
Anti-Angiogenic Therapy: Albumin-Binding Proteins Could Mediate Mechanisms Underlying the Accumulation of Small Molecule Receptor Tyrosine Kinase Inhibitors in Normal Tissues with Potential Harmful Effects on Health


Abstract
:Simple Summary
Abstract
1. Introduction
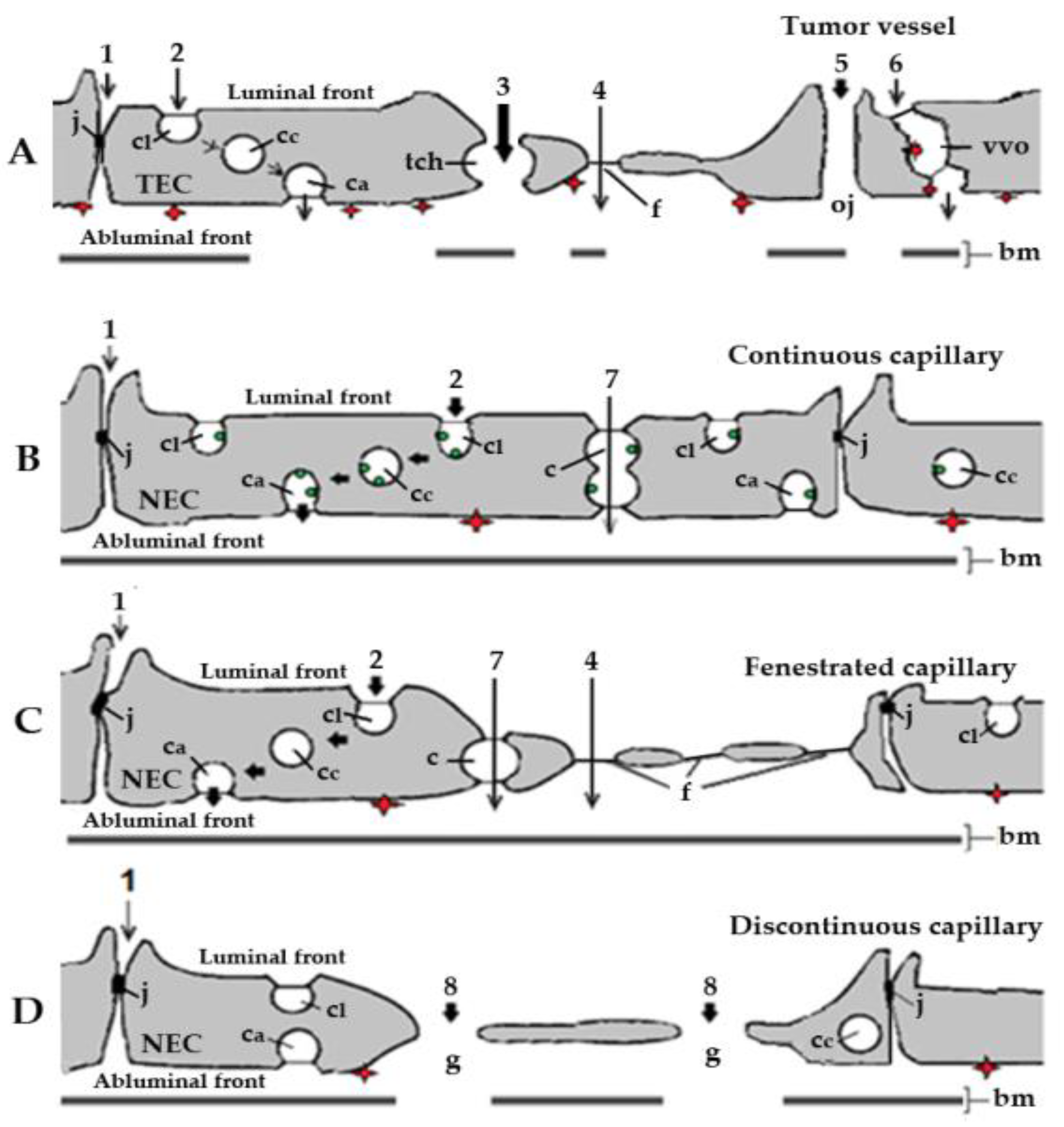
2. How Anti-Angiogenics Reach Their Target in Cancer: Cellular and Molecular Mechanisms Involved in the Accumulation of Drugs in Tumors
3. Diffusion and ABP-Mediated Mechanisms Could Underlie the Accumulation of RTKIs Bound to Albumin in Healthy Organs and Tissues
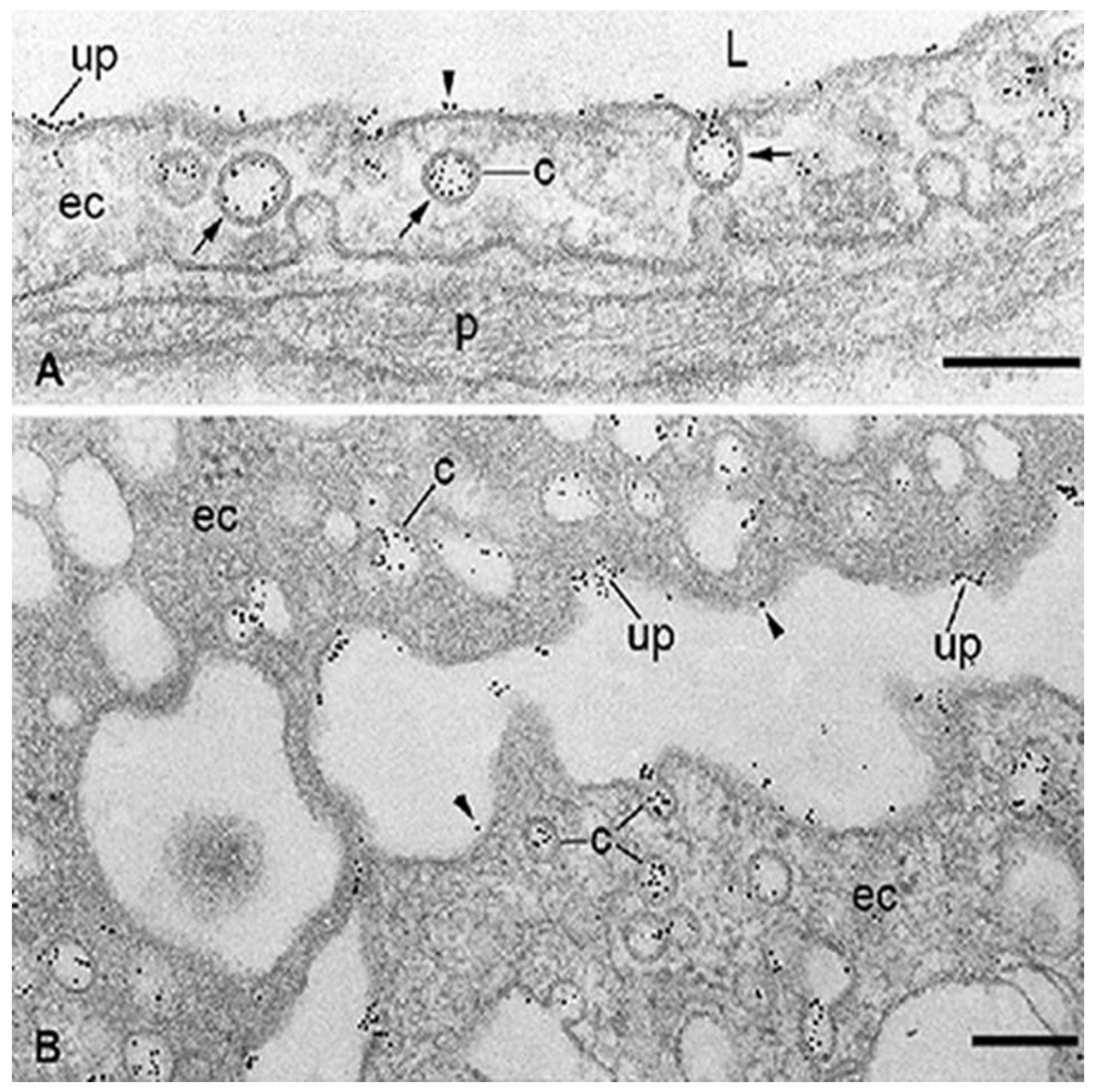
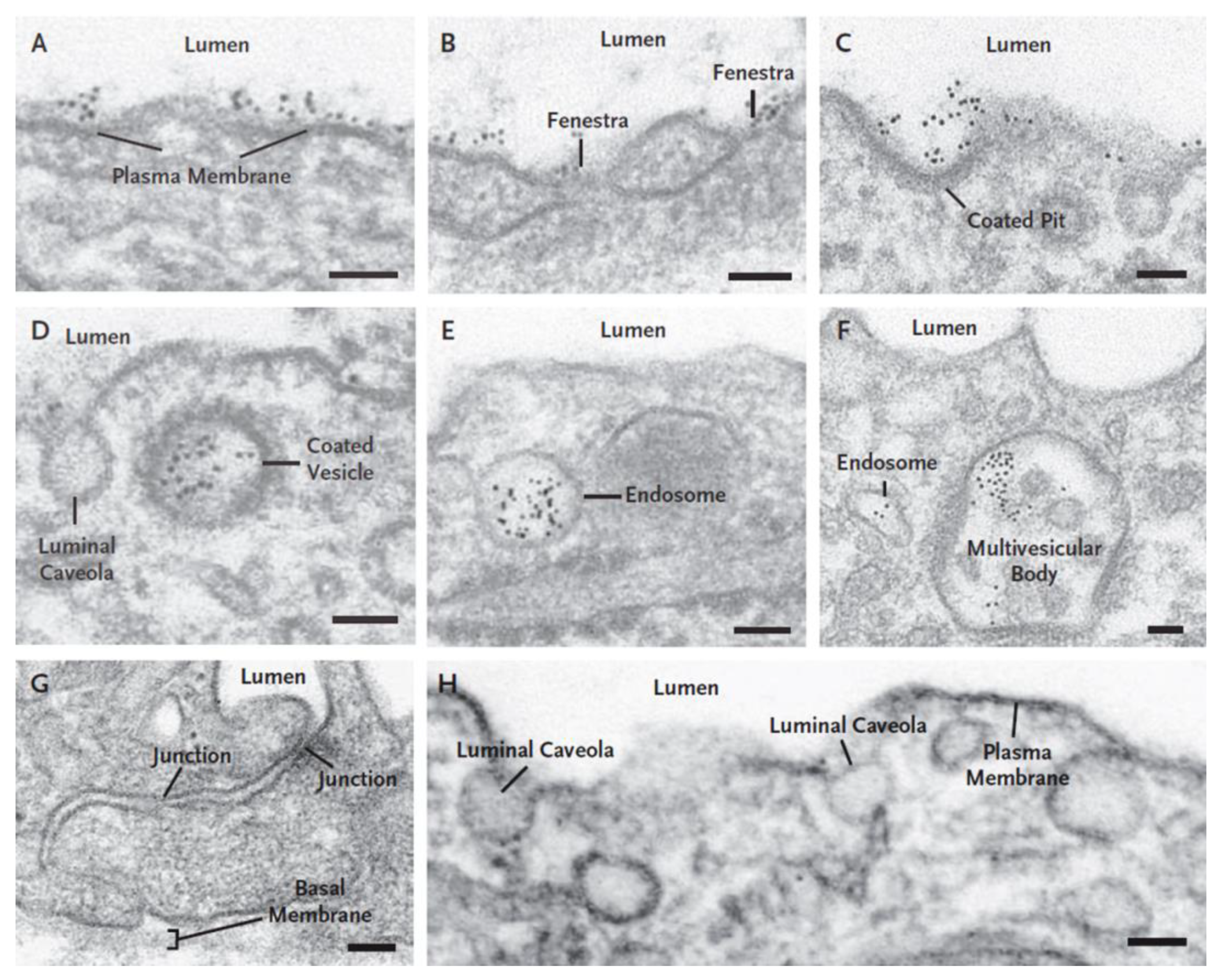
3.1. Endothelial Cell Pathways that May Be Involved in Transport of RTKIs Bound to Albumin
3.2. ABP-Mediated Accumulation of RTKIs Bound to Albumin in Healthy Organs and Tissues Could Be Responsible for Side Effects and Toxicity
4. Perspectives
Funding
Institutional Review Board Statement
Informed Consent Statement
Data Availability Statement
Conflicts of Interest
References
- Folkman, J. Tumor angiogenesis factor. Cancer Res. 1974, 34, 2109–2113. [Google Scholar]
- Bouïs, D.; Kusumanto, Y.; Meijer, C.; Mulder, N.H.; Hospers, G.A. A review on pro- and anti-angiogenic factors as targets of clinical intervention. Pharmacol. Res. 2006, 53, 89–103. [Google Scholar] [CrossRef]
- Raica, M.; Cimpean, A.M. Platelet-derived growth factor (PDGF)/PDGF receptors (PDGFR) axis as target for antitumor and anti-angiogenic therapy. Pharmaceuticals 2010, 3, 572–599. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Korc, M.; Friesel, R.E. The role of fibroblast growth factors in tumor growth. Curr. Cancer Drug Target 2009, 9, 639–651. [Google Scholar] [CrossRef]
- Presta, M.; Dell’Era, P.; Mitola, S.; Moroni, E.; Ronca, R.; Rusnati, M. Fibroblast growth factor/fibroblast growth factor receptor system in angiogenesis. Cytokine Growth Factor Rev. 2005, 16, 159–178. [Google Scholar] [CrossRef] [Green Version]
- Holash, J.; Maisonpierre, P.C.; Compton, D.; Boland, P.; Alexander, C.R.; Zagzag, D.; Yancopoulos, G.D.; Wiegand, S.J. Vessel cooption, regression, and growth in tumors mediated by angiopoietins and VEGF. Science 1999, 284, 1994–1998. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Weis, S.M.; Cheresh, D.A. Tumor angiogenesis: Molecular pathways and therapeutic targets. Nat. Med. 2011, 17, 1359–1370. [Google Scholar] [CrossRef] [PubMed]
- Folkman, J. Tumor angiogenesis: Therapeutic implications. N. Engl. J. Med. 1971, 285, 1182–1186. [Google Scholar]
- Folkman, J. Antiangiogenesis: New concept for therapy of solid tumors. Ann. Surg. 1972, 175, 409–416. [Google Scholar] [CrossRef]
- Boehm, T.; Folkman, J.; Browder, T.; O’Reilly, M.S. Anti-angiogenic therapy of experimental cancer does not induce acquired drug resistance. Nature 1997, 390, 404–407. [Google Scholar] [CrossRef]
- Eberhard, A.; Kahlert, S.; Goede, V.; Hemmerlein, B.; Plate, K.H.; Augustin, H.G. Heterogeneity of angiogenesis and blood vessel maturation in human tumors: Implications for anti-angiogenic tumor therapies. Cancer Res. 2000, 60, 1388–1393. [Google Scholar] [PubMed]
- Gotink, K.J.; Verheu, H.M.W. Anti-angiogenic tyrosine kinase inhibitors: What is their mechanism of action? Angiogenesis 2010, 13, 1–14. [Google Scholar] [CrossRef] [Green Version]
- van Beijnum, J.R.; Nowak-Sliwinska, P.; Huijbers, E.J.M.; Thijssen, V.L.; Griffioen, A.W. The great escape; the hallmarks of resistance to anti-angiogenic therapy. Pharmacol. Rev. 2015, 67, 441–461. [Google Scholar] [CrossRef] [Green Version]
- Ellis, L.M.; Hicklin, D.J. VEGF-target therapies: Mechanisms of anti-tumor therapy. Nat. Rev. Cancer 2008, 8, 579–591. [Google Scholar] [CrossRef]
- Carmeliet, P.; Jain, R.K. Molecular mechanisms and clinical applications of angiogenesis. Nature 2011, 473, 298–307. [Google Scholar] [CrossRef] [Green Version]
- Zhang, Y.; Toneri, M.; Ma, H.; Yang, Z.; Bouvet, M.; Goto, Y.; Seki, N.; Hoffman, R.M. Real-Time GFP intravital imaging of the differences in cellular and angiogenic behavior of subcutaneous and orthotropic nude-mouse models of human PC-3 prostate cancer. J. Cell Biochem. 2016, 117, 2546–2551. [Google Scholar] [CrossRef] [PubMed]
- Ruoslahti, E. Drug targeting to specific vascular sites. Drug Discov. Today 2002, 7, 1138–1143. [Google Scholar] [CrossRef]
- Algire, G.H.; Chalkley, H.W. Vascular reactions of normal and malignant tissues in vivo. l. Vascular reactions of mice to wounds and to normal and neoplastic transplants. J. Natl. Cancer Inst. 1945, 6, 73–85. [Google Scholar] [CrossRef]
- Dvorak, H.F. Tumor stroma, tumor blood vessels, and antiangiogenesis therapy. Cancer J. 2015, 21, 237–243. [Google Scholar] [CrossRef] [PubMed]
- Vartanian, R.K.; Weidner, N. Endothelial cell proliferation in prostatic carcinoma and prostatic hyperplasia: Correlation with Gleason’s score, microvessel density, and epithelial cell proliferation. Lab. Inv. 1995, 73, 844–850. [Google Scholar]
- Baldewijns, M.M.; Thijssen, V.L.; Van den Eynden, G.G.; Van Laere, S.J.; Bluekens, A.M.; Roskams, T.; van Poppel, H.; De Bruïne, A.P.; Griffioen, A.W.; Vermeulen, P.B. High-grade clear cell renal cell carcinoma has a higher angiogenic activity than low-grade renal cell carcinoma based on histomorphological quantification and qRT–PCR mRNA expression profile. Br. J. Cancer 2007, 96, 1888–1895. [Google Scholar] [CrossRef] [PubMed]
- Fox, S.B.; Gatter, K.C.; Bicknell, R.; Going, J.J.; Stanton, P.; Cooke, T.G.; Harris, A.L. Relationship of endothelial cell proliferation to tumor vascularity in human breast cancer. Cancer Res. 1993, 53, 4161–4163. [Google Scholar]
- Vermeulen, P.B.; Verhoeven, D.; Hubens, G.; Van Marck, E.; Goovaerts, G.; Huyghe, M.; De Bruijn, E.A.; Van Oosterom, A.T.; Dirix, L.Y. Microvessel density, endothelial cell proliferation and tumour cell proliferation in human colorectal adenocarcinomas. Ann. Oncol. 1995, 6, 59–64. [Google Scholar] [CrossRef]
- Kim, Y.B.; Park, Y.N.; Park, C. Increased proliferation activities of vascular endothelial cells and tumor cells in residual hepatocellular carcinoma following transcatheter arterial embolization. Histopathology 2001, 38, 160–166. [Google Scholar] [CrossRef] [PubMed]
- Bergers, G.; Hanahan, D. Modes of resistance to anti-angiogenic therapy. Nat. Rev. Cancer 2008, 8, 592–603. [Google Scholar] [CrossRef] [Green Version]
- Ribatti, D.; Vacca, A.; Dammacco, F. New non-angiogenesis dependent pathways for tumour growth. Eur. J. Cancer 2003, 39, 1835–1841. [Google Scholar] [CrossRef]
- Kuczynski, E.A.; Reynolds, A.R. Vessel co-option and resistance to anti-angiogenic therapy. Angiogenesis 2020, 13, 55–74. [Google Scholar] [CrossRef]
- Kuczynski, E.A.; Vermeulen, P.B.; Pezzella, F.; Kerbel, R.S.; Reynolds, A.R. Vessel co-option in cancer. Nat. Rev. Clin. Oncol. 2019, 16, 469–493. [Google Scholar] [CrossRef]
- Bridgeman, V.L.; Vermeulen, P.B.; Foo, S.; Bilecz, A.; Daley, F.; Kostaras, E.; Nathan, M.R.; Wan, E.; Frentzas, S.; Schweiger, T.; et al. Vessel co-option is common in human lung metastases and mediates resistance to anti-angiogenic therapy in preclinical lung metastases models. J. Pathol. 2017, 241, 362–374. [Google Scholar] [CrossRef] [PubMed]
- Qian, C.-N. Hijacking the vasculature in ccRCC—co-option, remodeling and angiogenesis. Nat. Rev. Urol. 2013, 10, 300–304. [Google Scholar] [CrossRef] [PubMed]
- Frentzas, S.; Simoneau, E.; Bridgeman, V.L.; Vermeulen, P.B.; Fao, S.; Kostaras, E.; Nathan, M.; Wotherspoon, A.; Gao, Z.H.; Shi, Y.; et al. Vessel co-option mediates resistance to anti-angiogenic therapy in liver metastases. Nat. Med. 2016, 22, 1294–1302. [Google Scholar] [CrossRef] [Green Version]
- Montemagno, C.; Pagès, G. Resistance to anti-angiogenic therapies: A mechanism depending on the time of exposure to the drugs. Front. Cell Dev. Biol. 2020. [Google Scholar] [CrossRef]
- Haibe, Y.; Kreidieh, M.; El Khalief, I.; Mukherji, D.; Temraz, S.; Shamseddine, A. Resistance mechanisms to anti-angiogenic therapies in cancer. Front. Oncol. 2020, 10, 221. [Google Scholar] [CrossRef] [Green Version]
- De Sande, L.V.; Cosyns, S.; Willaert, W.; Ceelen, W. Albumin-based cancer therapeutics for intraperitoneal drug delivery: A review. Drug Deliv. 2020, 1, 40–53. [Google Scholar] [CrossRef]
- Sleep, D.; Cameron, J.; Evans, L.R. Albumin as a versatile platform for drug half-life expansions. Biochim. Biophys. Acta 2013, 1830, 5526–5534. [Google Scholar] [CrossRef] [PubMed]
- Yamasaki, K.; Chuang, V.T.; Maruyama, T.; Otagiri, M. Albumin-drug interaction and its clinical implication. Biochim. Biophys. Acta 2013, 1830, 5435–5443. [Google Scholar] [CrossRef] [PubMed]
- Hashizume, H.; Baluk, P.; Morikawa, S.; McLean, J.W.; Thurston, G.; Roberge, S.; Jain, R.K.; McDonald, D.M. Openings between defective endothelial cells explains tumor vessel leakiness to tumor cells. Am. J. Pathol. 2000, 156, 1363–1380. [Google Scholar] [CrossRef] [Green Version]
- Roberts, W.G.; Palade, G.E. Neovasculature induced by vascular endothelial growth factor is fenestrated. Cancer Res. 1997, 57, 765–772. [Google Scholar]
- Dvorak, A.M.; Kohn, S.; Morgan, E.S.; Fox, P.; Nagy, J.A.; Dvorak, H.F. The vesiculo-vacuolar organelle (VVO): A distinct endothelial cell structure that provides a transcellular pathway for macromolecular extravasation. Leukoc. Biol. 1996, 59, 100–115. [Google Scholar] [CrossRef]
- Sindhwani, S.; Syed, A.M.; Ngai, J.; Kingston, B.R.; Maiorino, L.; Rothschild, J.; Presley Mc, M.; Zhang, Y.; Rajesh, N.U.; Wu, T.; et al. The entry of nanoparticles into solid tumours. Nat. Mater. 2020, 19, 566–575. [Google Scholar] [CrossRef] [PubMed]
- Ghinea, N.; Milgrom, E. Transport of protein hormones through the vascular endothelium. J. Endocrinol. 1995, 145, 1–9. [Google Scholar] [CrossRef] [PubMed]
- Jain, R.K. Transport of molecules across tumor vasculature. Cancer Metastasis Rev. 1987, 6, 559–593. [Google Scholar] [CrossRef]
- Abdel-Qadir, H.; Ethier, J.L.; Lee, D.S.; Thavendiranathan, P.; Amir, E. Cardiovascular toxicity of angiogenesis inhibitors in treatment of malignancy: A systematic review and meta-analysis. Cancer Treat. Rev. 2017, 53, 120–127. [Google Scholar] [CrossRef]
- Dy, G.K.; Adjei, A.A. Understanding, recognizing, and managing of targeted anticancer therapies. Calif. Cancer J. Clin. 2013, 63, 249–279. [Google Scholar] [CrossRef]
- van Heeckeren, W.J.; Ortiz, J.; Cooney, M.M.; Remick, S.C. Hypertension, proteinuria, and antagonism of vascular endothelial growth factor signaling: Clinical toxicity, therapeutic target, or novel biomarker? J. Clin. Oncol. 2007, 25, 2993–2995. [Google Scholar] [CrossRef]
- Simionescu, M.; Ghinea, N. The use of tracers in transport studies. In Models of Lung Disease. Microscopy and Structural Methods; Gill, J., Ed.; Marcel Dekker, Inc.: New York, NY, USA, 1990; pp. 359–408. [Google Scholar]
- Larson, S.M.; Carrasquillo, J.A.; Reynolds, J.C. Radioimmunodetection and radioimmunotherapy. Cancer Inv. 1984, 2, 358–381. [Google Scholar] [CrossRef] [PubMed]
- Powers, M.R.; Bell, D.R. Initial equilibration of albumin and IgG in rabbit hind paw skin and lymph. Microvasc. Res. 1990, 40, 230–245. [Google Scholar] [CrossRef]
- Ghitescu, L.; Bendayan, M. Transendothelial transport of serum albumin. A quantitative immunocytochemical study. J. Cell Biol. 1992, 117, 745–755. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Karnovsky, M.J. The ultrastructural basis of capillary permeability studied with peroxidase as a tracer. J. Cell Biol. 1967, 35, 213–236. [Google Scholar] [CrossRef]
- Wissig, S.L.; Williams, M.C. Permeability of muscle capillaries to microperoxidase. J. Cell Biol. 1978, 76, 341–359. [Google Scholar] [CrossRef] [Green Version]
- Bruns, R.R.; Palade, G.E. Studies on blood capillaries. II. Transport of ferritin molecules across the wall of muscle capillaries. J. Cell Biol. 1968, 37, 277–299. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Simionescu, N.; Simionescu, M.; Palade, G.E. Permeability of muscle capillaries to small heme-peptides. Evidence for the existence of patent transendothelial channels. J. Cell Biol. 1975, 64, 586–607. [Google Scholar] [CrossRef]
- Clementi, F.; Palade, G.E. Intestinal capillaries. I. Permeability to peroxidase and ferritin. J. Cell Biol. 1969, 41, 33–58. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Simionescu, N.; Simionescu, M.; Palade, G.E. Open junctions in the endothelium of postcapillary venules of the diaphragm. J. Cell Biol. 1978, 79, 27–44. [Google Scholar] [CrossRef] [PubMed]
- Reese, T.S.; Karnovsky, M.J. Fine structural localization of a blood-brain barrier to exogenous peroxidase. J. Cell Biol. 1967, 34, 207–217. [Google Scholar] [CrossRef] [Green Version]
- Majno, G.; Palade, G.E. Studies on inflammation. I. The effect of histamine and serotonin on vascular permeability: An electron microscopic study. J. Biophys. Biochem. Cytol. 1961, 11, 607–626. [Google Scholar] [CrossRef] [Green Version]
- Ghinea, N.; Simionescu, N. Anionized and cationized hemeundecapeptides as probes for cell surface charge and permeability studies: Differentiated labeling of endothelial plasmalemmal vesicles. J. Cell Biol. 1985, 100, 606–612. [Google Scholar] [CrossRef] [Green Version]
- Ghinea, N.; Hasu, M. Charge effect on binding, uptake and transport of ferritin through fenestrated endothelium. J. Submicrosc. Cytol. 1986, 18, 647–659. [Google Scholar]
- Hervé, F.; Ghinea, N.; Scherrmann, J.M. CNS delivery via adsorptive transcytosis. AAPS J. 2008, 10, 455–472. [Google Scholar] [CrossRef] [Green Version]
- Ghitescu, L.; Fixman, A.; Simionescu, M.; Simionescu, N. Specific binding sites for albumin restricted to plasmalemmal vesicles of continuous capillary endothelium: Receptor-mediated transcytosis. J. Cell Biol. 1986, 102, 1304–1311. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Ghinea, N.; Fixman, A.; Alexandru, D.; Popov, D.; Hasu, M.; Ghitescu, L.; Eskenasy, M.; Simionescu, M.; Simionescu, N. Identification of albumin-binding proteins in capillary endothelial cells. J. Cell Biol. 1988, 107, 231–239. [Google Scholar] [CrossRef] [Green Version]
- Ghinea, N.; Eskenasy, M.; Simionescu, M.; Simionescu, N. Endothelial albumin binding proteins are membrane-associated components exposed on the cell surface. J. Biol. Chem. 1989, 264, 4755–4758. [Google Scholar] [CrossRef]
- Schnitzer, J.E.; Carley, W.W.; Palade, G.E. Albumin interacts specifically with a 60-kDa microvascular glycoprotein. Proc. Natl. Acad. Sci. USA 1988, 85, 6773–6777. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Simionescu, M.; Simionescu, N. Endothelial transport of macromolecules: Transcytosis and endocytosis. A look from cell biology. Cell Biol. Rev. 1991, 25, 1–78. [Google Scholar]
- Frokjaer-Jensen, J. The vesicle controversy. Progress Appl. Microcirc. 1985, 9, 21–42. [Google Scholar]
- Bundgaard, M.; Frokjaer-Jensen, J.; Crone, C. Endothelial plasmalemmal vesicular profiles as elements in a system of branching invaginations from the cell surface. Proc. Natl. Acad. Sci. USA 1979, 76, 6439–6442. [Google Scholar] [CrossRef] [Green Version]
- Stan, R.V.; Tse, D.; Deharvengt, S.J.; Smits, N.C.; Xu, Y.; Luciano, M.R.; McGarry, C.L.; Buitendijk, M.; Nemani, K.V.; Elgueta, R.; et al. The diaphragms of fenestrated endothelia—gatekeepers of vascular permeability and blood composition. Dev. Cell 2012, 23, 1203–1218. [Google Scholar] [CrossRef] [Green Version]
- Simionescu, M.; Simionescu, N.; Silbert, J.E.; Palade, G.E. Differentiated microdomains on the luminal surface of the capillary endothelium. II. Partial characterization of their anionic sites. J. Cell Biol. 1981, 90, 614–621. [Google Scholar] [CrossRef] [Green Version]
- Pino, R.M. The cell surface of a restrictive fenestrated endothelium. II. Dynamics of cationic ferritin binding and the identification of heparin and heparan sulfate domains on the choriocapillaries. Cell Tissue Res. 1986, 243, 157–164. [Google Scholar] [CrossRef]
- Wang, B.; Chen, K.; Xu, W.; Chen, D.; Tang, W.; Xia, T.S. Integrative genomic analyses of secreted protein acidic and rich in cysteine and its role in cancer prediction. Mol. Med. Rep. 2014, 10, 1461–1468. [Google Scholar] [CrossRef]
- Sage, H.; Vernon, R.B.; Funk, S.E.; Everitt, E.A.; Angello, J. SPARC, a secreted protein associated with cellular proliferation, inhibits cell spreading in vitro and exhibits Ca+2-dependent binding to the extracellular matrix. J. Cell Biol. 1989, 109, 341–356. [Google Scholar] [CrossRef] [PubMed]
- Schnitzer, J.E.; Bravo, J. High affinity binding, endocytosis, and degradation of conformationally modified albumins. Potential role of gp30 and gp18 as novel scavenger receptors. J. Biol. Chem. 1993, 268, 7562–7750. [Google Scholar] [CrossRef]
- Schnitzer, J.; Sung, A.; Horvat, R.; Bravo, J. Preferential interaction of albumin-binding proteins, gp30 and gp18, with conformationally modified albumins. Presence in many cells and tissues with possible role in catabolism. J. Biol. Chem. 1992, 267, 24544–24553. [Google Scholar] [CrossRef]
- Jussing, E.; Lu, L.; Grafström, J.; Tegnebratt, T.; Arnberg, F.; Walllberg Rosik, H.; Wennborg, A.; Holmin, S.; Feldwisch, J.; Stone-Elander, S. [68Ga]ABY-028: An albumin-binding domain (ABD) protein-based imaging tracer for positron emission tomography (PET) studies of altered vascular permeability and predictions of albumin-drug conjugate transport. EJNMMI Res. 2020, 10, 106. [Google Scholar] [CrossRef] [PubMed]
- Park, J.Y.; Song, M.G.; Kim, W.H.; Kim, K.W.; Lodhi, N.A.; Choi, J.Y.; Kim, Y.J.; Kim, J.Y.; Chung, H.; Oh, C.; et al. Versatile and finely tuned albumin nanoplatform based on click chemistry. Theranostics 2019, 9, 3398–3409. [Google Scholar] [CrossRef]
- Zhong, C.C.; Chen, F.; Yang, J.L.; Li, L.; Cheng, C.; Du, F.F.; Zhang, S.P.; Xie, C.Y.; Zhang, N.T.; Olaleye, O.E.; et al. Pharmacokinetics and disposition of anlotinib, an oral tyrosine kinase inhibitor, in experimental animal species. Acta Pharmacol. Sin. 2018, 39, 1048–1063. [Google Scholar] [CrossRef] [Green Version]
- Yedgar, S.; Carew, T.E.; Pittman, R.C.; Beltz, W.F.; Steinberg, D. Tissue sites of catabolism of albumin in rabbits. Am. J. Physiol. 1983, 244, E101–E107. [Google Scholar] [CrossRef]
- Bodnar, R.J. Anti-angiogenic drugs: Involvement in cutaneous side effects and wound-healing complication. Adv. Would Care 2014, 3, 635–646. [Google Scholar] [CrossRef] [Green Version]
- Dobbin, S.J.H.; Cameron, A.C.; Petrie, M.C.; Jones, R.J.; Touyz, R.M.; Lang, N.N. Toxicity of cancer therapy: What the cardiologist needs to know about angiogenesis inhibitors. Heart 2018, 104, 1995–2002. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Randrup Hansen, C.; Grimm, D.; Bauer, J.; Wehland, M.; Magnusson, N.E. Effects and side effects of using sorafenib and sunitinib in the treatment of metastatic renal cell carcinoma. Int. J. Mol. Sci. 2017, 18, 461. [Google Scholar] [CrossRef] [PubMed]
- Semeniuk-Wojtaś, A.; Lubas, A.; Stec, R.; Szczylik, C.; Niemczyk, S. Influence of tyrosine kinase inhibitors on hypertension and nephrotoxicity in metastatic renal cell cancer patients. Int. J. Mol. Sci. 2016, 17, 2073. [Google Scholar] [CrossRef] [Green Version]
- Sun, Y.; Niu, W.; Du, F.; Du, C.; Li, S.; Wang, J.; Li, L.; Wang, F.; Hao, Y.; Li, C.; et al. Safety, pharmacokinetics, and antitumor properties of anlotinib, an oral multi-target tyrosine kinase inhibitor, in patients with advanced refractory solid tumors. J. Hematol. Oncol. 2016, 9, 105. [Google Scholar] [CrossRef] [Green Version]
- Fasano, M.; Curry, S.; Terreno, E.; Galliano, M.; Fanali, G.; Narciso, P.; Notari, S.; Ascenzi, P. The extraordinary ligand binding properties of human serum albumin. IUBMB Life 2005, 57, 787–796. [Google Scholar] [CrossRef]
- Ebos, J.M.; Lee, C.R.; Cruz-Munoz, W.; Bjarnason, G.A.; Christensen, J.G.; Kerbel, R.S. Accelerated metastasis after short-term treatment with a potent inhibitor of tumor angiogenesis. Cancer Cell 2009, 15, 232–239. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Wang, D.; Xiao, F.; Feng, Z.; Li, M.; Kong, L.; Huang, L.; Wei, Y.; Li, H.; Liu, F.; Zang, H.; et al. Sunitinib facilitates metastatic breast cancer spreading by inducing endothelial cell senescence. Brest Cancer Res. 2020, 22, 103. [Google Scholar] [CrossRef]
- Zhai, W.; Li, S.; Zhang, J.; Chen, Y.; Ma, J.; Kong, W.; Gong, D.; Zheng, J.; Xue, W.; Xu, Y. Sunitinib-suppressed miR-452-5p facilitates renal cancer cell invasion and metastasis through modulating SMAD4/SMAD7 signals. Mol. Cancer 2018, 17, 157. [Google Scholar] [CrossRef] [Green Version]
- Jászai, J.; Schmidt, M.H.H. Trends and challenges in tumor anti-angiogenic therapies. Cells 2019, 8, 1102. [Google Scholar] [CrossRef] [Green Version]
- Ferrara, N.; Adamis, A.P. Ten years of anti-vascular endothelial growth factor therapy. Nat. Rev. Drug Discov. 2016, 15, 385–403. [Google Scholar] [CrossRef] [Green Version]
- Neri, D.; Bickell, R. Tumor vascular targeting. Nat. Rev. Cancer 2005, 5, 436–446. [Google Scholar] [CrossRef] [PubMed]
- Ghinea, N. Vascular endothelial receptor, a target of interest for cancer therapy. Endocrinology 2018, 159, 3268–3274. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Yang, W.P.; Green, K.; Pinz-Sweeney, S.; Briones, A.T.; Burton, D.R.; Barbas III, C.F. CDR walking mutagenesis for the affinity maturation of a potent human anti-HIV-1 antibody into picomolar range. J. Mol. Biol. 1995, 254, 392–403. [Google Scholar] [CrossRef] [Green Version]
- Trachel, E.; Neri, D. Antibodies for angiogenesis inhibition, vascular targeting and endothelial cell transcytosis. Adv. Drug Deliv. Rev. 2006, 58, 735–754. [Google Scholar] [CrossRef]
- Radu, A.; Pichon, C.; Camparo, P.; Antoine, M.; Allory, Y.; Couvelard, A.; Fromont, G.; Vu Hai, M.T.; Ghinea, N. Expression of follicle-stimulating hormone receptor in tumor blood vessels. N. Engl. J. Med. 2010, 363, 1621–1630. [Google Scholar] [CrossRef]
- Siraj, A.; Desestret, V.; Antoine, M.; Fromont, G.; Huerre, M.; Sanson, M.; Camparo, P.; Pichon, C.; Planeix, F.; Gonin, J.; et al. Expression of follicle-stimulating hormone receptor by the vascular endothelium in tumor metastases. BMC Cancer 2013, 13, 246. [Google Scholar] [CrossRef] [Green Version]
- Planeix, F.; Siraj, M.A.; Bidard, F.C.; Robin, B.; Pichon, C.; Sastre-Garau, X.; Antoine, M.; Ghinea, N. Endothelial follicle-stimulating hormone receptor expression in invasive breast cancer and vascular remodeling at tumor periphery. J. Exp. Clin. Cancer Res. 2015, 34, 12. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Siraj, M.A.; Pichon, C.; Radu, A.; Ghinea, N. Endothelial follicle stimulating hormone receptor in primary kidney cancer correlates with subsequent response to sunitinib. J. Cell. Mol. Med. 2012, 16, 2010–2016. [Google Scholar] [CrossRef]
- Ghinea, N.; Robin, B.; Pichon, C.; Leclere, R.; Nicolas, A.; Chnecker, C.; Côté, J.F.; Guillonneau, B.; Radu, A. Vasa nervorum angiogenesis in prostate cancer with perineural invasion. Prostate 2019, 79, 640–646. [Google Scholar] [CrossRef] [PubMed]
- Renner, M.; Goeppert, B.; Siraj, M.A.; Radu, A.; Penzel, R.; Wardelmann, E.; Lehner, B.; Ulrich, A.; Stenzinger, A.; Warth, A.; et al. Follicle-stimulating hormone receptor expression in soft tissue sarcomas. Histopathology 2013, 63, 29–35. [Google Scholar] [CrossRef] [PubMed]
- Cheung, J.; Lokman, N.A.; Abraham, R.D.; Macpherson, A.M.; Lee, E.; Frank Grutzner, F.; Ghinea, N.; Oehler, M.K.; Ricciardelli, C. Reduced gonadotrophin receptor expression is associated with a more aggressive ovarian cancer phenotype. Int. J. Mol. Sci. 2021, 22, 71. [Google Scholar] [CrossRef]
- Möker, N.; Möker, N.; Solveig Peters, S.; Rauchenberger, R.; Ghinea, N.; Kunz, C. Antibody selection for cancer target validation of FSH-receptor in immunohistochemical settings. Antibodies 2017, 6, 15. [Google Scholar] [CrossRef] [Green Version]
- Zeng, H.; Zhao, D.; Yang, S.; Datta, K.; Mukhopadhyay, D. Heterotrimeric G alpha q/G alpha 11 proteins function upstream of vascular endothelial growth factor (VEGF) receptor-2 (KDR) phosphorylation in vascular permeability factor/VEGF signaling. J. Biol. Chem. 2003, 278, 20738–20745. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Vannier, B.; Loosfelt, H.; Meduri, G.; Pichon, C.; Milgrom, E. Anti-human FSH receptor monoclonal antibodies: Immunochemical and immunocytochemical characterization of the receptor. Biochemistry 1996, 35, 1358–1366. [Google Scholar] [CrossRef] [PubMed]
- Ghinea, N. Antibodies Against the Human FSHR Extracellular Domain. Available online: https//www.WO2018/172078 A1 (accessed on 9 April 2021).
- Urbanska, K.; Stashwick, C.; Poussin, M.; Powell, D.J., Jr. Follicle-stimulating hormone receptor as a target in the redirected t-cell therapy for cancer. Cancer Immunol. Res. 2015, 3, 1130–1137. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Perales-Puchalt, A.; Svoranos, N.; Rutkovsky, M.R.; Allegrezza, M.J.; Tesone, A.J.; Payne, K.K.; Wickramasinghe, J.; Nguyen, J.M.; O’Brien, S.W.; Gumireddy, K.; et al. Follicle-stimulating hormone receptor is expressed by most ovarian cancer subtypes and is a safe and effective immunotherapeutic target. Clin. Cancer Res. 2016, 23, 441–453. [Google Scholar] [CrossRef] [Green Version]




{kind=link}
{kind=link}
{kind=link}
| RTKI c | Molecular Weight | Albumin Binding | RTK |
|---|---|---|---|
| (Da) | (%) | ||
| Anlotinib | 407.4 | 93.0 | VEGFR, PDGFR, FGFR |
| Apatinib | 397.5 | 92.4 | VEGFR, PDGFR |
| Axitinib | 386.5 | 99.0 | VEGFR, PDGFR |
| Cabozatinib | 501.5 | 99.7 | VEGFR |
| Cediranib | 450.5 | 99.8 | VEGFR, PDGFR |
| Leflunomide | 270.2 | 99.8 | PDGFR |
| Lenvatinib | 523.0 | 99.0 | VEGFR |
| Nilotinib | 529.5 | 93.9 | PDGFR |
| Pazopanib | 437.5 | 99.9 | VEGFR, PDGFR |
| Ponatinib | 532.6 | 99.0 | VEGFR, FGFR |
| Regrorafinib | 482.0 | 99.0 | VEGFR |
| Sorafenib | 464.8 | 99.5 | VEGFR, PDGFR |
| Sunitinib | 398.5 | 95.0 | VEGFR |
| Tivozanib | 454.9 | 99.0 | VEGFR |
| Vandetanib | 475.4 | 90.0 | VEGFR, EGFR |
Publisher’s Note: MDPI stays neutral with regard to jurisdictional claims in published maps and institutional affiliations. |
© 2021 by the author. Licensee MDPI, Basel, Switzerland. This article is an open access article distributed under the terms and conditions of the Creative Commons Attribution (CC BY) license (https://creativecommons.org/licenses/by/4.0/).
Share and Cite
Ghinea, N. Anti-Angiogenic Therapy: Albumin-Binding Proteins Could Mediate Mechanisms Underlying the Accumulation of Small Molecule Receptor Tyrosine Kinase Inhibitors in Normal Tissues with Potential Harmful Effects on Health. Diseases 2021, 9, 28. https://doi.org/10.3390/diseases9020028
Ghinea N. Anti-Angiogenic Therapy: Albumin-Binding Proteins Could Mediate Mechanisms Underlying the Accumulation of Small Molecule Receptor Tyrosine Kinase Inhibitors in Normal Tissues with Potential Harmful Effects on Health. Diseases. 2021; 9(2):28. https://doi.org/10.3390/diseases9020028
Chicago/Turabian StyleGhinea, Nicolae. 2021. "Anti-Angiogenic Therapy: Albumin-Binding Proteins Could Mediate Mechanisms Underlying the Accumulation of Small Molecule Receptor Tyrosine Kinase Inhibitors in Normal Tissues with Potential Harmful Effects on Health" Diseases 9, no. 2: 28. https://doi.org/10.3390/diseases9020028
APA StyleGhinea, N. (2021). Anti-Angiogenic Therapy: Albumin-Binding Proteins Could Mediate Mechanisms Underlying the Accumulation of Small Molecule Receptor Tyrosine Kinase Inhibitors in Normal Tissues with Potential Harmful Effects on Health. Diseases, 9(2), 28. https://doi.org/10.3390/diseases9020028