Implications of PI3K/AKT/PTEN Signaling on Superoxide Dismutases Expression and in the Pathogenesis of Alzheimer’s Disease

 ,
, 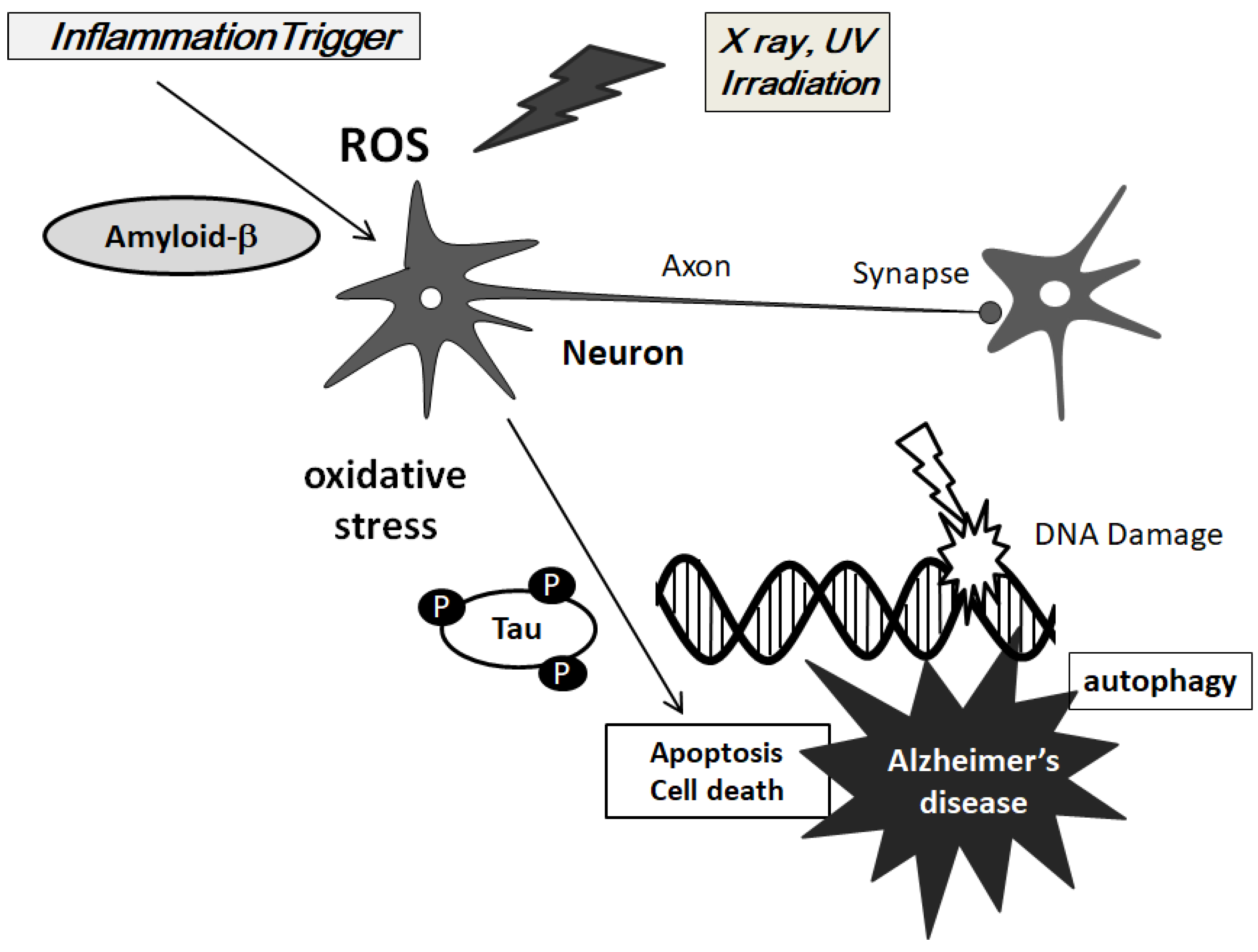{kind=link}
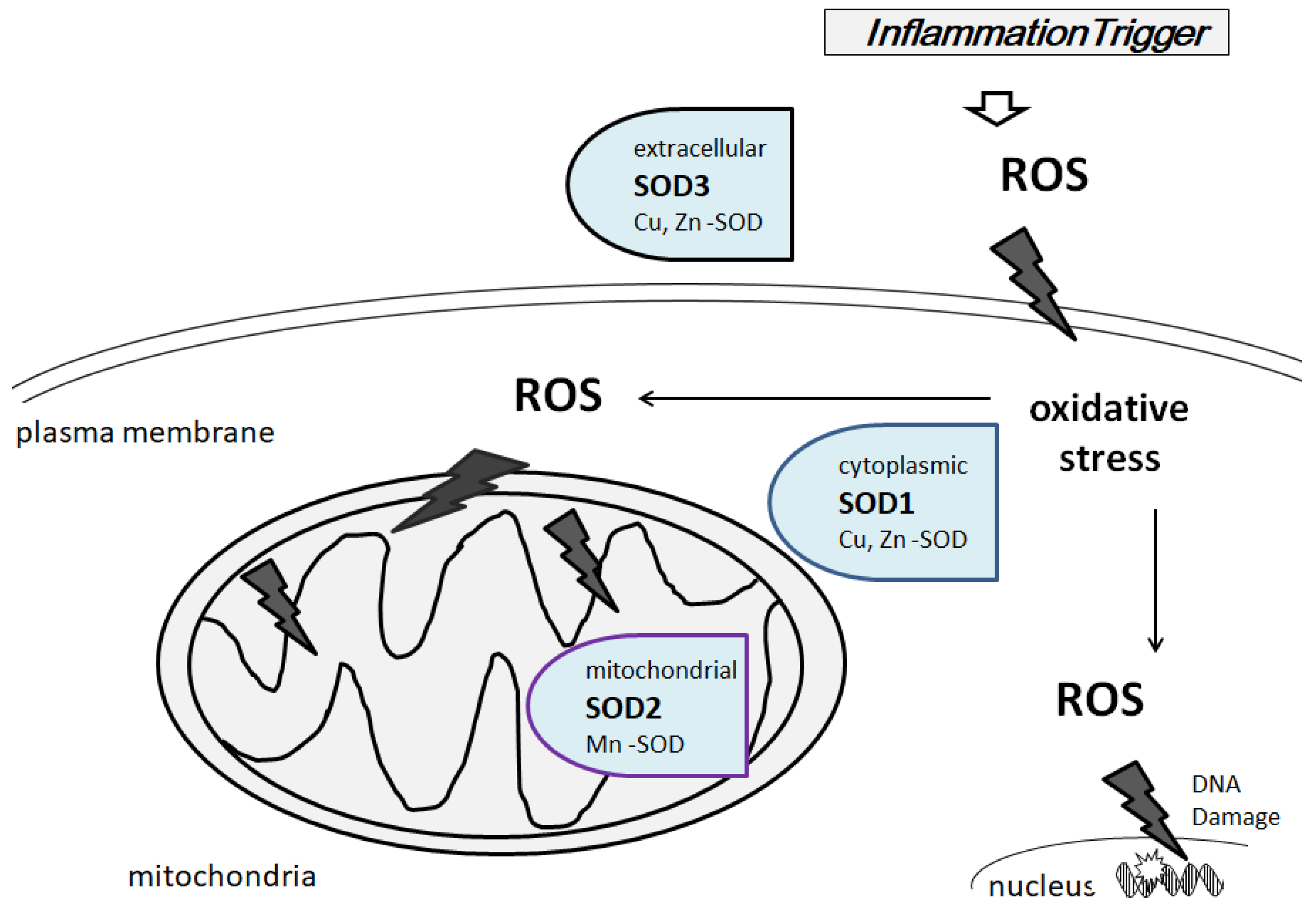{kind=link}
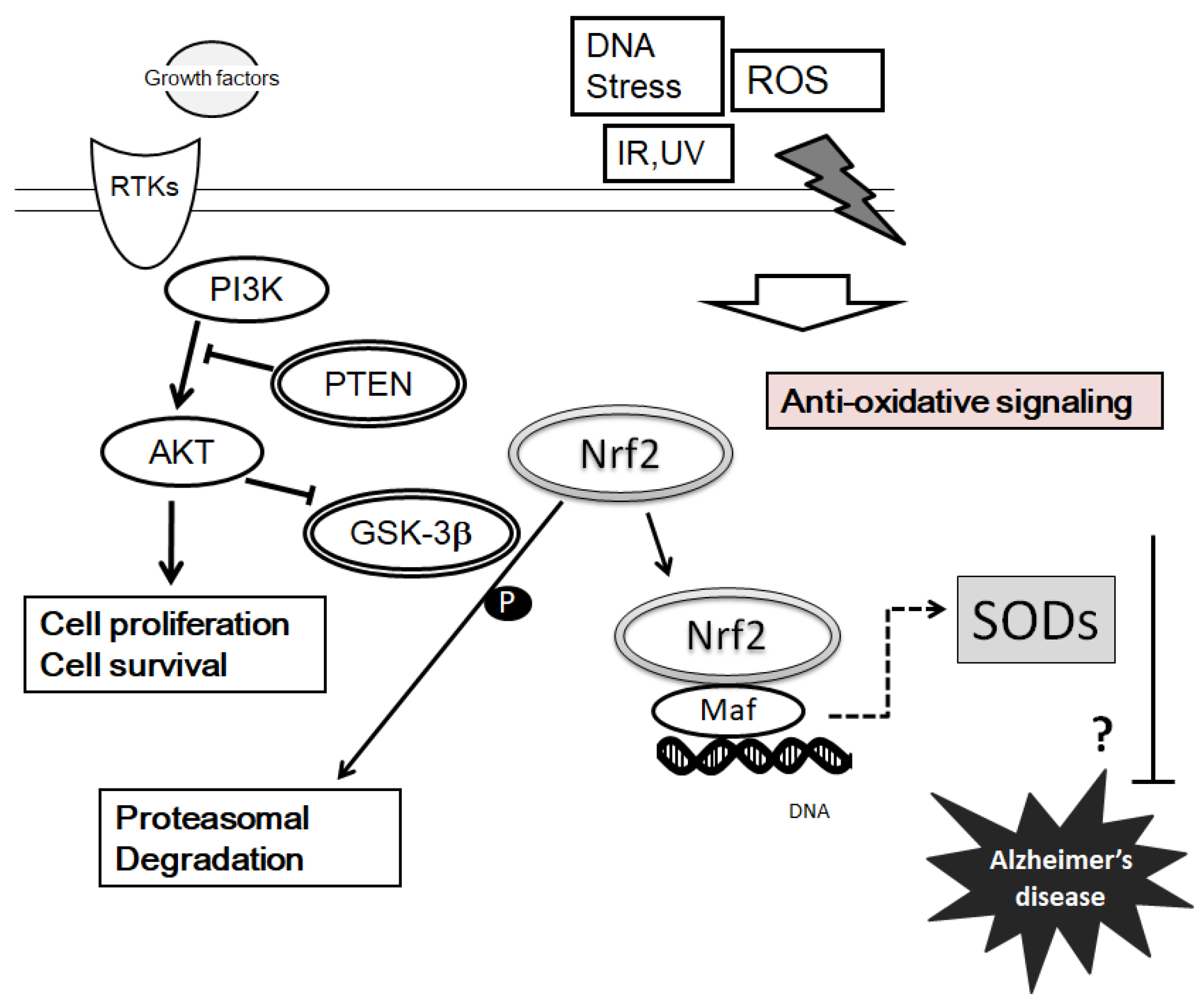{kind=link}
Abstract
:1. Introduction
2. ROS is Involved in the Pathogenesis of AD
3. Characterization of Superoxide Dismutases
4. Suggestions of PI3K/AKT/PTEN Signaling on SOD Expression and Pathogenesis of AD
5. Diet Approach for Neuronal Cellular Protection in AD Prevention
6. Future Perspectives
Acknowledgments
Author Contributions
Conflicts of Interest
Abbreviations
| AD | Alzheimer’s disease |
| Apo A-IV | apoliprotein A-IV |
| ARE | antioxidant response elements |
| ATF2 | activating transcription factor 2 |
| ATP | adenosine triphosphate |
| BBB | blood brain barrier |
| CNS | central nervous system |
| DHA | docosahexaenoic acid |
| DNA | deoxyribonucleic acid |
| EPA | eicosapentaenoic acid |
| GSK3β | glycogen synthase kinase-3β |
| HIF1α | hypoxia inducible factor-1α |
| Maf | macrophage activating factor |
| MAPK | mitogen-activated protein kinase |
| Nrf2 | NF-E2-related factor-2 |
| PIP3 | phosphatidylinositol 3,4,5-triphosphate |
| PIP2 | phosphatidylinositol 4,5- bisphosphate |
| PI3K | phosphoinositide-3 kinase |
| PKC | protein kinase c |
| PPAR | peroxisome proliferator-activated receptor |
| PPREs | PPAR response elements |
| PS | presenilin |
| PTEN | phosphatase and tensin homologue deleted on chromosome 10 |
| PUFAs | polyunsaturated fatty acids |
| ROS | reactive oxygen species |
| SODs | superoxide dismutases |
References
- Hajipour, M.J.; Santoso, M.R.; Rezaee, F.; Aghaverdi, H.; Mahmoudi, M.; Perry, G. Advances in Alzheimer’s Diagnosis and Therapy: The Implications of Nanotechnology. Trends Biotechnol. 2017, 35, 937–953. [Google Scholar] [CrossRef] [PubMed]
- Hess, N.C.; Smart, N.A. Isometric Exercise Training for Managing Vascular Risk Factors in Mild Cognitive Impairment and Alzheimer’s Disease. Front. Aging Neurosci. 2017, 9, 48. [Google Scholar] [CrossRef] [PubMed]
- Mathys, Z.K.; White, A.R. Copper and Alzheimer’s Disease. Adv. Neurobiol. 2017, 18, 199–216. [Google Scholar] [PubMed]
- Takahashi, R.H.; Nagao, T.; Gouras, G.K. Plaque formation and the intraneuronal accumulation of β-amyloid in Alzheimer’s disease. Pathol. Int. 2017, 67, 185–193. [Google Scholar] [CrossRef] [PubMed]
- Vale, C.; Alonso, E.; Rubiolo, J.A.; Vieytes, M.R.; LaFerla, F.M.; Giménez-Llort, L.; Botana, L.M. Profile for amyloid-beta and tau expression in primary cortical cultures from 3xTg-AD mice. Cell Mol. Neurobiol. 2010, 30, 577–590. [Google Scholar] [CrossRef] [PubMed]
- Grizzanti, J.; Lee, H.G.; Camins, A.; Pallas, M.; Casadesus, G. The therapeutic potential of metabolic hormones in the treatment of age-related cognitive decline and Alzheimer’s disease. Nutr. Res. 2016, 36, 1305–1315. [Google Scholar] [CrossRef] [PubMed]
- Head, E. Oxidative damage and cognitive dysfunction: Antioxidant treatments to promote healthy brain aging. Neurochem. Res. 2009, 34, 670–678. [Google Scholar] [CrossRef] [PubMed]
- Mukherjee, K.; Clark, H.R.; Chavan, V.; Benson, E.K.; Kidd, G.J.; Srivastava, S. Analysis of Brain Mitochondria Using Serial Block-Face Scanning Electron Microscopy. J. Vis. Exp. 2016, 113, 54214. [Google Scholar] [CrossRef] [PubMed]
- Redza-Dutordoir, M.; Averill-Bates, D.A. Activation of apoptosis signalling pathways by reactive oxygen species. Biochim. Biophys. Acta 2016, 1863, 2977–2992. [Google Scholar] [CrossRef] [PubMed]
- André-Lévigne, D.; Modarressi, A.; Pepper, M.S.; Pittet-Cuénod, B. Reactive Oxygen Species and NOX Enzymes Are Emerging as Key Players in Cutaneous Wound Repair. Int. J. Mol. Sci. 2017, 18, E2149. [Google Scholar] [CrossRef] [PubMed]
- Case, A.J. On the Origin of Superoxide Dismutase: An Evolutionary Perspective of Superoxide-Mediated Redox Signaling. Antioxidants 2017, 6, E82. [Google Scholar] [CrossRef] [PubMed]
- Vasilaki, A.; Jackson, M.J. Role of reactive oxygen species in the defective regeneration seen in aging muscle. Free Radic. Biol. Med. 2013, 65, 317–323. [Google Scholar] [CrossRef] [PubMed]
- Robbins, D.; Zhao, Y. Manganese superoxide dismutase in cancer prevention. Antioxid. Redox. Signal. 2014, 20, 1628–1645. [Google Scholar] [CrossRef] [PubMed]
- Venigalla, M.; Sonego, S.; Gyengesi, E.; Sharman, M.J.; Münch, G. Novel promising therapeutics against chronic neuroinflammation and neurodegeneration in Alzheimer’s disease. Neurochem. Int. 2016, 95, 63–74. [Google Scholar] [CrossRef] [PubMed]
- Ajith, T.A.; Padmajanair, G. Mitochondrial Pharmaceutics: A New Therapeutic Strategy to Ameliorate Oxidative Stress in Alzheimer’s Disease. Curr. Aging Sci. 2015, 8, 235–240. [Google Scholar] [CrossRef] [PubMed]
- Wu, M.F.; Yin, J.H.; Hwang, C.S.; Tang, C.M.; Yang, D.I. NAD attenuates oxidative DNA damages induced by amyloid beta-peptide in primary rat cortical neurons. Free Radic. Res. 2014, 48, 794–805. [Google Scholar] [CrossRef] [PubMed]
- Mittal, M.; Siddiqui, M.R.; Tran, K.; Reddy, S.P.; Malik, A.B. Reactive oxygen species in inflammation and tissue injury. Antioxid. Redox. Signal. 2014, 20, 1126–1167. [Google Scholar] [CrossRef] [PubMed]
- Barone, E.; Di Domenico, F.; Mancuso, C.; Butterfield, D.A. The Janus face of the heme oxygenase/biliverdin reductase system in Alzheimer disease: It’s time for reconciliation. Neurobiol. Dis. 2014, 62, 144–159. [Google Scholar] [CrossRef] [PubMed]
- Weinreb, O.; Mandel, S.; Bar-Am, O.; Yogev-Falach, M.; Avramovich-Tirosh, Y.; Amit, T.; Youdim, M.B. Multifunctional neuroprotective derivatives of rasagiline as anti-Alzheimer’s disease drugs. Neurotherapeutics 2009, 6, 163–174. [Google Scholar] [CrossRef] [PubMed]
- Poprac, P.; Jomova, K.; Simunkova, M.; Kollar, V.; Rhodes, C.J.; Valko, M. Targeting Free Radicals in Oxidative Stress-Related Human Diseases. Trends Pharmacol. Sci. 2017, 38, 592–607. [Google Scholar] [CrossRef] [PubMed]
- Shafi, O. Inverse relationship between Alzheimer’s disease and cancer, and other factors contributing to Alzheimer’s disease: A systematic review. BMC Neurol. 2016, 16, 236. [Google Scholar] [CrossRef] [PubMed]
- Pinto, M.; Pickrell, A.M.; Fukui, H.; Moraes, C.T. Mitochondrial DNA damage in a mouse model of Alzheimer’s disease decreases amyloid beta plaque formation. Neurobiol. Aging 2013, 34, 2399–2407. [Google Scholar] [CrossRef] [PubMed]
- Zhang, C.; Rissman, R.A. Corticotropin-releasing factor receptor-1 modulates biomarkers of DNA oxidation in Alzheimer’s disease mice. PLoS ONE 2017, 12, e0181367. [Google Scholar] [CrossRef] [PubMed]
- Huang, Y.N.; Yang, L.Y.; Greig, N.H.; Wang, Y.C.; Lai, C.C.; Wang, J.Y. Neuroprotective effects of pifithrin-α against traumatic brain injury in the striatum through suppression of neuroinflammation, oxidative stress, autophagy, and apoptosis. Sci. Rep. 2018, 8, 2368. [Google Scholar] [CrossRef] [PubMed]
- Kapay, N.A.; Isaev, N.K.; Stelmashook, E.V.; Popova, O.V.; Zorov, D.B.; Skrebitsky, V.G.; Skulachev, V.P. In vivo injected mitochondria-targeted plastoquinone antioxidant SkQR1 prevents β-amyloid-induced decay of long-term potentiation in rat hippocampal slices. Biochemistry 2011, 76, 1367–1370. [Google Scholar] [CrossRef] [PubMed]
- Melone, M.A.B.; Dato, C.; Paladino, S.; Coppola, C.; Trebini, C.; Giordana, M.T.; Perrone, L. Verapamil Inhibits Ser202/Thr205 Phosphorylation of Tau by Blocking TXNIP/ROS/p38 MAPK Pathway. Pharm. Res. 2018, 35, 44. [Google Scholar] [CrossRef] [PubMed]
- Kang, S.W.; Kim, S.J.; Kim, M.S. Oxidative stress with tau hyperphosphorylation in memory impaired 1,2-diacetylbenzene-treated mice. Toxicol. Lett. 2017, 279, 53–59. [Google Scholar] [CrossRef] [PubMed]
- Yu, C.H.; Si, T.; Wu, W.H.; Hu, J.; Du, J.T.; Zhao, Y.F.; Li, Y.M. O-GlcNAcylation modulates the self-aggregation ability of the fourth microtubule-binding repeat of tau. Biochem. Biophys. Res. Commun. 2008, 375, 59–62. [Google Scholar] [CrossRef] [PubMed]
- Ando, K.; Maruko-Otake, A.; Ohtake, Y.; Hayashishita, M.; Sekiya, M.; Iijima, K.M. Stabilization of Microtubule-Unbound Tau via Tau Phosphorylation at Ser262/356 by Par-1/MARK Contributes to Augmentation of AD-Related Phosphorylation and Aβ42-Induced Tau Toxicity. PLoS Genet. 2016, 12, e1005917. [Google Scholar] [CrossRef] [PubMed]
- Guo, J.; Cheng, J.; North, B.J.; Wei, W. Functional analyses of major cancer-related signaling pathways in Alzheimer’s disease etiology. Biochim. Biophys. Acta 2017, 1868, 341–358. [Google Scholar] [CrossRef] [PubMed]
- Mao, P.; Reddy, P.H. Aging and Amyloid beta-induced oxidative DNA damage and mitochondrial dysfunction in Alzheimer’s Disease: Implications for early intervention and therapeutics. Biochim. Biophys. Acta 2011, 1812, 1359–1370. [Google Scholar] [CrossRef] [PubMed]
- Birnbaum, J.H.; Wanner, D.; Gietl, A.F.; Saake, A.; Kündig, T.M.; Hock, C.; Nitsch, R.M.; Tackenberg, C. Oxidative stress and altered mitochondrial protein expression in the absence of amyloid-β and tau pathology in iPSC-derived neurons from sporadic Alzheimer’s disease patients. Stem Cell Res. 2018, 27, 121–130. [Google Scholar] [CrossRef] [PubMed]
- Wu, W.; Bromberg, P.A.; Samet, J.M. Zinc ions as effectors of environmental oxidative lung injury. Free Radic. Biol. Med. 2013, 65, 57–69. [Google Scholar] [CrossRef] [PubMed]
- Ge, Q.; Wang, Z.; Wu, Y.; Huo, Q.; Qian, Z.; Tian, Z.; Ren, W.; Zhang, X.; Han, J. High salt diet impairs memory-related synaptic plasticity via increased oxidative stress and suppressed synaptic protein expression. Mol. Nutr. Food Res. 2017. [CrossRef] [PubMed]
- Kölker, S.; Burgard, P.; Sauer, S.W.; Okun, J.G. Current concepts in organic acidurias: Understanding intra- and extracerebral disease manifestation. J. Inherit. Metab. Dis. 2013, 36, 635–644. [Google Scholar] [CrossRef] [PubMed]
- Belaidi, A.A.; Bush, A.I. Iron neurochemistry in Alzheimer’s disease and Parkinson’s disease: Targets for therapeutics. J. Neurochem. 2016, 139, 179–197. [Google Scholar] [CrossRef] [PubMed]
- He, L.; He, T.; Farrar, S.; Ji, L.; Liu, T.; Ma, X. Antioxidants Maintain Cellular Redox Homeostasis by Elimination of Reactive Oxygen Species. Cell Physiol. Biochem. 2017, 44, 532–553. [Google Scholar] [CrossRef] [PubMed]
- Hossain, M.S.; ElSayed, A.I.; Moore, M.; Dietz, K.J. Redox and Reactive Oxygen Species Network in Acclimation for Salinity Tolerance in Sugar Beet. J. Exp. Bot. 2017, 68, 1283–1298. [Google Scholar] [CrossRef] [PubMed]
- Ebadi, M.; Iversen, P.L.; Hao, R.; Cerutis, D.R.; Rojas, P.; Happe, H.K.; Murrin, L.C.; Pfeiffer, R.F. Expression and regulation of brain metallothionein. Neurochem. Int. 1995, 27, 1–22. [Google Scholar] [CrossRef]
- Fukai, T.; Ushio-Fukai, M. Superoxide dismutases: Role in redox signaling, vascular function, and diseases. Antioxid. Redox. Signal. 2011, 15, 1583–1606. [Google Scholar] [CrossRef] [PubMed]
- Son, M.; Elliott, J.L. Mitochondrial defects in transgenic mice expressing Cu, Zn superoxide dismutase mutations: The role of copper chaperone for SOD1. J. Neurol. Sci. 2014, 336, 1–7. [Google Scholar] [CrossRef] [PubMed]
- Van Zundert, B.; Brown, R.H., Jr. Silencing strategies for therapy of SOD1-mediated ALS. Neurosci. Lett. 2017, 636, 32–39. [Google Scholar] [CrossRef] [PubMed]
- Sangwan, S.; Eisenberg, D.S. Perspective on SOD1 mediated toxicity in Amyotrophic Lateral Sclerosis. Postepy Biochem. 2016, 62, 362–369. [Google Scholar] [PubMed]
- Saberi, S.; Stauffer, J.E.; Schulte, D.J.; Ravits, J. Neuropathology of Amyotrophic Lateral Sclerosis and Its Variants. Neurol. Clin. 2015, 33, 855–876. [Google Scholar] [CrossRef] [PubMed]
- Wang, R.; Yin, C.; Li, X.X.; Yang, X.Z.; Yang, Y.; Zhang, M.Y.; Wang, H.Y.; Zheng, X.F. Reduced SOD2 expression is associated with mortality of hepatocellular carcinoma patients in a mutant p53-dependent manner. Aging 2016, 8, 1184–1200. [Google Scholar] [CrossRef] [PubMed]
- O'Brien, K.M.; Dirmeier, R.; Engle, M.; Poyton, R.O. Mitochondrial protein oxidation in yeast mutants lacking manganese-(MnSOD) or copper- and zinc-containing superoxide dismutase (CuZnSOD): Evidence that MnSOD and CuZnSOD have both unique and overlapping functions in protecting mitochondrial proteins from oxidative damage. J. Biol. Chem. 2004, 279, 51817–51827. [Google Scholar] [PubMed]
- Flynn, J.M.; Melov, S. SOD2 in mitochondrial dysfunction and neurodegeneration. Free Radic. Biol. Med. 2013, 62, 4–12. [Google Scholar] [CrossRef] [PubMed]
- Lapointe, J.; Hughes, B.G.; Bigras, E.; Hekimi, S. Compensatory elevation of voluntary activity in mouse mutants with impaired mitochondrial energy metabolism. Physiol. Rep. 2014, 2, e12214. [Google Scholar] [CrossRef] [PubMed]
- Zhong, Q.; Kowluru, R.A. Epigenetic modification of Sod2 in the development of diabetic retinopathy and in the metabolic memory: Role of histone methylation. Investig. Ophthalmol. Vis. Sci. 2013, 54, 244–250. [Google Scholar] [CrossRef] [PubMed]
- Silva, J.P.; Shabalina, I.G.; Dufour, E.; Petrovic, N.; Backlund, E.C.; Hultenby, K.; Wibom, R.; Nedergaard, J.; Cannon, B.; Larsson, N.G. SOD2 overexpression: Enhanced mitochondrial tolerance but absence of effect on UCP activity. EMBO J. 2005, 24, 4061–4070. [Google Scholar] [CrossRef] [PubMed]
- Cha, M.Y.; Cho, H.J.; Kim, C.; Jung, Y.O.; Kang, M.J.; Murray, M.E.; Hong, H.S.; Choi, Y.J.; Choi, H.; Choi, D.K.K.H.; et al. Mitochondrial ATP synthase activity is impaired by suppressed O-GlcNAcylation in Alzheimer’s disease. Hum. Mol. Genet. 2015, 24, 6492–6504. [Google Scholar] [CrossRef] [PubMed]
- Ongali, B.; Nicolakakis, N.; Tong, X.K.; Aboulkassim, T.; Papadopoulos, P.; Rosa-Neto, P.; Lecrux, C.; Imboden, H.; Hamel, E. Angiotensin II type 1 receptor blocker losartan prevents and rescues cerebrovascular, neuropathological and cognitive deficits in an Alzheimer’s disease model. Neurobiol. Dis. 2014, 68, 126–136. [Google Scholar] [CrossRef] [PubMed]
- Tong, X.K.; Nicolakakis, N.; Kocharyan, A.; Hamel, E. Vascular remodeling versus amyloid beta-induced oxidative stress in the cerebrovascular dysfunctions associated with Alzheimer’s disease. J. Neurosci. 2005, 25, 11165–11174. [Google Scholar] [CrossRef] [PubMed]
- Lob, H.E.; Marvar, P.J.; Guzik, T.J.; Sharma, S.; McCann, L.A.; Weyand, C.; Gordon, F.J.; Harrison, D.G. Induction of hypertension and peripheral inflammation by reduction of extracellular superoxide dismutase in the central nervous system. Hypertension 2010, 55, 277–283. [Google Scholar] [CrossRef] [PubMed]
- Lakshmi, S.V.; Naushad, S.M.; Reddy, C.A.; Saumya, K.; Rao, D.S.; Kotamraju, S.; Kutala, V.K. Oxidative stress in coronary artery disease: Epigenetic perspective. Mol. Cell Biochem. 2013, 374, 203–211. [Google Scholar] [CrossRef] [PubMed]
- Priyadarshini, M.; Tuimala, J.; Chen, Y.C.; Panula, P. A zebrafish model of PINK1 deficiency reveals key pathway dysfunction including HIF signaling. Neurobiol. Dis. 2013, 54, 127–138. [Google Scholar] [CrossRef] [PubMed]
- Hattan, N.; Chilian, W.M.; Park, F.; Rocic, P. Restoration of coronary collateral growth in the Zucker obese rat: Impact of VEGF and ecSOD. Basic Res. Cardiol. 2007, 102, 217–223. [Google Scholar] [CrossRef] [PubMed]
- Perveen, S.; Patel, H.; Arif, A.; Younis, S.; Codipilly, C.N.; Ahmed, M. Role of EC-SOD overexpression in preserving pulmonary angiogenesis inhibited by oxidative stress. PLoS ONE 2012, 7, e51945. [Google Scholar] [CrossRef] [PubMed]
- Singh, B.; Bhat, H.K. Superoxide dismutase 3 is induced by antioxidants, inhibits oxidative DNA damage and is associated with inhibition of estrogen-induced breast cancer. Carcinogenesis 2012, 33, 2601–2610. [Google Scholar] [CrossRef] [PubMed]
- Janyou, A.; Wicha, P.; Jittiwat, J.; Suksamrarn, A.; Tocharus, C.; Tocharus, J. Dihydrocapsaicin Attenuates Blood Brain Barrier and Cerebral Damage in Focal Cerebral Ischemia/Reperfusion via Oxidative Stress and Inflammatory. Sci. Rep. 2017, 7, 10556. [Google Scholar] [CrossRef] [PubMed]
- Nakanishi, A.; Minami, A.; Kitagishi, Y.; Ogura, Y.; Matsuda, S. BRCA1 and p53 tumor suppressor molecules in Alzheimer’s disease. Int. J. Mol. Sci. 2015, 16, 2879–2892. [Google Scholar] [CrossRef] [PubMed]
- Rodríguez-Sureda, V.; Vilches, Á.; Sánchez, O.; Audí, L.; Domínguez, C. Intracellular oxidant activity, antioxidant enzyme defense system, and cell senescence in fibroblasts with trisomy 21. Oxid. Med. Cell Longev. 2015, 2015, 509241. [Google Scholar] [CrossRef] [PubMed]
- McLoughlin, N.M.; Mueller, C.; Grossmann, T.N. The Therapeutic Potential of PTEN Modulation: Targeting Strategies from Gene to Protein. Cell Chem. Biol. 2018, 25, 19–29. [Google Scholar] [CrossRef] [PubMed]
- Gupta, A.; Dey, C.S. PTEN, a widely known negative regulator of insulin/PI3K signaling, positively regulates neuronal insulin resistance. Mol. Biol. Cell. 2012, 23, 3882–3898. [Google Scholar] [CrossRef] [PubMed]
- Carnero, A. The PKB/AKT pathway in cancer. Curr. Pharm. Des. 2010, 16, 34–44. [Google Scholar] [CrossRef] [PubMed]
- Kriplani, N.; Hermida, M.A.; Brown, E.R.; Leslie, N.R. Class I PI 3-kinases: Function and evolution. Adv. Biol. Regul. 2015, 59, 53–64. [Google Scholar] [CrossRef] [PubMed]
- D'Antoni, S.; Ranno, E.; Spatuzza, M.; Cavallaro, S.; Catania, M.V. Endothelin-1 Induces Degeneration of Cultured Motor Neurons Through a Mechanism Mediated by Nitric Oxide and PI3K/Akt Pathway. Neurotox. Res. 2017, 32, 58–70. [Google Scholar] [CrossRef] [PubMed]
- Knafo, S.; Esteban, J.A. PTEN: Local and Global Modulation of Neuronal Function in Health and Disease. Trends Neurosci. 2017, 40, 83–91. [Google Scholar] [CrossRef] [PubMed]
- Mechlovich, D.; Amit, T.; Bar-Am, O.; Mandel, S.; Youdim, M.B.; Weinreb, O. The novel multi-target iron chelator, M30 modulates HIF-1α-related glycolytic genes and insulin signaling pathway in the frontal cortex of APP/PS1 Alzheimer’s disease mice. Curr. Alzheimer. Res. 2014, 11, 119–127. [Google Scholar] [CrossRef] [PubMed]
- Baki, L.; Neve, R.L.; Shao, Z.; Shioi, J.; Georgakopoulos, A.; Robakis, N.K. Wild-type but not FAD mutant presenilin-1 prevents neuronal degeneration by promoting phosphatidylinositol 3-kinase neuroprotective signaling. J. Neurosci. 2008, 28, 483–490. [Google Scholar] [CrossRef] [PubMed]
- Shang, Y.; Wu, Y.; Yao, S.; Wang, X.; Feng, D.; Yang, W. Protective effect of erythropoietin against ketamine-induced apoptosis in cultured rat cortical neurons: Involvement of PI3K/Akt and GSK-3 beta pathway. Apoptosis 2007, 12, 2187–2195. [Google Scholar] [CrossRef] [PubMed]
- Cavallini, A.; Brewerton, S.; Bell, A.; Sargent, S.; Glover, S.; Hardy, C.; Moore, R.; Calley, J.; Ramachandran, D.; Poidinger, M.; et al. An unbiased approach to identifying tau kinases that phosphorylate tau at sites associated with Alzheimer disease. J. Biol. Chem. 2013, 288, 23331–23347. [Google Scholar] [CrossRef] [PubMed]
- Hernandez, F.; Lucas, J.J.; Avila, J. GSK3 and tau: Two convergence points in Alzheimer’s disease. J. Alzheimers Dis. 2013, 33, S141–S144. [Google Scholar] [PubMed]
- Wang, Y.; Wu, C.; Han, B.; Xu, F.; Mao, M.; Guo, X.; Wang, J. Dexmedetomidine attenuates repeated propofol exposure-induced hippocampal apoptosis, PI3K/Akt/Gsk-3β signaling disruption, and juvenile cognitive deficits in neonatal rats. Mol. Med. Rep. 2016, 14, 769–775. [Google Scholar] [CrossRef] [PubMed]
- Zhang, J.; Shu, Y.; Qu, Y.; Zhang, L.; Chu, T.; Zheng, Y.; Zhao, H. C-myb Plays an Essential Role in the Protective Function of IGF-1 on Cytotoxicity Induced by Aβ25-35 via the PI3K/Akt Pathway. J. Mol. Neurosci. 2017, 63, 412–418. [Google Scholar] [CrossRef] [PubMed]
- Nakanishi, A.; Wada, Y.; Kitagishi, Y.; Matsuda, S. Link between PI3K/AKT/PTEN Pathway and NOX Proteinin Diseases. Aging Dis. 2014, 5, 203–211. [Google Scholar] [CrossRef] [PubMed]
- Kim, K.C.; Lee, I.K.; Kang, K.A.; Cha, J.W.; Cho, S.J.; Na, S.Y.; Chae, S.; Kim, H.S.; Kim, S.; Hyun, J.W. 7,8-Dihydroxyflavone suppresses oxidative stress-induced base modification in DNA via induction of the repair enzyme 8-oxoguanine DNA glycosylase-1. Biomed. Res. Int. 2013, 2013, 863720. [Google Scholar] [CrossRef] [PubMed]
- Surh, Y.J.; Kundu, J.K.; Na, H.K. Nrf2 as a master redox switch in turning on the cellular signaling involved in the induction of cytoprotective genes by some chemopreventive phytochemicals. Planta Med. 2008, 74, 1526–1539. [Google Scholar] [CrossRef] [PubMed]
- Wu, L.; Xu, H.; Cao, L.; Li, T.; Li, R.; Feng, Y.; Chen, J.; Ma, J. Salidroside Protects against MPP+-Induced Neuronal Injury through DJ-1-Nrf2 Antioxidant Pathway. Evid. Based Complement. Alternat. Med. 2017, 2017, 5398542. [Google Scholar] [CrossRef] [PubMed]
- Kim, K.C.; Lee, I.K.; Kang, K.A.; Piao, M.J.; Ryu, M.J.; Kim, J.M.; Lee, N.H.; Hyun, J.W. Triphlorethol-A from Ecklonia cava up-regulates the oxidant sensitive 8-oxoguanine DNA glycosylase 1. Mar. Drug 2014, 12, 5357–5371. [Google Scholar] [CrossRef] [PubMed]
- Zhao, Y.; Song, W.; Wang, Z.; Wang, Z.; Jin, X.; Xu, J.; Bai, L.; Li, Y.; Cui, J.; Cai, L. Resveratrol attenuates testicular apoptosis in type 1 diabetic mice: Role of Akt-mediated Nrf2 activation and p62-dependent Keap1 degradation. Redox. Biol. 2018, 14, 609–617. [Google Scholar] [CrossRef] [PubMed]
- Yamamoto, T.; Suzuki, T.; Kobayashi, A.; Wakabayashi, J.; Maher, J.; Motohashi, H.; Yamamoto, M. Physiological Significance of Reactive Cysteine Residues of Keap1 in Determining Nrf2 Activity. Mol. Cell Biol. 2008, 28, 2758–2770. [Google Scholar] [CrossRef] [PubMed]
- Kannappan, R.; Gupta, S.C.; Kim, J.H.; Reuter, S.; Aggarwal, B.B. Neuroprotection by spice-derived nutraceuticals: You are what you eat! Mol. Neurobiol. 2011, 44, 142–159. [Google Scholar] [CrossRef] [PubMed]
- Dou, J.T.; Chen, M.; Dufour, F.; Alkon, D.L.; Zhao, W.Q. Insulin receptor signaling in long-term memory consolidation following spatial learning. Learn Mem. 2005, 12, 646–655. [Google Scholar] [CrossRef] [PubMed]
- Craft, S.; Claxton, A.; Baker, L.D.; Hanson, A.J.; Cholerton, B.; Trittschuh, E.H.; Dahl, D.; Caulder, E.; Neth, B.; Montine, T.J.; et al. Effects of Regular and Long-Acting Insulin on Cognition and Alzheimer’s Disease Biomarkers: A Pilot Clinical Trial. J. Alzheimers Dis. 2017, 57, 1325–1334. [Google Scholar] [CrossRef] [PubMed]
- Zhang, Y.; Huang, N.Q.; Yan, F.; Jin, H.; Zhou, S.Y.; Shi, J.S.; Jin, F. Diabetes mellitus and Alzheimer’s disease: GSK-3β as a potential link. Behav. Brain Res. 2018, 339, 57–65. [Google Scholar] [CrossRef] [PubMed]
- He, Y.; Wang, P.; Wei, P.; Feng, H.; Ren, Y.; Yang, J.; Rao, Y.; Shi, J.; Tian, J. Effects of curcumin on synapses in APPswe/PS1dE9 mice. Int. J. Immunopathol. Pharmacol. 2016, 29, 217–225. [Google Scholar] [CrossRef] [PubMed]
- Feng, H.L.; Dang, H.Z.; Fan, H.; Chen, X.P.; Rao, Y.X.; Ren, Y.; Yang, J.D.; Shi, J.; Wang, P.W.; Tian, J.Z. Curcumin ameliorates insulin signalling pathway in brain of Alzheimer’s disease transgenic mice. Int. J. Immunopathol. Pharmacol. 2016, 29, 734–741. [Google Scholar] [CrossRef] [PubMed]
- Yan, Z.; Dai, Y.; Fu, H.; Zheng, Y.; Bao, D.; Yin, Y.; Chen, Q.; Nie, X.; Hao, Q.; Hou, D.; Cui, Y. Curcumin Exerts a Protective Effect against Premature Ovarian Failure in Mice. J. Mol. Endocrinol. 2018, 60, 261–271. [Google Scholar] [CrossRef] [PubMed]
- Ma, Q.L.; Zuo, X.; Yang, F.; Ubeda, O.J.; Gant, D.J.; Alaverdyan, M.; Teng, E.; Hu, S.; Chen, P.P.; Maiti, P.; Teter, B.; Cole, G.M.; Frautschy, S.A. Curcumin suppresses soluble tau dimers and corrects molecular chaperone, synaptic, and behavioral deficits in aged human tau transgenic mice. J. Biol. Chem. 2013, 288, 4056–4065. [Google Scholar] [CrossRef] [PubMed]
- Shen, L.; Lo, C.C.; Woollett, L.A.; Liu, M. Apolipoprotein A-IV exerts its anorectic action through a PI3K/Akt signaling pathway in the hypothalamus. Biochem. Biophys. Res. Commun. 2017, 494, 152–157. [Google Scholar] [CrossRef] [PubMed]
- Calon, F. Omega-3 polyunsaturated fatty acids in Alzheimer’s disease: Key questions and partial answers. Curr. Alzheimer Res. 2011, 8, 470–478. [Google Scholar] [CrossRef] [PubMed]
- Kitagishi, Y.; Matsuda, S. Diets involved in PPAR and PI3K/AKT/PTEN pathway may contribute to neuroprotection in a traumatic brain injury. Alzheimers. Res. Ther. 2013, 5, 42. [Google Scholar] [CrossRef] [PubMed]
- Yoshida, H.; Okumura, N.; Kitagishi, Y.; Nishimura, Y.; Matsuda, S. Ethanol extract of Rosemary repressed PTEN expression in K562 culture cells. Int. J. Appl. Biol. Pharm. Technol. 2011, 2, 316–322. [Google Scholar]
- Lee, Y.R.; Yu, H.N.; Noh, E.M.; Kim, J.S.; Song, E.K.; Han, M.K.; Kim, B.S.; Lee, S.H.; Parkd, J. Peroxisome Proliferator-Activated Receptor γ and Retinoic Acid Receptor Synergistically Up-Regulate the Tumor Suppressor PTEN in Human Promyeloid Leukemia Cells. Int. J. Hematol. 2007, 85, 231–237. [Google Scholar] [CrossRef] [PubMed]
- Motawi, T.M.; Hashem, R.M.; Rashed, L.A.; El-Razek, S.M. Comparative study between the effect of the peroxisome proliferator activated receptor-alpha ligands fenofibrate and n-3 polyunsaturated fatty acids on activation of 5'-AMP-activated protein kinase-alpha1 in high-fat fed rats. J. Pharm. Pharmacol. 2009, 61, 1339–1346. [Google Scholar] [CrossRef] [PubMed]
- Tuller, E.R.; Beavers, C.T.; Lou, J.R.; Ihnat, M.A.; Benbrook, D.M.; Ding, W.Q. Docosahexaenoic acid inhibits superoxide dismutase 1 gene transcription in human cancer cells: The involvement of peroxisome proliferator-activated receptor alpha and hypoxia-inducible factor-2alpha signaling. Mol. Pharmacol. 2009, 76, 588–595. [Google Scholar] [CrossRef] [PubMed]
- Fu, J.; Zhang, X.W.; Liu, K.; Li, Q.S.; Zhang, L.R.; Yang, X.H.; Zhang, Z.M.; Li, C.Z.; Luo, Y.; He, Z.X.; Zhu, H.L. Hypolipidemic activity in Sprague-Dawley rats and constituents of a novel natural vegetable oil from Cornus wilsoniana fruits. J. Food Sci. 2012, 77, H160–H169. [Google Scholar] [CrossRef] [PubMed]
- Capiralla, H.; Vingtdeux, V.; Zhao, H.; Sankowski, R.; Al-Abed, Y.; Davies, P.; Marambaud, P. Resveratrol mitigates lipopolysaccharide- and Aβ-mediated microglial inflammation by inhibiting the TLR4/NF-κB/STAT signaling cascade. J. Neurochem. 2012, 120, 461–472. [Google Scholar] [CrossRef] [PubMed]
- Luo, S.; Lan, T.; Liao, W.; Zhao, M.; Yang, H. Genistein inhibits Aβ₂₅₋₃₅ -induced neurotoxicity in PC12 cells via PKC signaling pathway. Neurochem. Res. 2012, 37, 2787–2794. [Google Scholar] [CrossRef] [PubMed]
- Qian, Y.; Guan, T.; Huang, M.; Cao, L.; Li, Y.; Cheng, H.; Jin, H.; Yu, D. Neuroprotection by the soy isoflavone, genistein, via inhibition of mitochondria-dependent apoptosis pathways and reactive oxygen induced-NF-κB activation in a cerebral ischemia mouse model. Neurochem. Int. 2012, 60, 759–767. [Google Scholar] [CrossRef] [PubMed]
- Hwang, S.; Lim, J.W.; Kim, H. Inhibitory Effect of Lycopene on Amyloid-β-Induced Apoptosis in Neuronal Cells. Nutrients 2017, 9, E883. [Google Scholar] [CrossRef] [PubMed]
- Bayer, T.A.; Schäfer, S.; Simons, A.; Kemmling, A.; Kamer, T.; Tepest, R.; Eckert, A.; Schüssel, K.; Eikenberg, O.; Sturchler-Pierrat, C.; et al. Dietary Cu stabilizes brain superoxide dismutase 1 activity and reduces amyloid Abeta production in APP23 transgenic mice. Proc. Nat. Acad. Sci. USA 2003, 100, 14187–14192. [Google Scholar] [CrossRef] [PubMed]
- Shi, X.; Bai, Y.; Zhang, G.; Liu, Y.; Xiao, H.; Liu, X.; Zhang, W. Effects of over-expression of SOD2 in bone marrow-derived mesenchymal stem cells on traumatic brain injury. Cell Tissue Res. 2017, 1, 67–75. [Google Scholar] [CrossRef] [PubMed]
- Esposito, L.; Raber, J.; Kekonius, L.; Yan, F.; Yu, G.Q.; Bien-Ly, N.; Puoliväli, J.; Scearce-Levie, K.; Masliah, E.; Mucke, L. Reduction in mitochondrial superoxide dismutase modulates Alzheimer’s disease-like pathology and accelerates the onset of behavioral changes in human amyloid precursor protein transgenic mice. J. Neurosci. 2006, 26, 5167–5179. [Google Scholar] [CrossRef] [PubMed]



© 2018 by the authors. Licensee MDPI, Basel, Switzerland. This article is an open access article distributed under the terms and conditions of the Creative Commons Attribution (CC BY) license (http://creativecommons.org/licenses/by/4.0/).
Share and Cite
Matsuda, S.; Nakagawa, Y.; Tsuji, A.; Kitagishi, Y.; Nakanishi, A.; Murai, T. Implications of PI3K/AKT/PTEN Signaling on Superoxide Dismutases Expression and in the Pathogenesis of Alzheimer’s Disease. Diseases 2018, 6, 28. https://doi.org/10.3390/diseases6020028
Matsuda S, Nakagawa Y, Tsuji A, Kitagishi Y, Nakanishi A, Murai T. Implications of PI3K/AKT/PTEN Signaling on Superoxide Dismutases Expression and in the Pathogenesis of Alzheimer’s Disease. Diseases. 2018; 6(2):28. https://doi.org/10.3390/diseases6020028
Chicago/Turabian StyleMatsuda, Satoru, Yukie Nakagawa, Ai Tsuji, Yasuko Kitagishi, Atsuko Nakanishi, and Toshiyuki Murai. 2018. "Implications of PI3K/AKT/PTEN Signaling on Superoxide Dismutases Expression and in the Pathogenesis of Alzheimer’s Disease" Diseases 6, no. 2: 28. https://doi.org/10.3390/diseases6020028
APA StyleMatsuda, S., Nakagawa, Y., Tsuji, A., Kitagishi, Y., Nakanishi, A., & Murai, T. (2018). Implications of PI3K/AKT/PTEN Signaling on Superoxide Dismutases Expression and in the Pathogenesis of Alzheimer’s Disease. Diseases, 6(2), 28. https://doi.org/10.3390/diseases6020028