reads
2.5. Analysis of Differentially Expressed Genes
2.6. Enrichment Analysis
2.7. Protein-Protein Interaction Network Construction
2.8. Quantitative Real-Time PCR (qRT-PCR) for Verification of DEGs
3. Results
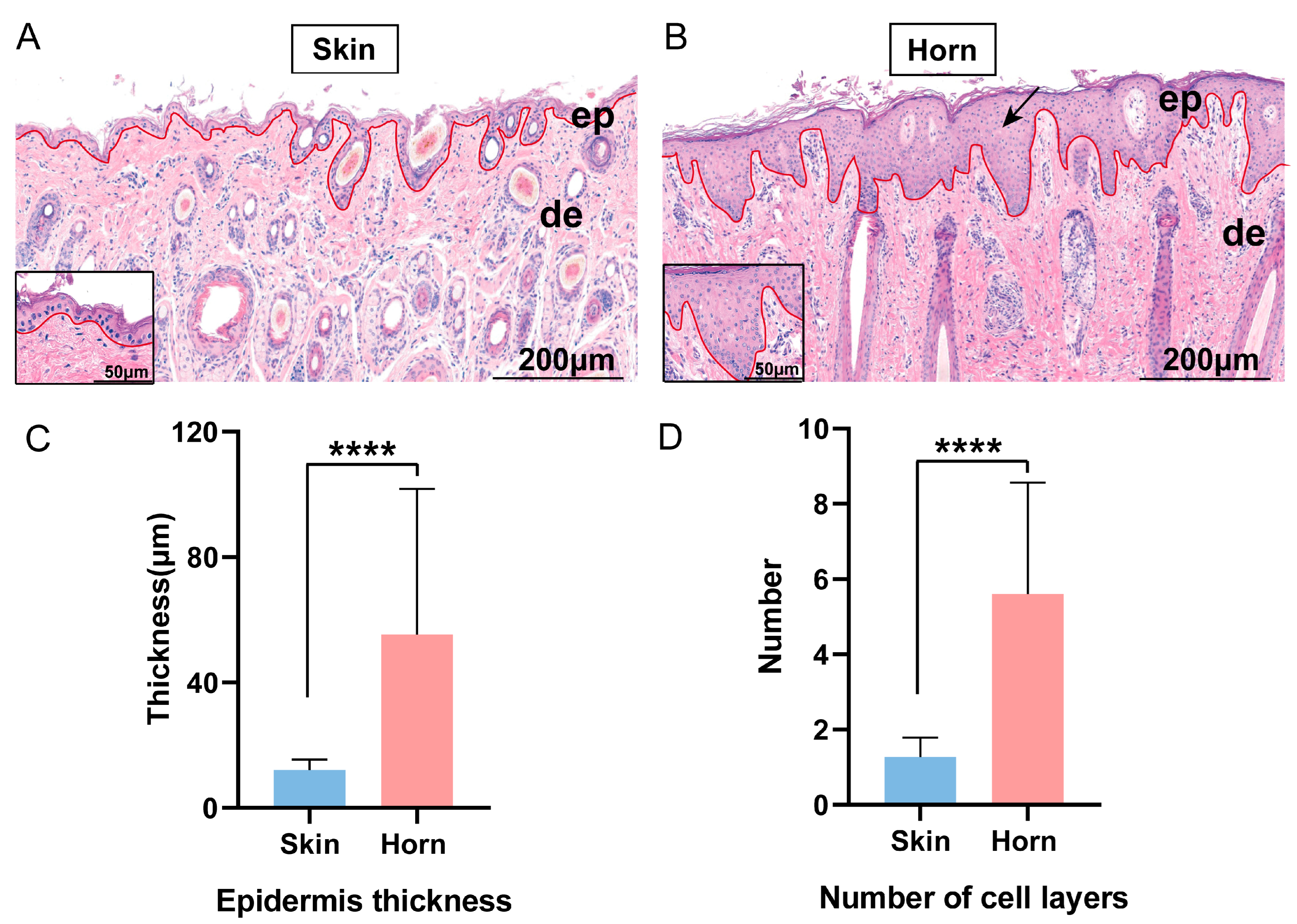
3.1. Histological Appearance
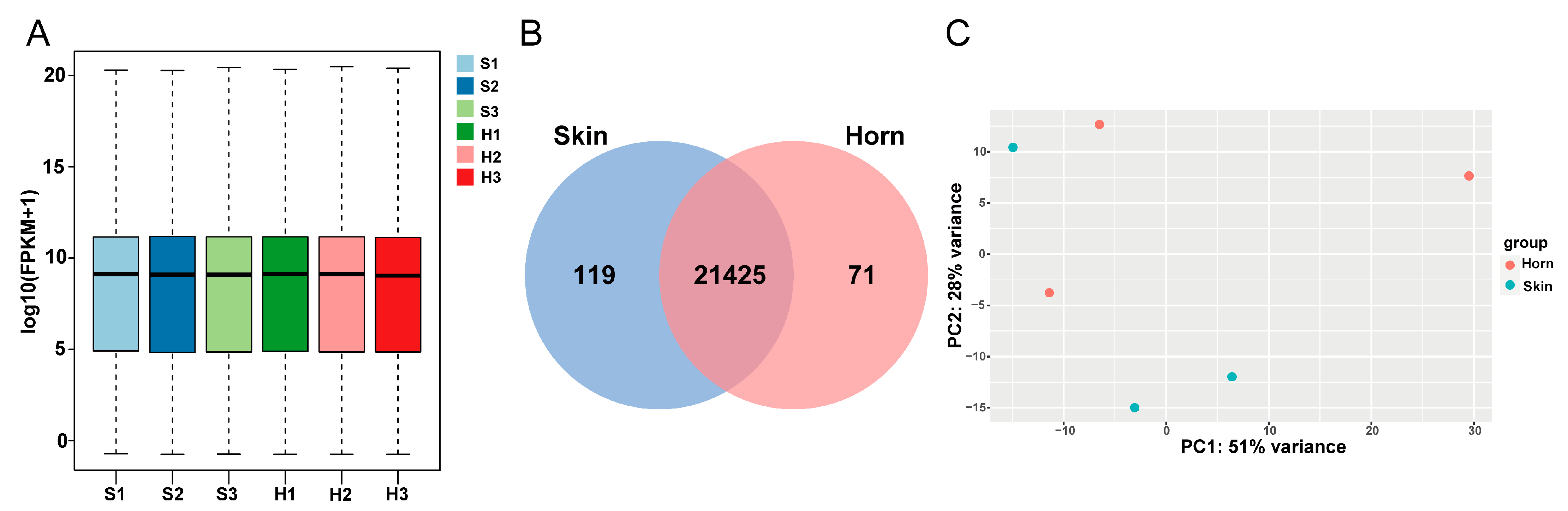
3.2. Overview of Transcriptomics Data
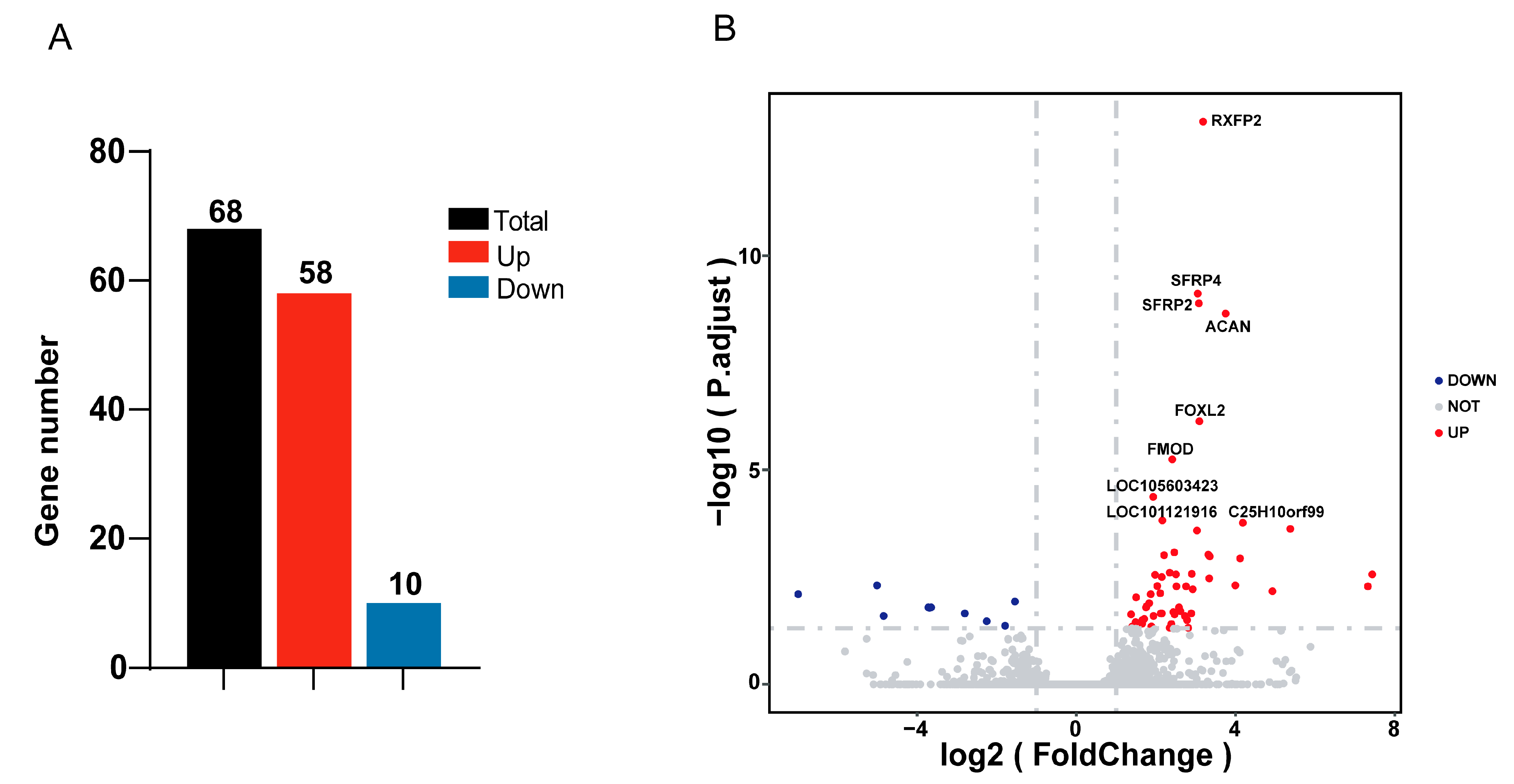
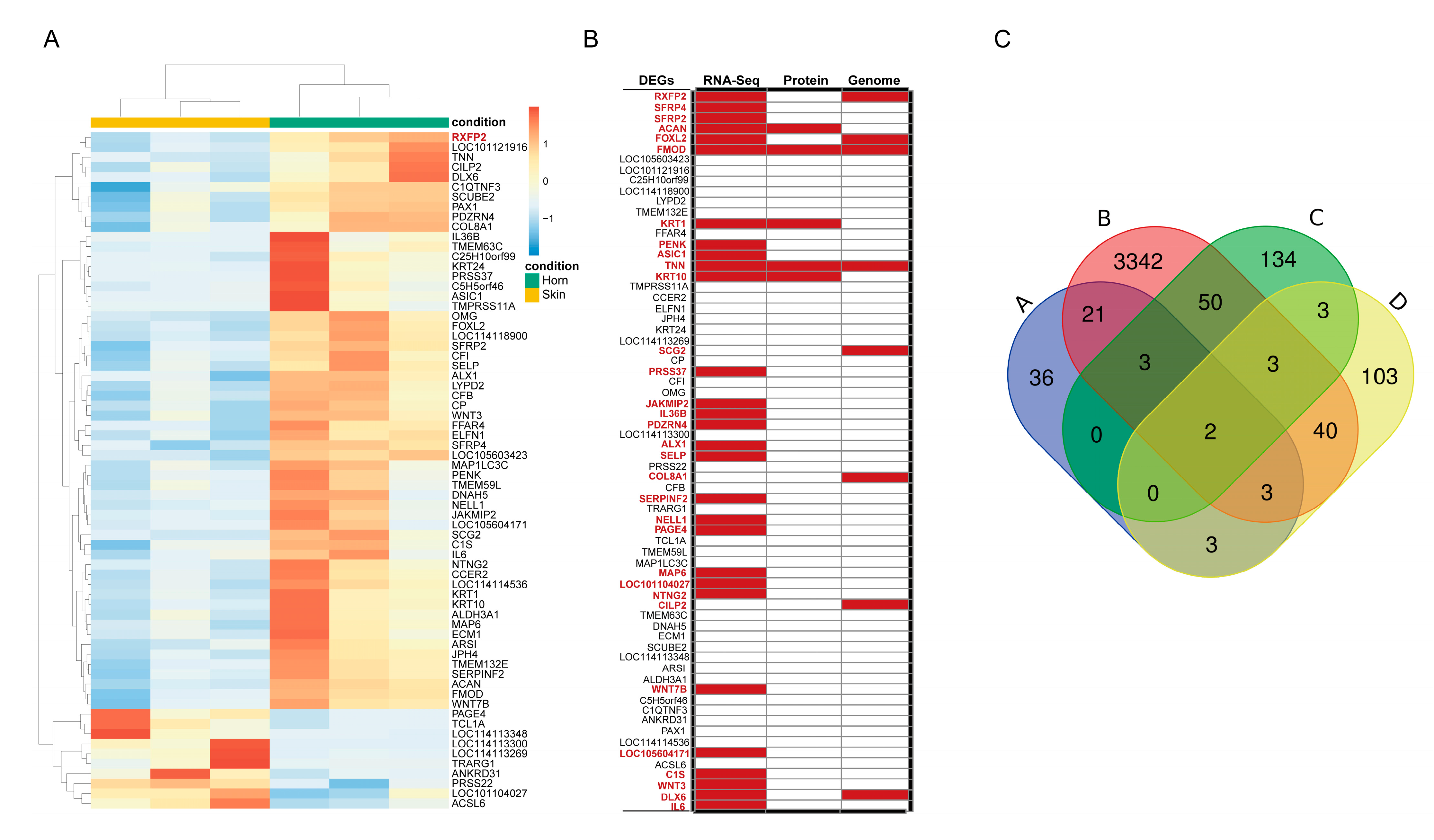
3.3. Differentially Expressed Genes Analysis
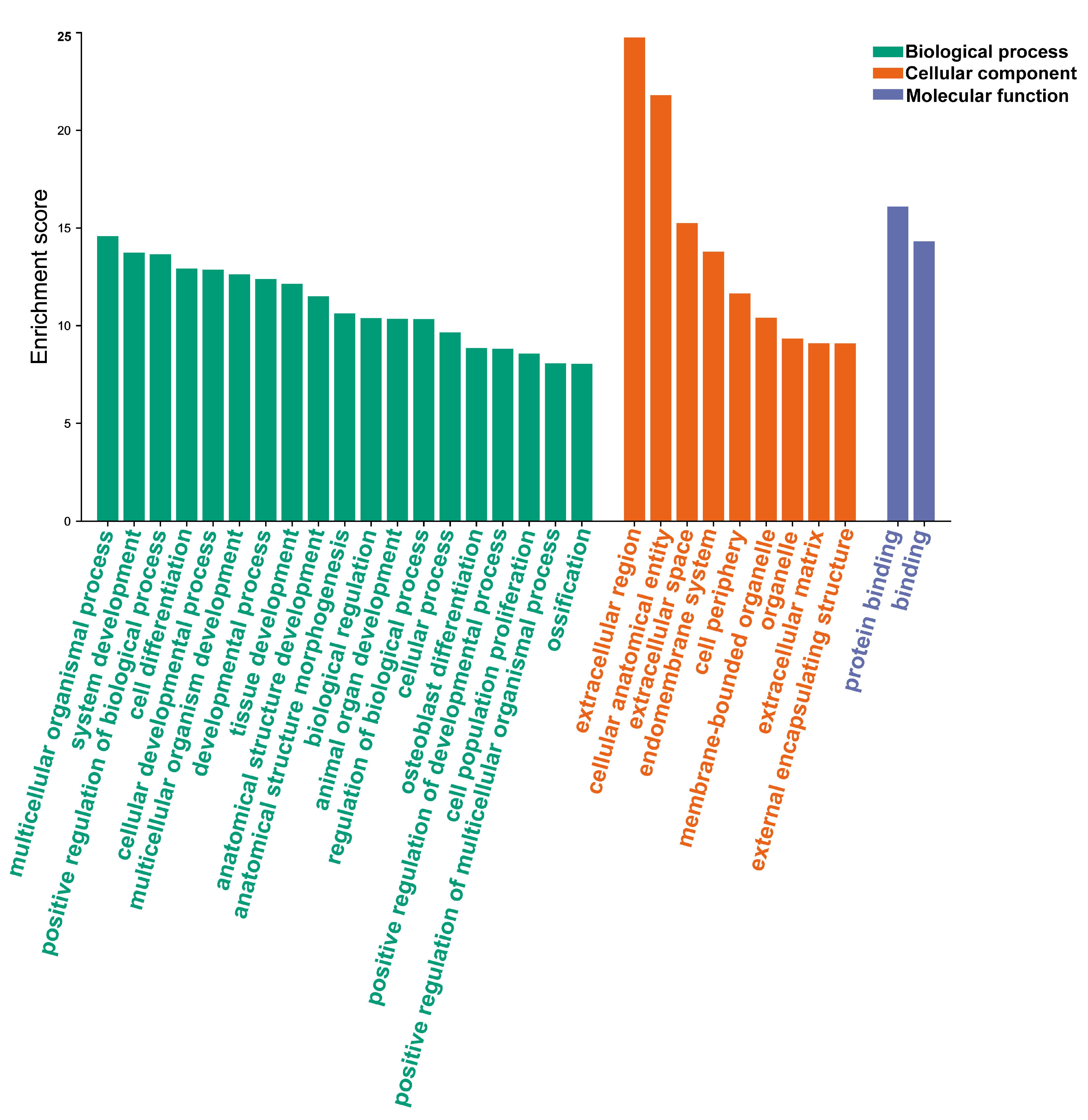
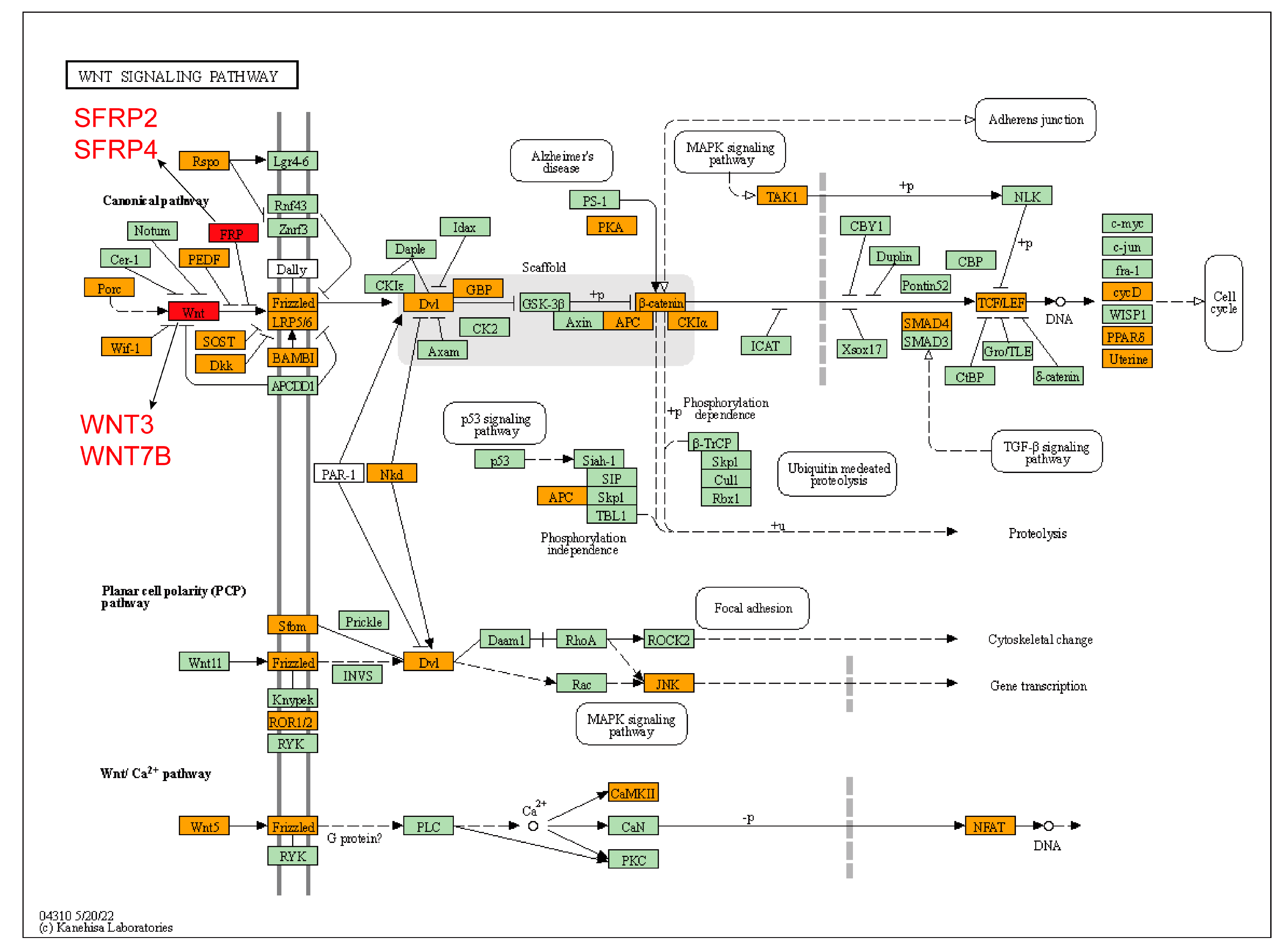
3.4. Analysis of Gene Function Enrichment
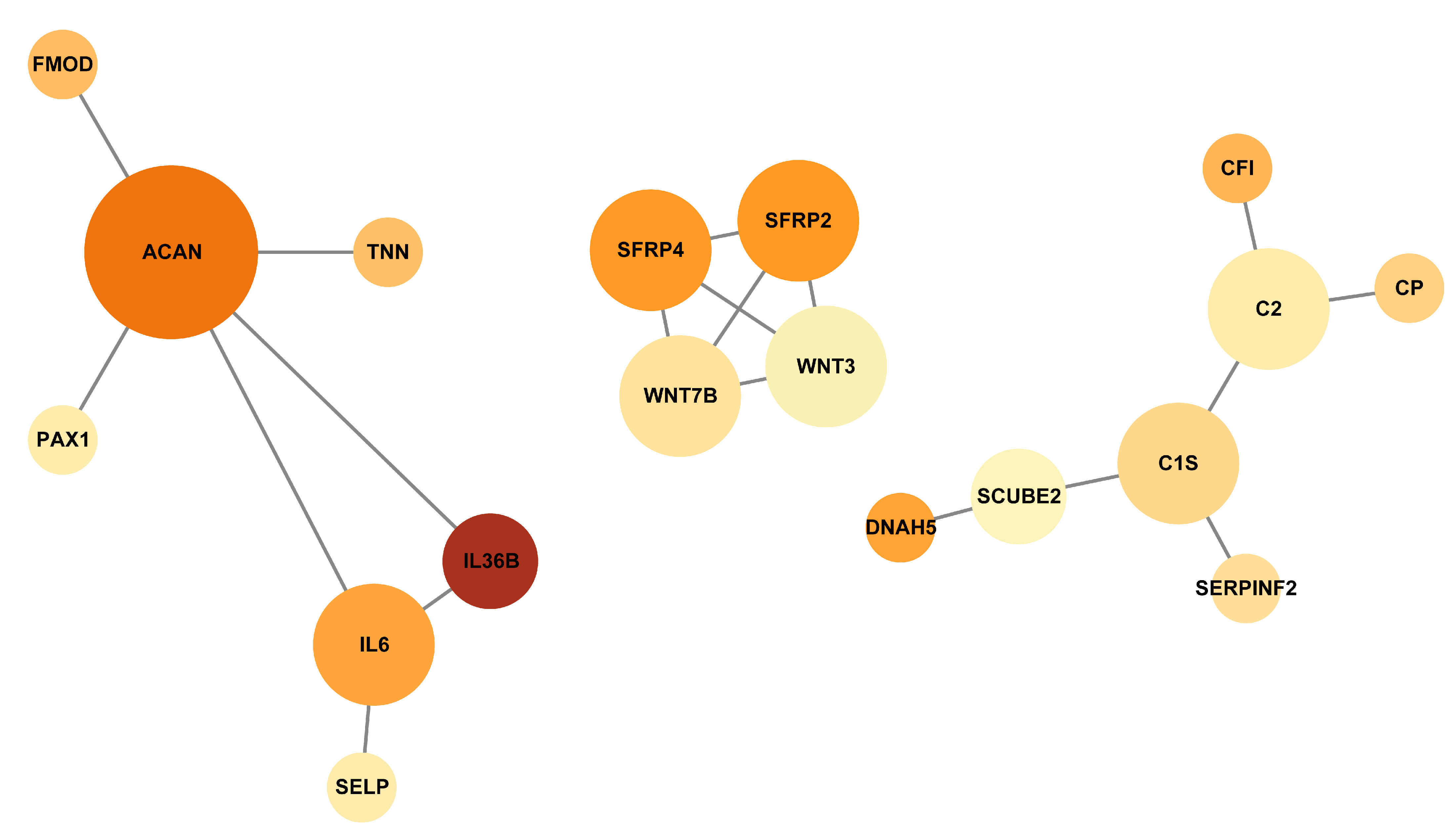
3.5. Protein–Protein Interaction Networks of the DEGs
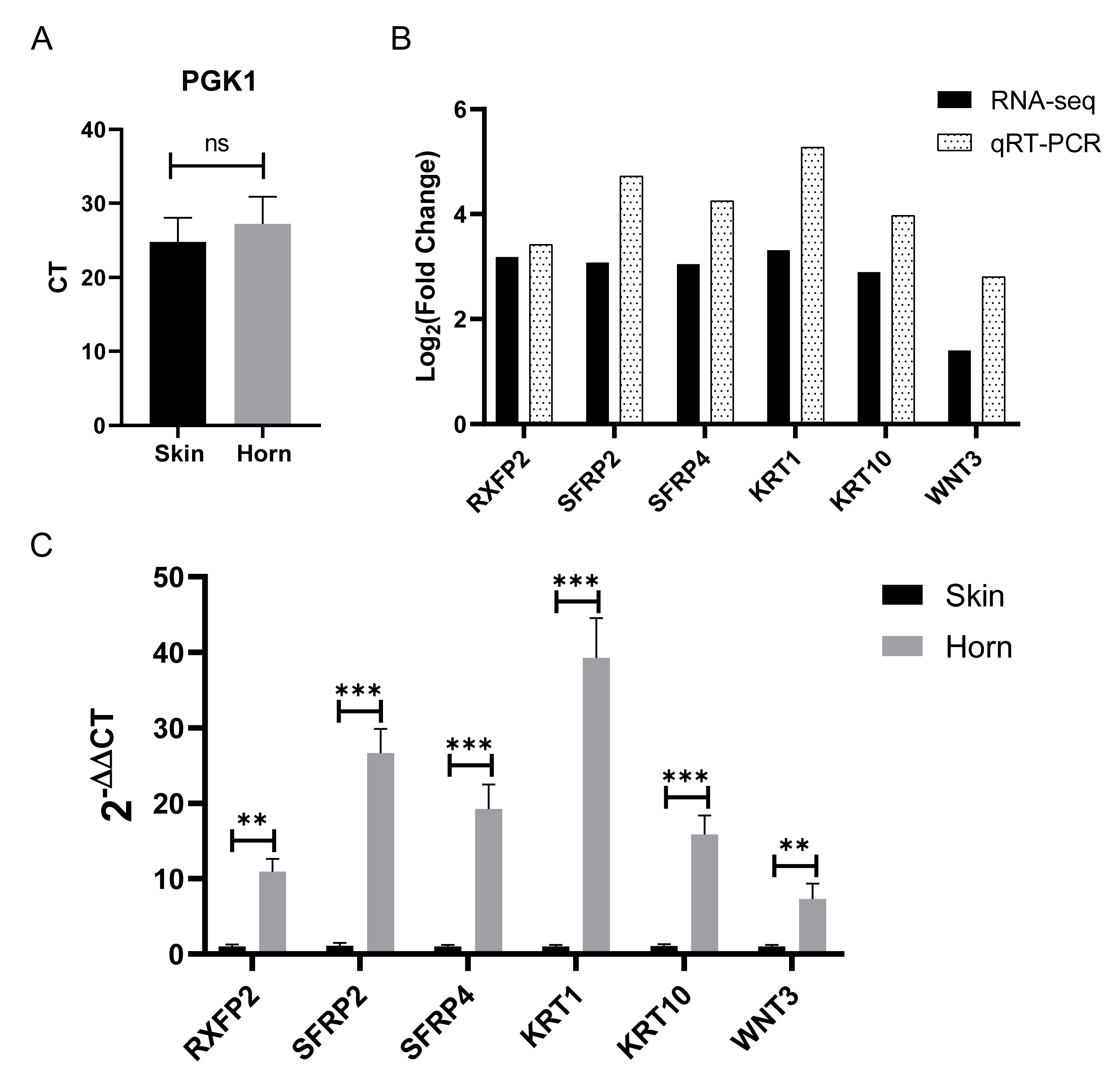
3.6. Validation of RNA-Seq Data
4. Discussion
5. Conclusions
Supplementary Materials
Author Contributions
Funding
Institutional Review Board Statement
Informed Consent Statement
Data Availability Statement
Conflicts of Interest
References
- Li, X.; Yang, J.; Shen, M.; Xie, X.-L.; Liu, G.-J.; Xu, Y.-X.; Lv, F.-H.; Yang, H.; Yang, Y.-L.; Liu, C.-B. Whole-genome resequencing of wild and domestic sheep identifies genes associated with morphological and agronomic traits. Nat. Commun. 2020, 11, 2815. [Google Scholar] [CrossRef]
- Davis, E.B.; Brakora, K.A.; Lee, A.H. Evolution of ruminant headgear: A review. Proc. R. Soc. B Biol. Sci. 2011, 278, 2857–2865. [Google Scholar] [CrossRef] [PubMed]
- Wang, Y.; Zhang, C.; Wang, N.; Li, Z.; Heller, R.; Liu, R.; Zhao, Y.; Han, J.; Pan, X.; Zheng, Z. Genetic basis of ruminant headgear and rapid antler regeneration. Science 2019, 364, eaav6335. [Google Scholar] [CrossRef] [PubMed]
- Poissant, J.; Davis, C.; Malenfant, R.; Hogg, J.; Coltman, D. QTL mapping for sexually dimorphic fitness-related traits in wild bighorn sheep. Heredity 2012, 108, 256–263. [Google Scholar] [CrossRef] [PubMed]
- Kardos, M.; Luikart, G.; Bunch, R.; Dewey, S.; Edwards, W.; McWilliam, S.; Stephenson, J.; Allendorf, F.W.; Hogg, J.T.; Kijas, J. Whole-genome resequencing uncovers molecular signatures of natural and sexual selection in wild bighorn sheep. Mol. Ecol. 2015, 24, 5616–5632. [Google Scholar] [CrossRef] [PubMed]
- Martin, A.; Festa-Bianchet, M.; Coltman, D.; Pelletier, F. Demographic drivers of age-dependent sexual selection. J. Evol. Biol. 2016, 29, 1437–1446. [Google Scholar] [CrossRef] [PubMed]
- Johnston, S.; Beraldi, D.; McRae, A.; Pemberton, J.; Slate, J. Horn type and horn length genes map to the same chromosomal region in Soay sheep. Heredity 2010, 104, 196–205. [Google Scholar] [CrossRef] [PubMed]
- Johnston, S.E.; McEwan, J.C.; Pickering, N.K.; Kijas, J.W.; Beraldi, D.; Pilkington, J.G.; Pemberton, J.M.; Slate, J. Genome-wide association mapping identifies the genetic basis of discrete and quantitative variation in sexual weaponry in a wild sheep population. Mol. Ecol. 2011, 20, 2555–2566. [Google Scholar] [CrossRef]
- Johnston, S.E.; Gratten, J.; Berenos, C.; Pilkington, J.G.; Clutton-Brock, T.H.; Pemberton, J.M.; Slate, J. Life history trade-offs at a single locus maintain sexually selected genetic variation. Nature 2013, 502, 93–95. [Google Scholar] [CrossRef]
- Montgomery, G.; Henry, H.; Dodds, K.; Beattie, A.; Wuliji, T.; Crawford, A. Mapping the Horns (Ho) locus in sheep: A further locus controlling horn development in domestic animals. J. Hered. 1996, 87, 358–363. [Google Scholar] [CrossRef]
- Beraldi, D.; McRae, A.F.; Gratten, J.; Slate, J.; Visscher, P.M.; Pemberton, J.M. Development of a linkage map and mapping of phenotypic polymorphisms in a free-living population of Soay sheep (Ovis aries). Genetics 2006, 173, 1521–1537. [Google Scholar] [CrossRef] [PubMed]
- Kijas, J.W.; Lenstra, J.A.; Hayes, B.; Boitard, S.; Porto Neto, L.R.; San Cristobal, M.; Servin, B.; McCulloch, R.; Whan, V.; Gietzen, K. Genome-wide analysis of the world’s sheep breeds reveals high levels of historic mixture and strong recent selection. PLoS Biol. 2012, 10, e1001258. [Google Scholar] [CrossRef] [PubMed]
- Wiedemar, N.; Drögemüller, C. A 1.8-kb insertion in the 3′-UTR of RXFP2 is associated with polledness in sheep. Anim. Genet. 2015, 46, 457–461. [Google Scholar] [CrossRef]
- Lühken, G.; Krebs, S.; Rothammer, S.; Küpper, J.; Mioč, B.; Russ, I.; Medugorac, I. The 1.78-kb insertion in the 3′-untranslated region of RXFP2 does not segregate with horn status in sheep breeds with variable horn status. Genet. Sel. Evol. 2016, 48, 1–14. [Google Scholar] [CrossRef]
- He, X.; Zhou, Z.; Pu, Y.; Chen, X.; Ma, Y.; Jiang, L. Mapping the four-horned locus and testing the polled locus in three Chinese sheep breeds. Anim. Genet. 2016, 47, 623–627. [Google Scholar] [CrossRef] [PubMed]
- Pan, Z.; Li, S.; Liu, Q.; Wang, Z.; Zhou, Z.; Di, R.; Miao, B.; Hu, W.; Wang, X.; Hu, X. Whole-genome sequences of 89 Chinese sheep suggest role of RXFP2 in the development of unique horn phenotype as response to semi-feralization. GigaScience 2018, 7, giy019. [Google Scholar] [CrossRef] [PubMed]
- HE, X.-h.; CHEN, X.-f.; PU, Y.-b.; GUAN, W.-j.; Shen, S.; ZHAO, Q.-j.; LI, X.-c.; Jiang, L. iTRAQ-based quantitative proteomic analysis reveals key pathways responsible for scurs in sheep (Ovis aries). J. Integr. Agric. 2018, 17, 1843–1851. [Google Scholar] [CrossRef]
- Simon, R.; Drögemüller, C.; Lühken, G. The complex and diverse genetic architecture of the absence of horns (polledness) in domestic ruminants, including goats and sheep. Genes 2022, 13, 832. [Google Scholar] [CrossRef] [PubMed]
- Lyne, A.; Hollis, D. Development of horns in merino sheep. Aust. J. Zool. 1973, 21, 153–169. [Google Scholar] [CrossRef]
- Li, X.; Zhang, C.; Gong, T.; Ni, X.; Li, J.e.; Zhan, D.; Liu, M.; Song, L.; Ding, C.; Xu, J. A time-resolved multi-omic atlas of the developing mouse stomach. Nat. Commun. 2018, 9, 4910. [Google Scholar] [CrossRef] [PubMed]
- Andersson, R.; Gebhard, C.; Miguel-Escalada, I.; Hoof, I.; Bornholdt, J.; Boyd, M.; Chen, Y.; Zhao, X.; Schmidl, C.; Suzuki, T. An atlas of active enhancers across human cell types and tissues. Nature 2014, 507, 455–461. [Google Scholar] [CrossRef]
- Wiener, D.J.; Wiedemar, N.; Welle, M.M.; Drögemüller, C. Novel features of the prenatal horn bud development in cattle (Bos taurus). PLoS ONE 2015, 10, e0127691. [Google Scholar] [CrossRef] [PubMed]
- Liao, Y.; Smyth, G.K.; Shi, W. featureCounts: An efficient general purpose program for assigning sequence reads to genomic features. Bioinformatics 2014, 30, 923–930. [Google Scholar] [CrossRef] [PubMed]
- Love, M.I.; Huber, W.; Anders, S. Moderated estimation of fold change and dispersion for RNA-seq data with DESeq2. Genome Biol. 2014, 15, 1–21. [Google Scholar] [CrossRef] [PubMed]
- Kanehisa, M.; Sato, Y.; Kawashima, M. KEGG mapping tools for uncovering hidden features in biological data. Protein Sci. 2022, 31, 47–53. [Google Scholar] [CrossRef]
- Kanehisa, M.; Furumichi, M.; Sato, Y.; Kawashima, M.; Ishiguro-Watanabe, M. KEGG for taxonomy-based analysis of pathways and genomes. Nucleic Acids Res. 2023, 51, D587–D592. [Google Scholar] [CrossRef]
- Zang, R.; Bai, J.; Xu, H.; Zhang, L.; Yang, J.; Yang, L.; Lu, J.; Wu, J. Selection of suitable reference genes for real-time quantitative PCR studies in Lanzhou fat-tailed sheep (Ovis aries). Asian J. Anim. Vet. Adv. 2011, 6, 789–804. [Google Scholar] [CrossRef]
- Livak, K.J.; Schmittgen, T.D. Analysis of relative gene expression data using real-time quantitative PCR and the 2−ΔΔCT method. Methods 2001, 25, 402–408. [Google Scholar] [CrossRef]
- Wang, X.; Zhou, G.; Li, Q.; Zhao, D.; Chen, Y. Discovery of SNPs in RXFP2 related to horn types in sheep. Small Rumin. Res. 2014, 116, 133–136. [Google Scholar] [CrossRef]
- Ren, X.; Yang, G.-L.; Peng, W.-F.; Zhao, Y.-X.; Zhang, M.; Chen, Z.-H.; Wu, F.-A.; Kantanen, J.; Shen, M.; Li, M.-H. A genome-wide association study identifies a genomic region for the polycerate phenotype in sheep (Ovis aries). Sci. Rep. 2016, 6, 21111. [Google Scholar] [CrossRef]
- Kijas, J.W.; Hadfield, T.; Naval Sanchez, M.; Cockett, N. Genome-wide association reveals the locus responsible for four-horned ruminant. Anim. Genet. 2016, 47, 258–262. [Google Scholar] [CrossRef]
- Pailhoux, E.; Vigier, B.; Chaffaux, S.; Servel, N.; Taourit, S.; Furet, J.-P.; Fellous, M.; Grosclaude, F.; Cribiu, E.P.; Cotinot, C. A 11.7-kb deletion triggers intersexuality and polledness in goats. Nat. Genet. 2001, 29, 453–458. [Google Scholar] [CrossRef]
- Pannetier, M.; Renault, L.; Jolivet, G.; Cotinot, C.; Pailhoux, E. Ovarian-specific expression of a new gene regulated by the goat PIS region and transcribed by a FOXL2 bidirectional promoter. Genomics 2005, 85, 715–726. [Google Scholar] [CrossRef]
- Boulanger, L.; Pannetier, M.; Gall, L.; Allais-Bonnet, A.; Elzaiat, M.; Le Bourhis, D.; Daniel, N.; Richard, C.; Cotinot, C.; Ghyselinck, N.B. FOXL2 is a female sex-determining gene in the goat. Curr. Biol. 2014, 24, 404–408. [Google Scholar] [CrossRef]
- Medugorac, I.; Seichter, D.; Graf, A.; Russ, I.; Blum, H.; Göpel, K.H.; Rothammer, S.; Förster, M.; Krebs, S. Bovine polledness–an autosomal dominant trait with allelic heterogeneity. PLoS ONE 2012, 7, e39477. [Google Scholar] [CrossRef]
- Allais-Bonnet, A.; Grohs, C.; Medugorac, I.; Krebs, S.; Djari, A.; Graf, A.; Fritz, S.; Seichter, D.; Baur, A.; Russ, I. Novel insights into the bovine polled phenotype and horn ontogenesis in Bovidae. PLoS ONE 2013, 8, e63512. [Google Scholar] [CrossRef] [PubMed]
- Qin, T.; Zhang, G.; Zheng, Y.; Li, S.; Yuan, Y.; Li, Q.; Hu, M.; Si, H.; Wei, G.; Gao, X. A population of stem cells with strong regenerative potential discovered in deer antlers. Science 2023, 379, 840–847. [Google Scholar] [CrossRef]
- Zhang, X.; Yin, M.; Zhang, L.-j. Keratin 6, 16 and 17—Critical barrier alarmin molecules in skin wounds and psoriasis. Cells 2019, 8, 807. [Google Scholar] [CrossRef]
- Osipowicz, K.; Wertheim-Tysarowska, K.; Kwiek, B.; Jankowska, E.; Gos, M.; Charzewska, A.; Woźniak, K.; Kowalewski, C. Bullous diseases caused by KRT1 gene mutations: From epidermolytic hyperkeratosis to a novel variant of epidermolysis bullosa simplex. Adv. Dermatol. Allergol. Postępy Dermatol. Alergol. 2021, 38, 1032–1038. [Google Scholar] [CrossRef] [PubMed]
- Ji, Y.; Bai, Y.; Wang, S.; Li, F. A KRT1 gene mutation related to epidermolytic ichthyosis in a Chinese family. Clin. Exp. Dermatol. 2015, 40, 879–882. [Google Scholar] [CrossRef] [PubMed]
- Calì, F.; Failla, P.; Vinci, M.; Siragusa, M.; Schepis, C. A de novo mutation of KRT1 in a baby girl causing epidermolytic ichthyosis with impressive epidermolytic palmoplantar keratoderma. Dermatol. Online J. 2020, 26. [Google Scholar] [CrossRef]
- Hotz, A.; Oji, V.; Bourrat, E.; Jonca, N.; Mazereeuw-Hautier, J.; Betz, R.C.; Blume-Peytavi, U.; Stieler, K.; Morice-Picard, F.; Schönbuchner, I. Expanding the clinical and genetic spectrum of KRT1, KRT2 and KRT10 mutations in keratinopathic ichthyosis. Acta Derm. Venereol. 2016, 96, 473–478. [Google Scholar] [CrossRef]
- Zheng, Z.; Jian, J.; Velasco, O.; Hsu, C.-y.; Zhang, K.; Levin, A.; Murphy, M.; Zhang, X.; Ting, K.; Soo, C. Fibromodulin enhances angiogenesis during cutaneous wound healing. Plast. Reconstr. Surg. Glob. Open 2014, 2, 171–184. [Google Scholar] [CrossRef] [PubMed]
- Adini, I.; Ghosh, K.; Adini, A.; Chi, Z.-L.; Yoshimura, T.; Benny, O.; Connor, K.M.; Rogers, M.S.; Bazinet, L.; Birsner, A.E. Melanocyte-secreted fibromodulin promotes an angiogenic microenvironment. J. Clin. Investig. 2014, 124, 425–436. [Google Scholar] [CrossRef] [PubMed]
- Kontos, C.D. More than skin deep: Connecting melanocyte pigmentation and angiogenic diseases. J. Clin. Investig. 2014, 124, 76–79. [Google Scholar] [CrossRef] [PubMed]
- Pourhanifeh, M.H.; Mohammadi, R.; Noruzi, S.; Hosseini, S.A.; Fanoudi, S.; Mohamadi, Y.; Hashemzehi, M.; Asemi, Z.; Mirzaei, H.R.; Salarinia, R. The role of fibromodulin in cancer pathogenesis: Implications for diagnosis and therapy. Cancer Cell Int. 2019, 19, 1–9. [Google Scholar] [CrossRef] [PubMed]
- Zheng, Z.; Nguyen, C.; Zhang, X.; Khorasani, H.; Wang, J.Z.; Zara, J.N.; Chu, F.; Yin, W.; Pang, S.; Le, A. Delayed wound closure in fibromodulin-deficient mice is associated with increased TGF-β3 signaling. J. Investig. Dermatol. 2011, 131, 769–778. [Google Scholar] [CrossRef]
- Zheng, Z.; Lee, K.S.; Zhang, X.; Nguyen, C.; Hsu, C.; Wang, J.Z.; Rackohn, T.M.; Enjamuri, D.R.; Murphy, M.; Ting, K. Fibromodulin-deficiency alters temporospatial expression patterns of transforming growth factor-β ligands and receptors during adult mouse skin wound healing. PLoS ONE 2014, 9, e90817. [Google Scholar] [CrossRef] [PubMed]
- Meng, F.-T.; Huang, M.; Shao, F.; Huang, Q. Upregulated FFAR4 correlates with the epithelial-mesenchymal transition and an unfavorable prognosis in human cholangiocarcinoma. Cancer Biomark. 2018, 23, 353–361. [Google Scholar] [CrossRef]
- Yasuda, S.; Morokawa, N.; Wong, G.W.; Rossi, A.; Madhusudhan, M.S.; Săli, A.; Askew, Y.S.; Adachi, R.; Silverman, G.A.; Krilis, S.A. Urokinase-type plasminogen activator is a preferred substrate of the human epithelium serine protease tryptase ϵ/PRSS22. Blood 2005, 105, 3893–3901. [Google Scholar] [CrossRef] [PubMed]
- Wong, G.W.; Yasuda, S.; Madhusudhan, M.S.; Li, L.; Yang, Y.; Krilis, S.A.; Săli, A.; Stevens, R.L. Human tryptase ε (PRSS22), a new member of the chromosome 16p13. 3 family of human serine proteases expressed in airway epithelial cells. J. Biol. Chem. 2001, 276, 49169–49182. [Google Scholar] [CrossRef]
- Lin, Y.-C.; Lee, Y.-C.; Li, L.-H.; Cheng, C.-J.; Yang, R.-B. Tumor suppressor SCUBE2 inhibits breast-cancer cell migration and invasion through the reversal of epithelial–mesenchymal transition. J. Cell Sci. 2014, 127, 85–100. [Google Scholar] [CrossRef] [PubMed]
- Chan, I. The role of extracellular matrix protein 1 in human skin. Clin. Exp. Dermatol. 2004, 29, 52–56. [Google Scholar] [CrossRef] [PubMed]
- Ehrlich, F.; Laggner, M.; Langbein, L.; Burger, P.; Pollreisz, A.; Tschachler, E.; Eckhart, L. Comparative genomics suggests loss of keratin K24 in three evolutionary lineages of mammals. Sci. Rep. 2019, 9, 10924. [Google Scholar] [CrossRef] [PubMed]
- Fuchs, E. Scratching the surface of skin development. Nature 2007, 445, 834–842. [Google Scholar] [CrossRef] [PubMed]
- Logan, C.Y.; Nusse, R. The Wnt signaling pathway in development and disease. Annu. Rev. Cell Dev. Biol. 2004, 20, 781–810. [Google Scholar] [CrossRef] [PubMed]
- Clevers, H.; Nusse, R. Wnt/β-catenin signaling and disease. Cell 2012, 149, 1192–1205. [Google Scholar] [CrossRef]
- Lim, X.; Nusse, R. Wnt signaling in skin development, homeostasis, and disease. Cold Spring Harb. Perspect. Biol. 2013, 5, a008029. [Google Scholar] [CrossRef]
- Veltri, A.; Lang, C.; Lien, W.-H. Concise review: Wnt signaling pathways in skin development and epidermal stem cells. Stem Cells 2018, 36, 22–35. [Google Scholar] [CrossRef]
- Zhou, Y.; Xu, J.; Luo, H.; Meng, X.; Chen, M.; Zhu, D. Wnt signaling pathway in cancer immunotherapy. Cancer Lett. 2022, 525, 84–96. [Google Scholar] [CrossRef]
- Yan, H.-C.; Sun, Y.; Zhang, M.-Y.; Zhang, S.-E.; Sun, J.-D.; Dyce, P.W.; Klinger, F.G.; De Felici, M.; Shen, W.; Cheng, S.-F. YAP regulates porcine skin-derived stem cells self-renewal partly by repressing Wnt/β-catenin signaling pathway. Histochem. Cell Biol. 2022, 157, 39–50. [Google Scholar] [CrossRef]
- Dorsky, R.I.; Moon, R.T.; Raible, D.W. Control of neural crest cell fate by the Wnt signalling pathway. Nature 1998, 396, 370–373. [Google Scholar] [CrossRef]
- Lee, H.-Y.; Kléber, M.; Hari, L.; Brault, V.; Suter, U.; Taketo, M.M.; Kemler, R.; Sommer, L. Instructive role of Wnt/ß-catenin in sensory fate specification in neural crest stem cells. Science 2004, 303, 1020–1023. [Google Scholar] [CrossRef]
- Hayat, R.; Manzoor, M.; Hussain, A. Wnt signaling pathway: A comprehensive review. Cell Biol. Int. 2022, 46, 863–877. [Google Scholar] [CrossRef] [PubMed]
- Ahmad, M.H.; Ghosh, B.; Rizvi, M.A.; Ali, M.; Kaur, L.; Mondal, A.C. Neural crest cells development and neuroblastoma progression: Role of Wnt signaling. J. Cell. Physiol. 2022, 238, 306–328. [Google Scholar] [CrossRef] [PubMed]
- Zhao, B.; Chen, Y.; Yang, N.; Chen, Q.; Bao, Z.; Liu, M.; Hu, S.; Li, J.; Wu, X. miR-218-5p regulates skin and hair follicle development through Wnt/β-catenin signaling pathway by targeting SFRP2. J. Cell. Physiol. 2019, 234, 20329–20341. [Google Scholar] [CrossRef] [PubMed]
- Kim, B.-K.; Yoon, S.K. Expression of sfrp2 is increased in catagen of hair follicles and inhibits keratinocyte proliferation. Ann. Dermatol. 2014, 26, 79–87. [Google Scholar] [CrossRef] [PubMed]
- Pawar, N.M.; Rao, P. Secreted frizzled related protein 4 (sFRP4) update: A brief review. Cell. Signal. 2018, 45, 63–70. [Google Scholar] [CrossRef]
- Haraguchi, R.; Kitazawa, R.; Mori, K.; Tachibana, R.; Kiyonari, H.; Imai, Y.; Abe, T.; Kitazawa, S. sFRP4-dependent Wnt signal modulation is critical for bone remodeling during postnatal development and age-related bone loss. Sci. Rep. 2016, 6, 1–14. [Google Scholar] [CrossRef]
- Maganga, R.; Giles, N.; Adcroft, K.; Unni, A.; Keeney, D.; Wood, F.; Fear, M.; Dharmarajan, A. Secreted Frizzled related protein-4 (sFRP4) promotes epidermal differentiation and apoptosis. Biochem. Biophys. Res. Commun. 2008, 377, 606–611. [Google Scholar] [CrossRef]
- Chen, R.; Baron, R.; Gori, F. Sfrp4 and the Biology of Cortical Bone. Curr. Osteoporos. Rep. 2022, 20, 153–161. [Google Scholar] [CrossRef] [PubMed]
- Jiang, S.; Li, Q.; Liu, Y.; Zhang, H.; Wang, Q.; Chen, Y.; Shi, X.; Li, J.; Zhang, H.; Zhang, Y. Activation of WNT7b autocrine eases metastasis of colorectal cancer via epithelial to mesenchymal transition and predicts poor prognosis. BMC Cancer 2021, 21, 1–11. [Google Scholar] [CrossRef]
- Hawkshaw, N.; Hardman, J.; Alam, M.; Jimenez, F.; Paus, R. Deciphering the molecular morphology of the human hair cycle: Wnt signalling during the telogen–anagen transformation. Br. J. Dermatol. 2020, 182, 1184–1193. [Google Scholar] [CrossRef] [PubMed]
- Andl, T.; Reddy, S.T.; Gaddapara, T.; Millar, S.E. WNT signals are required for the initiation of hair follicle development. Dev. Cell 2002, 2, 643–653. [Google Scholar] [CrossRef] [PubMed]










{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
| Samples | Raw Reads | Clean Reads | Q30(%) | Mapping Rate |
|---|---|---|---|---|
| S1 | 42,148,762 | 40,337,970 | 91.73; 90.53 | 95.17% |
| S2 | 36,713,688 | 35,483,446 | 91.87; 90.82 | 95.53% |
| S3 | 42,799,612 | 41,443,883 | 92.17; 91.07 | 95.41% |
| H1 | 48,177,097 | 46,171,913 | 92.09; 90.90 | 93.49% |
| H2 | 38,016,087 | 36,458,805 | 91.17; 87.15 | 94.13% |
| H3 | 39,607,975 | 38,257,622 | 93.77;92.89 | 96.10% |
Disclaimer/Publisher’s Note: The statements, opinions and data contained in all publications are solely those of the individual author(s) and contributor(s) and not of MDPI and/or the editor(s). MDPI and/or the editor(s) disclaim responsibility for any injury to people or property resulting from any ideas, methods, instructions or products referred to in the content. |
© 2023 by the authors. Licensee MDPI, Basel, Switzerland. This article is an open access article distributed under the terms and conditions of the Creative Commons Attribution (CC BY) license (https://creativecommons.org/licenses/by/4.0/).
Share and Cite
Luan, Y.; Wu, S.; Wang, M.; Pu, Y.; Zhao, Q.; Ma, Y.; Jiang, L.; He, X. Identification of Critical Genes for Ovine Horn Development Based on Transcriptome during the Embryonic Period. Biology 2023, 12, 591. https://doi.org/10.3390/biology12040591
Luan Y, Wu S, Wang M, Pu Y, Zhao Q, Ma Y, Jiang L, He X. Identification of Critical Genes for Ovine Horn Development Based on Transcriptome during the Embryonic Period. Biology. 2023; 12(4):591. https://doi.org/10.3390/biology12040591
Chicago/Turabian StyleLuan, Yuanyuan, Shangjie Wu, Mingkun Wang, Yabin Pu, Qianjun Zhao, Yuehui Ma, Lin Jiang, and Xiaohong He. 2023. "Identification of Critical Genes for Ovine Horn Development Based on Transcriptome during the Embryonic Period" Biology 12, no. 4: 591. https://doi.org/10.3390/biology12040591
APA StyleLuan, Y., Wu, S., Wang, M., Pu, Y., Zhao, Q., Ma, Y., Jiang, L., & He, X. (2023). Identification of Critical Genes for Ovine Horn Development Based on Transcriptome during the Embryonic Period. Biology, 12(4), 591. https://doi.org/10.3390/biology12040591