


Ecological Diversity of Bacterial Rhizomicrobiome Core during the Growth of Selected Wheat Cultivars

 , , , and
, , , and 
Abstract
:Simple Summary
Abstract
1. Introduction
2. Materials and Methods
2.1. Rhizospheric Soil Sampling and Description of Experimental Fields
2.2. DNA Isolation Procedure
2.3. Next-Generation Sequencing
2.4. Bioinformatic Analyses
3. Results
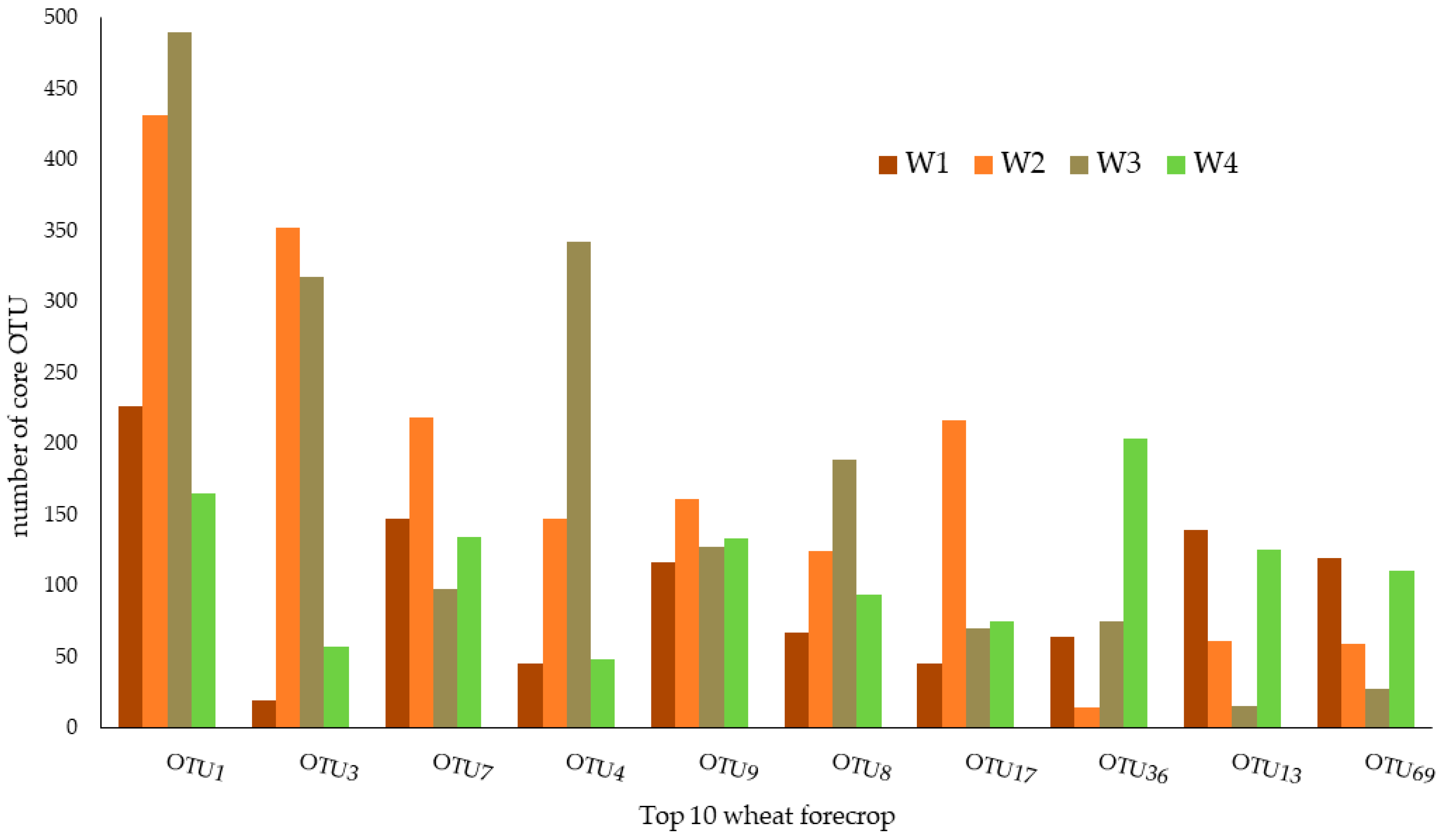
3.1. Top 10—Wheat Rhizospheric Core Microbiome Defined with the Membership-Based Method
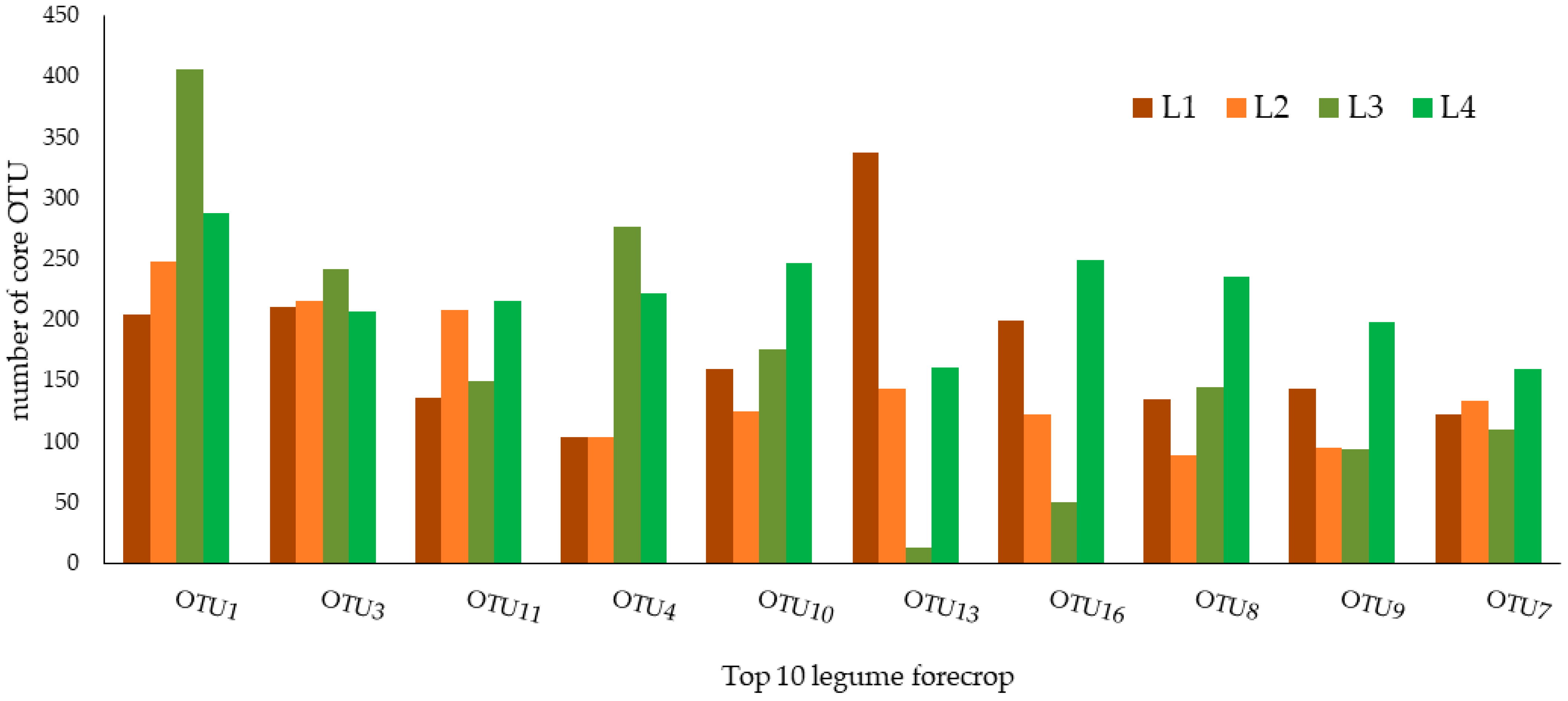
3.2. Core Microbiome Defined with the Composition-Based Method
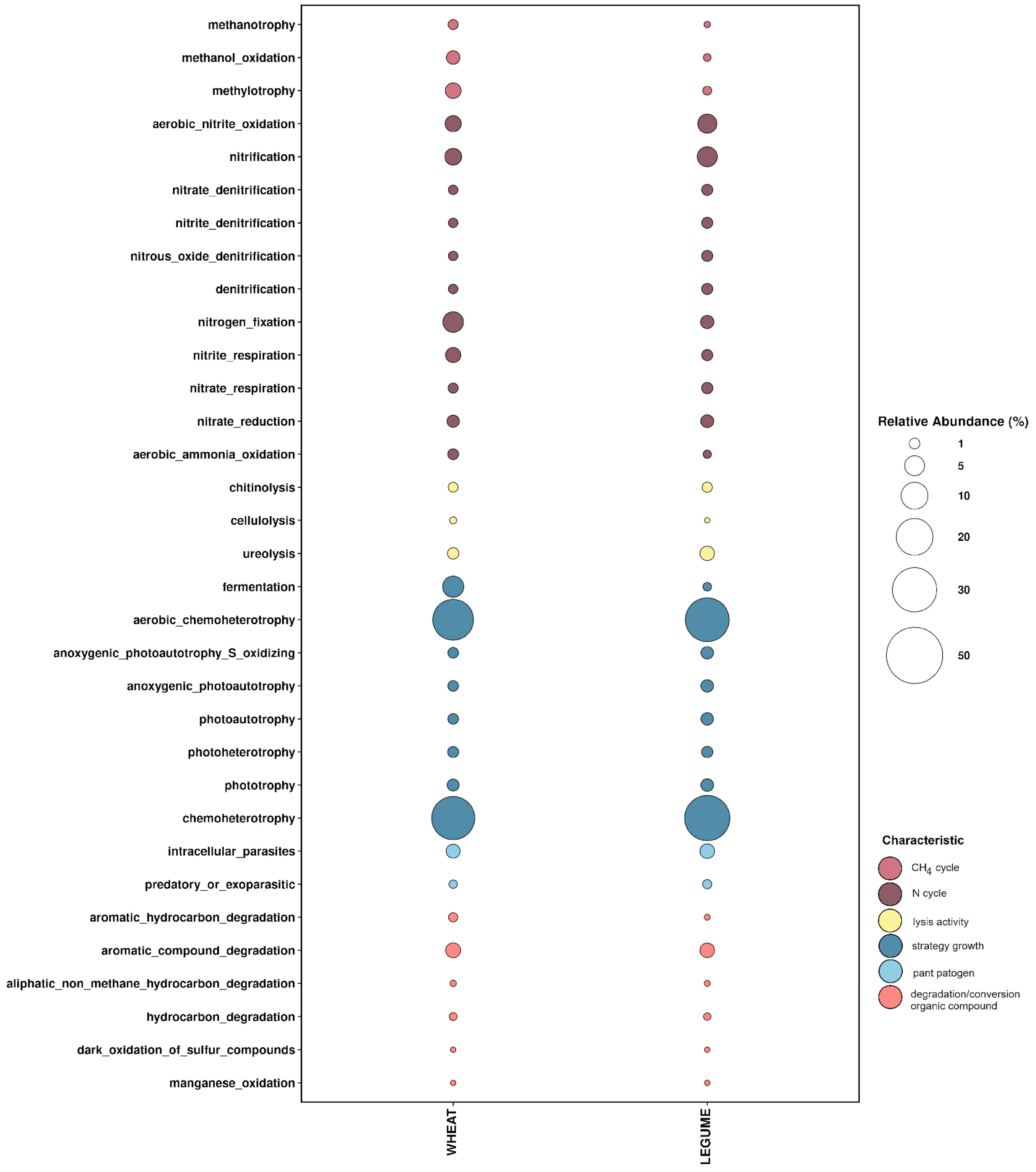
3.3. Core Microbiome Identified Based on Functional Redundancy
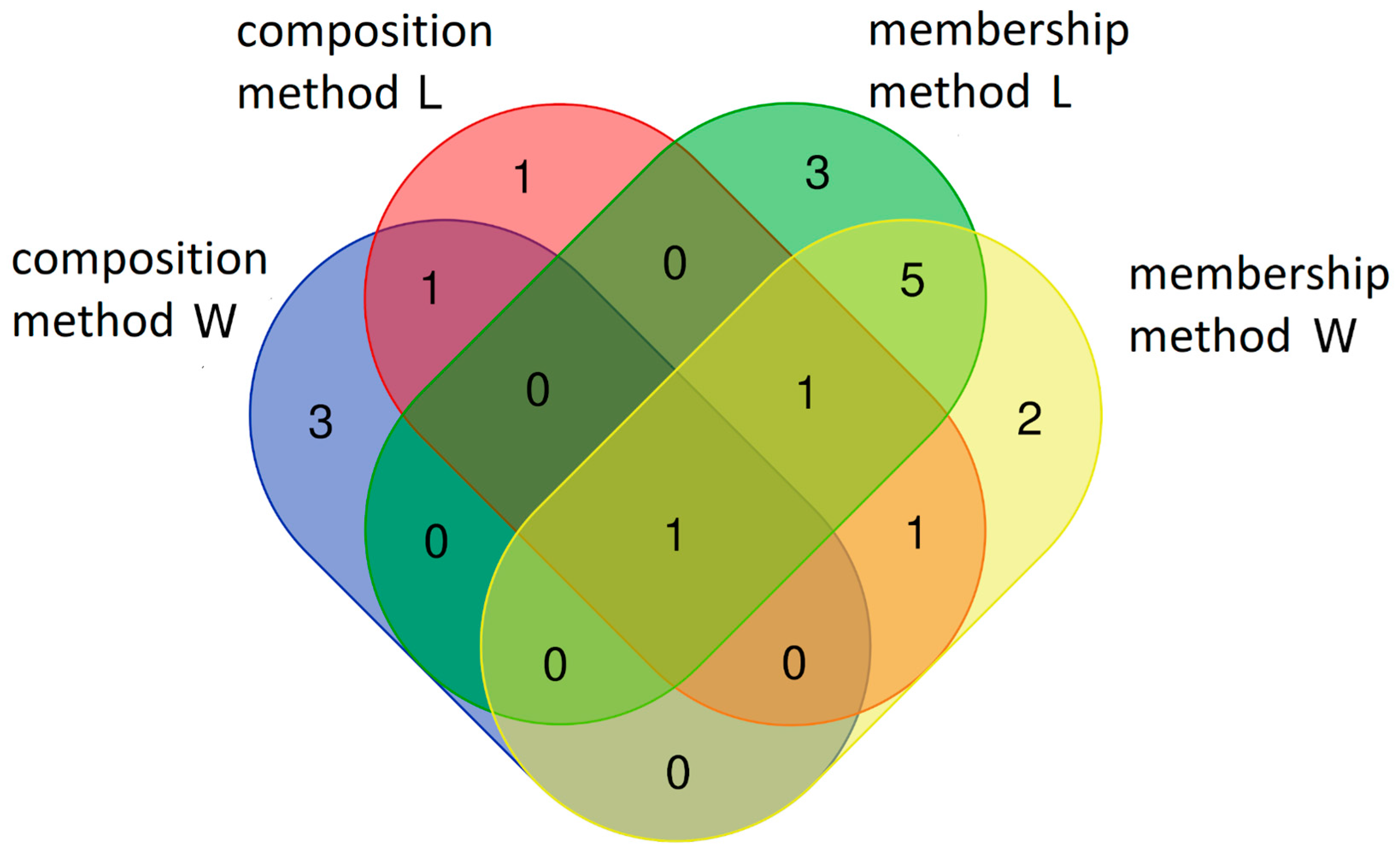
3.4. Summary of the Core Microbiome Composition Determined with the Different Methods
4. Discussion
5. Conclusions
Supplementary Materials
Author Contributions
Funding
Institutional Review Board Statement
Informed Consent Statement
Data Availability Statement
Acknowledgments
Conflicts of Interest
References
- Lee, J.; Kim, H.S.; Jo, H.Y.; Kwon, M.J. Revisiting soil bacterial counting methods: Optimal soil storage and pretreatment methods and comparison of culture-dependent and -independent methods. PLoS ONE 2021, 16, e0246142. [Google Scholar] [CrossRef]
- Aislabie, J.; Deslippe, J.R. Soil microbes and their contribution to soil services. In Ecosystem Services in New Zealand—Conditions and Trends: Conditions and Trends; Dymond, J.R., Ed.; Manaaki Whenua Press: Lincoln, UK, 2013; pp. 143–161. [Google Scholar]
- Furtak, K.; Grządziel, J.; Gałązka, A.; Niedźwiecki, J. Analysis of Soil Properties, Bacterial Community Composition, and Metabolic Diversity in Fluvisols of a Floodplain Area. Sustainability 2019, 11, 3929. [Google Scholar] [CrossRef] [Green Version]
- Canhilal, R.; Waeyenberge, L.; Yüksel, E.; Koca, A.S.; Deniz, Y.; İmren, M. Assessment of the natural presence of entomopathogenic nematodes in Kayseri soils, Turkey. Egypt. J. Biol. Pest Control 2017, 27, 237–244. [Google Scholar]
- Bickel, S.; Or, D. Soil bacterial diversity mediated by microscale aqueous-phase processes across biomes. Nat. Commun. 2020, 11, 116. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Jezierska-Tys, S.; Joniec, J.; Mocek-Płóciniak, A.; Gałązka, A.; Bednarz, J.; Furtak, K. Microbial activity and community level physiological profiles (CLPP) of soil under the cultivation of spring rape with the Roundup 360 SL herbicide. J. Environ. Health Sci. Eng. 2021, 19, 2013–2026. [Google Scholar] [CrossRef]
- Wolińska, A.; Kuźniar, A.; Zielenkiewicz, U.; Banach, A.; Błaszczyk, M. Indicators of arable soils fatigue—Bacterial families and genera: A metagenomic approach. Ecol. Indic. 2018, 93, 490–500. [Google Scholar] [CrossRef]
- Wolińska, A.; Podlewski, J.; Słomczewski, A.; Grządziel, J.; Gałązka, A.; Kuźniar, A. Fungal indicators of sensitivity and resistance to long-term maize monoculture: A culture-independent approach. Front. Microbiol. 2022, 12, 799378. [Google Scholar] [CrossRef]
- Kaiser, K.; Wemheuer, B.; Korolkow, V.; Wemheur, F.; Nacke, H.; Schoning, I.; Schrumpf, M.; Daniel, R. Driving forces of soil bacterial community structure, diversity, and function in temperate grasslands and forests. Sci. Rep. 2016, 6, 33696. [Google Scholar] [CrossRef] [Green Version]
- Wenjie, W.; Jiadan, T.; Yi, Y.Q.; Huangmei, H.; Huiqin, W.; Wenlong, Z.; Donglan, H. Responses of the rhizosphere bacterial community in acidic crop soil to pH: Changes in diversity, composition, interaction, and function. Sci. Total Environ. 2020, 700, 134418. [Google Scholar] [CrossRef]
- Huiyuan, C.; Bingde, W.; Mei, W.; Shu, W.; Xinshan, R.; Daolin, D.; Congyan, W. Changes in community structure and met-abolic function of soil bacteria depending on the type restoration processing in the degraded alpine grassland ecosystems in Northern Tibet. Sci. Total Environ. 2021, 755, 142619. [Google Scholar] [CrossRef]
- Furtak, K.; Gajda, A. Activity and variety of soil microorganisms depending on the diversity of the soil tillage system. In Sustainability of Agroecosystems; Intech Open: London, UK, 2018. [Google Scholar] [CrossRef] [Green Version]
- Wei, Z.; Qingli, G.; Zhiyuan, Z.; Jie, L.; Bingnian, Z.; Zhaohui, W.; Ziyan, L. Changes in the soil bacterial community structure and enzyme activities after intercrop mulch with cover crop for eight years in an orchard. Eur. J. Soil Biol. 2018, 86, 34–41. [Google Scholar] [CrossRef]
- Tecon, R.; Or, D. Biophysical processes supporting the diversity of microbial life in soil. FEMS Microbiol. Rev. 2017, 41, 599–623. [Google Scholar] [CrossRef] [Green Version]
- Annapurna, K.; Govindasamy, V.; Sharma, M.; Gosh, A.; Chikara, S.K. Whole genome shotgun sequence of Bacillus paralicheniformis strain KMS 80, a rhizobacterial endophyte isolated from rice (Oryza sativa L.). 3 Biotech 2018, 8, 223. [Google Scholar] [CrossRef]
- Chen, H.; Wu, H.; Yan, B.; Zhao, H.; Liu, F.; Zhang, H.; Sheng, Q.; Miao, F.; Liang, Z. Core microbiome of medicinal plant Salvia miltiorrhiza seed: A rich reservoir of beneficial microbes for secondary metabolism? Int. J. Mol. Sci. 2018, 19, 672. [Google Scholar] [CrossRef] [Green Version]
- Dong, C.; Shao, Q.; Zhang, Q.; Yao, T.; Huang, J.; Liang, Z.; Han, Y. Preferences for core microbiome composition and function by different definition methods: Evidence for the core microbiome of Eucommia ulmoides bark. Sci. Total Environ. 2021, 790, 148091. [Google Scholar] [CrossRef] [PubMed]
- Kavamura, V.N.; Mendes, R.; Bargaz, A.; Mauchline, T.H. Defining the wheat microbiome: Towards microbiome-facilitated crop production. Comput. Struct. Biotechnol. J. 2021, 19, 1200–1213. [Google Scholar] [CrossRef] [PubMed]
- Simonin, M.; Dasilva, C.; Terzi, V.; Ngonkeu, E.L.M.; Diouf, D.; Kane, A.; Bena, G.; Moulin, L. Influence of plant genotype and soil on the wheat rhizosphere microbiome: Evidence for a core microbiome across eight African and European soils. FEMS Microbiol. Ecol. 2020, 96, fiaa067. [Google Scholar] [CrossRef]
- Pfeiffer, S.; Mitter, B.; Oswald, A.; Schloter-Hai, B.; Schloter, M.; Declerck, S.; Sessitsch, A. Rhizosphere microbiomes of potato cultivated in the High Andes show stable and dynamic core microbiomes with different responses to plant development. FEMS Microbiol. Ecol. 2017, 93, fiw242. [Google Scholar] [CrossRef] [Green Version]
- Schlatter, D.C.; Yin, C.; Hulbert, S.; Paulitz, T.C. Core rhizosphere microbiomes of dryland wheat are influenced by location and land use history. Appl. Environ. Microbiol. 2020, 86, e02135-19. [Google Scholar] [CrossRef] [PubMed]
- Philippe, P.; Cuéllar, P.; Brunier-Coulin, F.; Luu, L.H.; Benahmed, N.; Bonelli, S.; Delenne, J.Y. Physics of soil erosion at the microscale. EPJ Web Conf. 2018, 140, 08014. [Google Scholar] [CrossRef]
- Nkongolo, K.K.; Narendrula-Kotha, R. Advances in monitoring soil microbial community dynamic and function. J. Appl. Gen. 2020, 61, 249–263. [Google Scholar] [CrossRef] [PubMed]
- Wolińska, A. Metagenomic achievements in microbial diversity determination in croplands: A review. In Microbial Diversity in the Genomic Era; Academic Press Elsevier: Cambridge, MA, USA, 2019; Volume 2, pp. 15–35. [Google Scholar]
- Kuźniar, A.; Włodarczyk, K.; Grządziel, J.; Woźniak, M.; Furtak, K.; Gałązka, A.; Dziadczyk, E.; Skórzyńska-Polit, E.; Wolińska, A. New insight into the composition of wheat seed microbiota. Int. J. Mol. Sci. 2020, 30, 4634. [Google Scholar] [CrossRef]
- Goodwin, S.; McPherson, J.D.; McCombie, W.R. Coming of age: Ten years of next-generation sequencing technologies. Nat. Rev. Genet. 2016, 17, 333–351. [Google Scholar] [CrossRef] [PubMed]
- Rossmann, M.; Perez-Jaramillo, J.E.; Kavamura, V.N.; Chiaramonte, J.B.; Dumack, K.; Fiore-Donno, A.M.; Mendes, L.W.; Ferreira, M.M.C.; Bonkowski, M.; Raaijmakers, J.M.; et al. Multitrophic interactions in the rhizosphere microbiome of wheat: From bacteria and fungi to protists. FEMS Microbiol. Ecol. 2020, 96, fiaa032. [Google Scholar] [CrossRef] [PubMed]
- Schlatter, D.; Kinkel, L.; Thomashow, L.; Weller, D.; Paulitz, T. Disease suppressive soils: New insights from the soil microbiome. Phytopathology 2017, 107, 1284–1297. [Google Scholar] [CrossRef] [Green Version]
- Wolińska, A.; Kuźniar, A.; Gałązka, A. Biodiversity in the rhizosphere of selected winter wheat (Triticum aestivum L.) cultivars—Genetic and catabolic fingerprinting. Agronomy 2020, 10, 953. [Google Scholar] [CrossRef]
- Mohanram, S.; Kumar, P. Rhizosphere microbiome: Revisiting the synergy of plant-microbe interactions. Ann. Microbiol. 2019, 69, 307–320. [Google Scholar] [CrossRef]
- Klindworth, A.; Pruesse, E.; Schweer, T.; Peplies, J.; Quast, C.; Horn, M.; Glöckner, F.O. Evaluation of general 16S ribosomal RNA gene PCR primers for classical and next-generation sequencing-based diversity studies. Nucleic Acids Res. 2013, 7, e1. [Google Scholar] [CrossRef]
- Sansupa, C.; Wahdan, S.F.M.; Hossen, S.; Disayathanoowat, T.; Wubet, T.; Purahong, W. Can we use functional annotation of prokaryotic taxa (FAPROTAX) to assign the ecological functions of soil bacteria? Appl. Sci. 2021, 11, 688. [Google Scholar] [CrossRef]
- Armada, E.; Leite, M.F.A.; Medina, A.; Rosario, A.; Kuramae, E.E. Native bacteria promote plant growth under drought stress conditions without impacting the rhizomicrobiome. FEMS Microbiol. Ecol. 2018, 94, fiy092. [Google Scholar] [CrossRef] [Green Version]
- Li, X.; Jousset, A.; de Boer, W.; Carrión, V.J.; Zhang, T.; Wang, X.; Kuramae, E.E. Legacy of land use history determines reprogram-ming of plant physiology by soil microbiome. ISME J. 2019, 13, 738–751. [Google Scholar] [CrossRef] [Green Version]
- Ling, N.; Wang, T.; Kuzyakov, Y. Rhizosphere bacteriome structure and functions. Nat. Commun. 2022, 13, 836. [Google Scholar] [CrossRef] [PubMed]
- Huang, M.; Tian, A.; Chen, J.; Cao, F.; Chen, Y.; Liu, L. Soil bacterial communities in three rice-based cropping systems differ in productivity. Sci. Rep. 2020, 17, 9867. [Google Scholar] [CrossRef]
- Hamonts, K.; Trivedi, P.; Garg, A.; Janitz, C.; Grinyer, J.; Holford, P.; Botha, F.C.; Anderson, I.C.; Singh, B.K. Field study reveals core plant microbiota and relative importance of their drivers. Environ. Microbiol. 2018, 20, 124–140. [Google Scholar] [CrossRef]
- Zhalnina, K.; Louie, K.B.; Hao, Z.; Mansoori, N.; da Rocha, U.N.; Shi, S.; Cho, H.; Karaoz, U.; Loqué, D.; Bowen, B.P.; et al. Dynamic root exudate chemistry and microbial substrate preferences drive patterns in rhizosphere microbial community assembly. Nat. Microbiol. 2018, 3, 470–480. [Google Scholar] [CrossRef] [Green Version]
- Jacquiod, S.; Raynaud, T.; Pimet, E.; Ducourtieux, C.; Casieri, L.; Wipf, D.; Blouin, M. Wheat rhizosphere microbiota respond to changes in plant genotype, chemical inputs, and plant phenotypic plasticity. Front. Ecol. Evol. 2022, 10, 903008. [Google Scholar] [CrossRef]
- Di Gregorio, L.; Tandoi, V.; Congestri, R.; Rossetti, S.; Di Pippo, F. Unravelling the core microbiome of biofilms in cooling tower systems. Biofouling 2017, 33, 793–806. [Google Scholar] [CrossRef] [PubMed]
- Nelkner, J.; Henke, C.; Lin, T.W.; Pätzold, W.; Hassa, J.; Jaenicke, S.; Grosch, R.; Pühler, A.; Sczyrba, A.; Schlüter, A. Effect of long-term farming practices on agricultural soil microbiome members represented by metagenomically assembled genomes (MAGs) and their predicted plant-beneficial genes. Genes 2019, 10, 424. [Google Scholar] [CrossRef] [Green Version]
- Shade, A.; Stopnisek, N. Abundance-occupancy distributions to prioritize plant core microbiome membership. Curr. Opin. Microbiol. 2019, 49, 50–58. [Google Scholar] [CrossRef]
- Shade, A.; Gilbert, J.A. Temporal patterns of rarity provide a more complete view of microbial diversity. Trends Microbiol. 2015, 23, 335–340. [Google Scholar] [CrossRef] [Green Version]
- Chaoyu, L.; Zi-Yang, H.; Hang-Wei, H.; Ji-Zheng, H. Niche specialization of comammox Nitrospira in terrestrial ecosystems: Oligotrophic or copiotrophic? Crit. Rev. Environ. Sci. Technol. 2023, 53, 161–176. [Google Scholar] [CrossRef]
- Jochum, M.D.; McWilliams, K.L.; Pierson, E.A.; Jo, Y.K. Host-mediated microbiome engineering (HMME) of drought tolerance in the wheat rhizosphere. PLoS ONE 2019, 14, e0225933. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Griebsch, A.; Matschiavelli, N.; Lewandowska, S.; Schmidtke, K. Presence of Bradyrhizobium sp. under continental conditions in Central Europe. Agriculture 2020, 10, 446. [Google Scholar] [CrossRef]
- Favero, V.O.; de Carvalho, R.H.; Leite, A.B.C.; dos Santos, D.M.T.; de Freitas, K.M.; Boddey, R.M.; Xavier, G.R.; Rumjanek, N.G.; Urquiaga, S. Bradyrhizobium strains from Brazilian tropical soils promote increases in nodulation, growth and nitrogen fixation in mung beans. Appl. Soil Ecol. 2022, 175, 104461. [Google Scholar] [CrossRef]
- William, A.; Walters, Z.J.; Youngblut, N.; Wallace, J.G.; Sutter, J.; Zhang, W.; González-Peña, A.; Peiffer, J.; Koren, O.; Shi, Q.; et al. Large-scale replicated field study of the maize rhizosphere identifies heritable microbes. Proc. Nat. Acad. Sci. USA 2018, 115, 201800918. [Google Scholar] [CrossRef] [Green Version]
- Maina, S.; Karuri, H.; Ng’endo, N.R. Nematode metabolic footprints, ecological and functional indices in tropical maize-beans agroecosystems under different farming practices. Acta Oecol. 2020, 108, 103622. [Google Scholar] [CrossRef]
- Peng, C.; Viana, T.; Petersen, M.A.; Larsen, F.H.; Arneborg, N. Metabolic footprint analysis of metabolites that discriminate single and mixed yeast cultures at two key time-points during mixed culture alcoholic fermentations. Metabolomics 2018, 4, 93. [Google Scholar] [CrossRef]
- Sriswasdi, S.; Yang, C.C.; Iwasaki, W. Generalist species drive microbial dispersion and evolution. Nat. Commun. 2017, 8, 1162. [Google Scholar] [CrossRef] [Green Version]





{kind=link}
{kind=link}
{kind=link}
{kind=link}
| OTU ID | Taxonomy | Relative Abundance |
|---|---|---|
| OTU1 | Hyphomicrobiaceae | 0.0334 |
| OTU2 | Sphingomonas sp. | 0.0328 |
| OTU14 | Bradyrhizobiaceae | 0.0213 |
| OTU5 | Burkholderiales | 0.0207 |
| OTU3 | Nitrospira sp. | 0.0199 |
| OTU7 | Rhizobiales | 0.0153 |
| OTU26 | Baekduia sp. | 0.0145 |
| OTU4 | Pseudarthrobacter sp. | 0.0144 |
| OTU9 | Bradyrhizobium sp. | 0.0135 |
| OTU12 | Bradyrhizobium sp. | 0.0118 |
| OTU ID | Taxonomy | Relative Abundance |
|---|---|---|
| OTU1 | Hyphomicrobiaceae | 0.0325 |
| OTU3 | Nitrospira sp. | 0.0248 |
| OTU11 | Bradyrhizobiaceae | 0.0206 |
| OTU4 | Pseudarthrobacter sp. | 0.0197 |
| OTU10 | Gp16 | 0.0195 |
| OTU6 | Rhizobiales | 0.0182 |
| OTU13 | Gp3 | 0.0173 |
| OTU16 | Bradyrhizobiaceae | 0.0169 |
| OTU8 | Pseudarthrobacter sp. | 0.0165 |
| OTU7 | Rhizobiales | 0.0149 |
| OTU ID | Wheat Forecrop | |
|---|---|---|
| Taxonomy | Relative Abundance (%) | |
| OTU1 | Hyphomicrobiaceae | 1.232 ± 0.1780 |
| OTU3 | Nitrospira sp. | 0.913 ± 0.2790 |
| OTU7 | Bradyrhizobiaceae | 0.3840 ± 0.0033 |
| OTU4 | Pseudarthrobacter sp. | 0.6180 ± 0.0021 |
| OTU9 | Bradyrhizobium sp. | 0.3380 ± 0.0016 |
| OTU8 | Pseudarthrobacter sp. | 0.2950 ± 0.0003 |
| OTU17 | Nitrospira sp. | 0.1350 ± 0.0005 |
| OTU36 | Streptosporangium sp. | 0.3041 ± 0.0023 |
| OTU13 | Acidobacteria_Gp3 | 0.2954 ± 0.0001 |
| OTU69 | Devosia sp. | 0.2575 ± 0.0001 |
| OTU ID | Legume Forecrop | |
|---|---|---|
| Taxonomy | Relative Abundance (%) | |
| OTU1 | Hyphomicrobiaceae | 1.0240 ± 0.1810 |
| OTU3 | Nitrospira sp. | 0.7360 ± 0.0930 |
| OTU11 | Rhodoplanes sp. | 0.6750 ± 0.1420 |
| OTU4 | Pseudarthrobacter sp. | 0.5760 ± 0.2663 |
| OTU10 | Acidobacteria_Gp16 | 0.5599 ± 0.1328 |
| OTU13 | Acidobacteria_Gp3 | 0.6360 ± 0.1460 |
| OTU16 | Rhizobiales | 0.4457 ± 0.0129 |
| OTU8 | Pseudarthrobacter sp. | 0.3634 ± 0.0500 |
| OTU9 | Nitrospira sp. | 0.3072 ± 0.0663 |
| OTU7 | Bradyrhizobiaceae | 0.3451 ± 0.1095 |
Disclaimer/Publisher’s Note: The statements, opinions and data contained in all publications are solely those of the individual author(s) and contributor(s) and not of MDPI and/or the editor(s). MDPI and/or the editor(s) disclaim responsibility for any injury to people or property resulting from any ideas, methods, instructions or products referred to in the content. |
© 2023 by the authors. Licensee MDPI, Basel, Switzerland. This article is an open access article distributed under the terms and conditions of the Creative Commons Attribution (CC BY) license (https://creativecommons.org/licenses/by/4.0/).
Share and Cite
Kuźniar, A.; Włodarczyk, K.; Jurczyk, S.; Maciejewski, R.; Wolińska, A. Ecological Diversity of Bacterial Rhizomicrobiome Core during the Growth of Selected Wheat Cultivars. Biology 2023, 12, 1067. https://doi.org/10.3390/biology12081067
Kuźniar A, Włodarczyk K, Jurczyk S, Maciejewski R, Wolińska A. Ecological Diversity of Bacterial Rhizomicrobiome Core during the Growth of Selected Wheat Cultivars. Biology. 2023; 12(8):1067. https://doi.org/10.3390/biology12081067
Chicago/Turabian StyleKuźniar, Agnieszka, Kinga Włodarczyk, Sara Jurczyk, Ryszard Maciejewski, and Agnieszka Wolińska. 2023. "Ecological Diversity of Bacterial Rhizomicrobiome Core during the Growth of Selected Wheat Cultivars" Biology 12, no. 8: 1067. https://doi.org/10.3390/biology12081067
APA StyleKuźniar, A., Włodarczyk, K., Jurczyk, S., Maciejewski, R., & Wolińska, A. (2023). Ecological Diversity of Bacterial Rhizomicrobiome Core during the Growth of Selected Wheat Cultivars. Biology, 12(8), 1067. https://doi.org/10.3390/biology12081067