
Cholinergic Modulation of the Immune System in Neuroinflammatory Diseases



Abstract
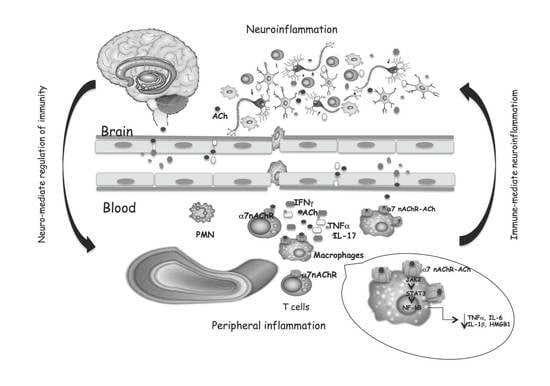
:
1. Cholinergic System: A Narrow Overview
Cholinergic System in Immune Cells
2. Cholinergic and Immune System Crosstalk in Widespread Neuro-Inflammatory Diseases
2.1. Multiple Sclerosis
2.2. Alzheimer’s Disease
2.3. Parkinson’s Disease
3. CholinomiRs: A Novel Approach for Neuroinflammatory Diseases
4. Genetic Polymorphisms of Cholinergic Components
5. Therapeutic Opportunities
6. Conclusions
Author Contributions
Funding
Conflicts of Interest
References
- Iversen, L.L.; Iversen, S.D.; Bloom, F.E.; Roth, R.H. Acetylcholine. In Introduction to Neuropsychopharmacology; Oxford University Press: New York, NY, USA, 2009; pp. 128–149. [Google Scholar]
- Okuda, T.; Haga, T. High-affinity choline transporter. Neurochem. Res. 2003, 28, 483–488. [Google Scholar] [CrossRef] [PubMed]
- Darvesh, S.; Hopkins, D.A.; Geula, C. Neurobiology of butyrylcholinesterase. Nat. Rev. Neurosci. 2003, 42, 131–138. [Google Scholar] [CrossRef] [PubMed]
- Soreq, H.; Seidman, S. Acetylcholinesterase--new roles for an old actor. Nat. Rev. Neurosci. 2001, 24, 294–302. [Google Scholar] [CrossRef]
- Ishii, M.; Kurachi, Y. Muscarinic acetylcholine receptors. Curr. Pharm. Des. 2006, 12, 3573–3581. [Google Scholar] [CrossRef] [PubMed]
- Gotti, C.; Clementi, F. Neuronal nicotinic receptors: From structure to pathology. Prog. Neurobiol. 2004, 74, 363–396. [Google Scholar] [CrossRef] [PubMed]
- Borovikova, L.V.; Ivanova, S.; Zhang, M.; Yang, H.; Botchkina, G.I.; Watkins, L.R.; Wang, H.; Abumrad, N.; Eaton, J.W.; Tracey, K.J. Vagus nerve stimulation attenuates the systemic inflammatory response to endotoxin. Nature 2000, 405, 458–462. [Google Scholar] [CrossRef]
- Marrero, M.B.; Bencherif, M. Convergence of alpha 7 nicotinic acetylcholine receptor-activated pathways for anti-apoptosis and anti-inflammation: Central role for JAK2 activation of STAT3 and NF-kappaB. Brain Res. 2009, 1256, 1–7. [Google Scholar] [CrossRef]
- Gergalova, G.L.; Likhmous, O.Y.; Skok, M.V. Possible Effect of Activation of α7-Nicotinic Acetylcholine Receptors in the Mitochondrial Membrane on the Development of Apoptosis. Neurophysiology 2011, 43, 195–197. [Google Scholar] [CrossRef]
- Skok, M.; Gergalova, G.L.; Lykhmus, O.Y.; Kalashnyk, O.; Koval, L.; Uspenska, K. Nicotinic acetylcholine receptors in mitochondria: Subunit composition, function and signalling. Neurotransmitter 2016, 3, e1290. [Google Scholar]
- Lykhmus, O.Y.; Gergalova, G.L.; Zouridakis, M.; Tzartos, S.; Komisarenko, S.; Skok, M. Inflammation decreases the level of alpha7 nicotinic acetylcholine receptors in the brain mitochondria and makes them more susceptible to apoptosis induction. Int. Immunopharmacol. 2015, 29, 148–151. [Google Scholar] [CrossRef]
- Lu, B.; Kwan, K.; Levine, Y.A.; Olofsson, P.S.; Yang, H.; Li, J.; Joshi, S.; Wang, H.; Andersson, U.; Chavan, S.S.; et al. α7 nicotinic acetylcholine receptor signaling inhibits inflammasome activation by preventing mitochondrial DNA release. Mol. Med. 2014, 20, 350–358. [Google Scholar] [CrossRef]
- Kawashima, K.; Fujii, T. Extraneuronal cholinergic system in lymphocytes. Pharmacol. Ther. 2000, 861, 29–48. [Google Scholar] [CrossRef]
- Kawashima, K.; Kajiyama, K.; Fujimoto, K.; Oohata, H.; Suzuki, T. Presence of acetylcholine in human blood and its localization in circulating mononuclear leukocytes. Biog. Amines 1993, 9, 251–258. [Google Scholar]
- Fujii, T.; Yamada, S.; Yamaguchi, N.; Fujimoto, K.; Suzuki, T.; Kawashima, K. Species differences in the concentration of acetylcholine, a neurotransmitter, in whole blood and plasma. Neurosci. Lett. 1995, 201, 207–210. [Google Scholar] [CrossRef]
- Strom, T.; Sytkowski, A.; Carpenter, C.; Merrill, J. Cholinergic augmentation of lymphocyte-mediated cytotoxicity. A study of the cholinergic receptor of cytotoxic T lymphocytes. Proc. Natl. Acad. Sci. USA 1974, 74, 1330–1333. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Bartha, E.; Rakonczay, Z.; Kása, P.; Hollán, S.; Gyévai, A. Molecular form of human lymphocyte membrane-bound acetylcholineesterase. Life Sci. 1987, 41, 1853–1860. [Google Scholar] [CrossRef]
- Szelényi, J.G.; Bartha, E.; Hollán, S.R. Acetylcholinesterase activity of lymphocytes: An enzyme characteristic of T-cells. Br. J. Haematol. 1982, 50, 241–245. [Google Scholar] [CrossRef] [PubMed]
- Tayebati, S.K.; El-Assouad, D.; Ricci, A.; Amenta, F. Immunochemical and immuno cyto chemical characterization of cholinergic markers in human peripheral blood lymphocytes. J. Neuroimmunol. 2002, 132, 147–155. [Google Scholar] [CrossRef]
- Fujii, T.; Mashimo, M.; Moriwaki, Y.; Misawa, H.; Ono, S.; Horiguchi, K.; Kawashima, K. Physiological functions of the cholinergic system in immune cells. J. Pharmacol. Sci. 2017, 134, 1–21. [Google Scholar] [CrossRef] [PubMed]
- Sato, K.Z.; Fujii, T.; Watanabe, Y.; Yamada, S.; Ando, T.; Fujimoto, K.; Kawashima, K. Diversity of mRNA expression for muscarinic acetylcholine receptor subtypes and neuronal nicotinic acetylcholine receptor subunits in human mononuclear leukocytes and leukemic cell lines. Neurosci. Lett. 1999, 266, 17–20. [Google Scholar] [CrossRef]
- Skok, M.V.; Grailhe, R.; Agenes, F.; Changeux, J.P. The role of nicotinic receptors in B-lymphocyte development and activation. Life Sci. 2007, 80, 2334–2336. [Google Scholar] [CrossRef] [PubMed]
- Rinner, I.; Kawashima, K.; Schauenstein, K. Rat lympocytes produce and secrete acetylcholine in dependence of differentiation and activation. J. Neuroimmunol. 1998, 81, 31–37. [Google Scholar] [CrossRef]
- Galitovskiy, V.; Qian, J.; Chernyavsky, A.I.; Marchenko, S.; Gindi, V.; Edwards, R.A.; Grando, S.A. Cytokine-induced alterations of alpha7 nicotinic receptor in colonic CD4 T cells mediate dichotomous response to nicotine in murine models of Th1/Th17- versus Th2-mediated colitis. J. Immunol. 2011, 187, 2677–2687. [Google Scholar] [CrossRef]
- Fujii, T.; Horiguchi, K.; Sunaga, H.; Moriwaki, Y.; Misawa, H.; Kasahara, T.; Tsuji, S.; Kawashima, K. SLURP-1, an endogenous alpha7 nicotinic acetylcholine receptor allosteric ligand, is expressed in CD205(+) dendritic cells in human tonsils and potentiates lymphocytic cholinergic activity. J. Neuroimmunol. 2014, 267, 43–49. [Google Scholar] [CrossRef] [PubMed]
- Rosas-Ballina, M.; Olofsson, P.S.; Ochani, M.; Valdés-Ferrer, S.I.; Levine, Y.A.; Reardon, C.; Tusche, M.W.; Pavlov, V.A.; Andersson, U.; Chavan, S.; et al. Acetylcholine-Synthesizing T Cells Relay Neural Signals in a Vagus Nerve Circuit. Science 2011, 334, 98–101. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- De Jonge, W.J.; Van Der Zanden, E.P.; The, F.O.; Bijlsma, M.F.; Van Westerloo, D.J.; Bennink, R.J.; Berthoud, H.-R.; Uematsu, S.; Akira, S.; Wijngaard, R.M.V.D.; et al. Stimulation of the vagus nerve attenuates macrophage activation by activating the Jak2-STAT3 signaling pathway. Nat. Immunol. 2005, 6, 844–851. [Google Scholar] [CrossRef] [PubMed]
- Olofsson, P.S.; Rosas-Ballina, M.; Levine, Y.A.; Tracey, K.J. Rethinking inflammation: Neural circuits in the regulation of immunity. Immunol. Rev. 2012, 248, 188–204. [Google Scholar] [CrossRef] [Green Version]
- Maldifassi, M.C.; Martín-Sánchez, C.; Atienza, G.; Cedillo, J.L.; Arnalich, F.; Bordas, A.; Zafra, F.; Giménez, C.; Extremera, M.; Renart, J.; et al. Interaction of the a7-nicotinic subunit with its human-specific duplicated dupa7 isoform in mammalian cells: Relevance in human inflammatory responses. J. Biol. Chem. 2018, 293, 13874–13888. [Google Scholar] [CrossRef] [Green Version]
- Chernyavsky, A.I.; Arredondo, J.; Skok, M.; Grando, S.A. Auto/paracrine control of inflammatory cytokines by acetylcholine in macrophage-like U937 cells through nicotinic receptors. Int. Immunopharmacol. 2010, 10, 308–315. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Strom, T.B.; Deisseroth, A.; Morganroth, J.; Carpenter, C.B.; Merrill, J.P. Alteration of the cytotoxic action of sensitized lymphocytes by cholinergic agents and activators of adenylate cyclase. Proc. Natl. Acad. Sci. USA 1972, 69, 2995–2999. [Google Scholar] [CrossRef] [Green Version]
- Nouri-Shirazi, M.; Guinet, E. Evidence for the immunosuppressive role of nicotine on human dendritic cell functions. Immunology 2003, 109, 365–373. [Google Scholar] [CrossRef]
- Qin, L.; Wu, X.; Block, M.L.; Liu, Y.; Breese, G.R.; Hong, J.S.; Knapp, D.J.; Crews, F.-T. Systemic LPS causes chronic neuroinflammation and progressive neurodegeneration. Glia 2007, 555, 453–462. [Google Scholar] [CrossRef] [Green Version]
- Bodea, L.G.; Wang, Y.; Linnartz-Gerlach, B.; Kopatz, J.; Sinkkonen, L.; Musgrove, R.; Kaoma, T.; Muller, A.; Vallar, L.; Di Monte, D.A.; et al. Neurodegeneration by activation of the microglial complement-phagosome pathway. J. Neurosci. 2014, 3425, 8546–8556. [Google Scholar] [CrossRef] [Green Version]
- Süβ, P.; Lana, A.J.; Schlachetzki, J.C.M. Chronic peripheral inflammation: A possible contributor to neurodegenerative diseases. Neural. Regen. Res. 2021, 169, 1711–1714. [Google Scholar] [CrossRef]
- Sandhu, J.K.; Kulka, M. Decoding Mast Cell-Microglia Communication in Neurodegenerative Diseases. Int. J. Mol. Sci. 2021, 223, 1093. [Google Scholar] [CrossRef]
- Guzman-Martinez, L.; Maccioni, R.B.; Andrade, V.; Navarrete, L.P.; Pastor, M.G.; Ramos-Escobar, N. Neuroinflammation as a Common Feature of Neurodegenerative Disorders. Front. Pharmacol. 2019, 10, 1008. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Block, M.L.; Hong, J.S. Chronic microglial activation and progressive dopaminergic neurotoxicity. Biochem. Soc. Trans. 2007, 35, 1127–1132. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Bachiller, S.; Jiménez-Ferrer, I.; Paulus, A.; Yang, Y.; Swanberg, M.; Deierborg, T.; Boza-Serrano, A. Microglia in Neurological Diseases: A Road Map to Brain-Disease Dependent-Inflammatory Response. Front. Cell Neurosci. 2018, 12, 488. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Brambilla, R. The contribution of astrocytes to the neuroinflammatory response in multiple sclerosis and experimental autoimmune encephalomyelitis. Acta Neuropathol. 2019, 137, 757–783. [Google Scholar] [CrossRef] [PubMed]
- Xie, L.; Choudhury, G.R.; Winters, A.; Yang, S.-H.; Jin, K. Cerebral regulatory T cells restrain microglia/macrophage-mediated inflammatory responses via IL-10. Eur. J. Immunol. 2015, 45, 180–191. [Google Scholar] [CrossRef]
- Korin, B.; Ben-Shaanan, T.L.; Schiller, M.; Dubovik, T.; Azulay-Debby, H.; Boshnak, N.T.; Koren, T.; Rolls, A. High-dimensional, single-cell characterization of the brain’s immune compartment. Nat. Neurosci. 2017, 20, 1300–1309. [Google Scholar] [CrossRef]
- Kipnis, J. Multifaceted interactions between adaptive immunity and the central nervous system. Science 2016, 353, 766–771. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Walsh, J.T.; Kipnis, J. Regulatory T cells in CNS injury: The simple, the complex and the confused. Trends Mol. Med. 2011, 17, 541–547. [Google Scholar] [CrossRef] [Green Version]
- Ziv, Y.; Ron, N.; Butovsky, O.; Landa, G.; Sudai, E.; Greenberg, N.; Cohen, H.; Kipnis, J.; Schwartz, M. Immune cells contribute to the maintenance of neurogenesis and spatial learning abilities in adulthood. Nat. Neurosci. 2006, 9, 268–275. [Google Scholar] [CrossRef]
- Radjavi, A.; Smirnov, I.; Kipnis, J. Brain antigen-reactive CD4+ T cells are sufficient to support learning behavior in mice with limited T cell repertoire. Brain Behav. Immun. 2014, 35, 58–63. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Morimoto, K.; Nakajima., K. Role of the Immune System in the Development of the Central Nervous System. Front. Neurosci. 2019, 13, 916. [Google Scholar] [CrossRef] [Green Version]
- Neumann, H.; Medana, I.M.; Bauer, J.; Lassmann, H. Cytotoxic T lymphocytes in autoimmune and degenerative CNS diseases. Trends Neurosci. 2002, 25, 313–319. [Google Scholar] [CrossRef]
- Larbi, A.; Pawelec, G.; Witkowski, J.M.; Schipper, H.M.; Derhovanessian, E.; Goldeck, D.; Fulop, T. Dramatic shifts in circulating CD4 but not CD8 T cell subsets in mild Alzheimer’s disease. J. Alzheimers Dis. 2009, 17, 91–103. [Google Scholar] [CrossRef] [PubMed]
- Quan, N. Immune-to-brain signaling: How important are the blood-brain barrier-independent pathways? Mol. Neurobiol. 2008, 372, 142–152. [Google Scholar] [CrossRef] [Green Version]
- Perry, V.H. The influence of systemic inflammation on inflammation in the brain: Implications for chronic neurodegenerative disease. Brain Behav. Immun. 2004, 18, 407–413. [Google Scholar] [CrossRef]
- Perry, V.H.; Cunningham, C.; Holmes, C. Systemic infections and inflammation affect chronic neurodegeneration. Nat. Rev. Immunol. 2007, 7, 161–167. [Google Scholar] [CrossRef]
- Yang, Q.; Wang, G.; Zhang, F. Role of Peripheral Immune Cells-Mediated Inflammation on the Process of Neurodegenerative Diseases. Front. Immunol. 2020, 11, 582825. [Google Scholar] [CrossRef]
- Cunningham, C.; Campion, S.; Lunnon, K.; Murray, C.L.; Woods, J.F.; Deacon, R.M.; Rawlins, J.N.P.; Perry, V.H. Systemic inflammation induces acute behavioral and cognitive changes and accelerates neurodegenerative disease. Biol. Psychiatry 2009, 65, 304–312. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Prinz, M.; Priller, J. The role of peripheral immune cells in the CNS in steady state and disease. Nat. Neurosci. 2017, 20, 136–144. [Google Scholar] [CrossRef] [PubMed]
- Kierdorf, K.; Masuda, T.; Jordao, M.J.C.; Prinz, M. Macrophages at CNS interfaces: Ontogeny and function in health and disease. Nat. Rev. Neurosci. 2019, 20, 547–562. [Google Scholar] [CrossRef] [PubMed]
- Kebir, H.; Kreymborg, K.; Ifergan, I.; Dodelet-Devillers, A.; Cayrol, R.; Bernard, M.; Giuliani, F.; Arbour, N.; Becher, B.; Part, A. Human TH17 lymphocytes promote blood-brain barrier disruption and central nervous system inflammation. Nat. Med. 2007, 13, 1173–1175. [Google Scholar] [CrossRef] [Green Version]
- Pacheco, R.; Riquelme, E.; Kalergis, A.M. Emerging evidence for the role of neurotransmitters in the modulation of T cell responses to cognate ligands. Cent. Nerv. Syst. Agents Med. Chem. 2010, 10, 65–83. [Google Scholar] [CrossRef]
- Gallowitsch-Puerta, M.; Pavlov, V.A. Neuro-immune interactions via the cholinergic anti-inflammatory pathway. Life Sci. 2007, 80, 2325–2329. [Google Scholar] [CrossRef] [Green Version]
- Wang, H.; Liao, H.; Ochani, M.; Justiniani, M.; Lin, X.; Yang, L.; Al-Abed, Y.; Wang, H.; Metz, C.; Miller, E.J.; et al. Cholinergic agonists inhibit HMGB1 release and improve survival in experimental sepsis. Nat. Med. 2004, 10, 1216–1221. [Google Scholar] [CrossRef]
- Lassmann, H.; Brück, W.; Lucchinetti, C.F. The immunopathology of multiple sclerosis: An overview. Brain Pathol. 2007, 17, 210–218. [Google Scholar] [CrossRef]
- Stys, P.K.; Zamponi, G.W.; Van Minnen, J.; Geurts, J.J. Will the real multiple sclerosis please stand up? Nat. Rev. Neurosci. 2012, 13, 507–514. [Google Scholar] [CrossRef] [PubMed]
- Hemmer, B.; Nessler, S.; Zhou, D.; Kieseier, B.; Hartung, H.P. Immunopathogenesis and immunotherapy of multiple sclerosis. Nat. Clin. Pract. Neurol. 2006, 2, 201–211. [Google Scholar] [CrossRef]
- Jamshidian, A.; Shaygannejad, V.; Pourazar, A.; Zarkesh-Esfahani, S.H.; Gharagozloo, M. Biased Treg/Th17 balance away from regulatory toward inflammatory phenotype in relapsed multiple sclerosis and its correlation with severity of symptoms. J. Neuroimmunol. 2013; 2621, 106–112. [Google Scholar] [CrossRef]
- Murphy, A.C.; Lalor, S.J.; Lynch, M.A.; Mills, K.H.G. Infiltration of Th1 and Th17 cells and activation of microglia in the CNS during the course of experimental autoimmune encephalomyelitis. Brain Behav. Immun. 2010, 24, 641–651. [Google Scholar] [CrossRef] [PubMed]
- Lu, J.; Wu, W. Cholinergic modulation of the immune syste -A novel therapeutic target for myocardial inflammation. Int. Immunopharmacol. 2021, 93, 107391. [Google Scholar] [CrossRef]
- Hao, J.; Simard, A.R.; Turner, G.H.; Wu, J.; Whiteaker, P.; Lukas, R.J.; Shi, F.D. Attenuation of CNS inflammatory responses by nicotine involves α7 and non-α7 nicotinic receptors. Exp. Neurol. 2011, 227, 110–119. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Wang, D.; Zhou, R.B.; Yao, Y.M.; Zhu, X.M.; Yin, Y.M.; Zhao, G.J.; Dong, N.; Sheng, Z.Y. Stimulation of α7 nicotinic acetylcholine receptor by nicotine increases suppressive capacity of naturally occurring CD4+CD25+ regulatory T cells in mice in vitro. J. Pharmacol. Exp. Ther. 2010, 335, 553–561. [Google Scholar] [CrossRef] [Green Version]
- De Rosa, M.J.; Dionisio, L.; Agriello, E.; Bouzat, C.; Del Carmen Esandi, M. Alpha 7nicotinic acetylcholine receptor modulates lymphocyte activation. Life Sci. 2009, 85, 444–449. [Google Scholar] [CrossRef] [PubMed]
- Nizri, E.; Irony-Tur-Sinai, M.; Lory, O.; Orr-Urtreger, A.; Lavi, E.; Brenner, T. Activation of the cholinergic anti-inflammatory system by nicotine attenuates neuroinflammation via suppression of Th1 and Th17 responses. J. Immunol. 2009, 183, 6681–6688. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Wang, H.; Yu, M.; Ochani, M.; Amella, C.A.; Tanovic, M.; Susarla, S.; Li, J.H.; Wang, H.; Yang, H.; Ulloa, L.; et al. Nicotinic acetylcholine receptor alpha7 subunit is anessential regulator of inflammation. Nature 2003, 421, 384–388. [Google Scholar] [CrossRef] [PubMed]
- Parrish, W.R.; Rosas-Ballina, M.; Gallowitsch-Puerta, M.; Ochani, M.; Ochani, K.; Yang, L.H.; Hudson, L.; Lin, X.; Patel, N.; Johnson, S.M.; et al. Modulation of TNF release by choline requires alpha7 subunit nicotinic acetylcholine receptormediated signaling. Mol. Med. 2008, 14, 567–574. [Google Scholar] [CrossRef]
- Saeed, R.W.; Varma, S.; Peng-Nemeroff, T.; Sherry, B.; Balakhaneh, D.; Huston, J.; Tracey, K.J.; Al-Abed, Y.; Metz, C.N. Cholinergic stimulation blocks endothelial cell activation and leukocyte recruitment during inflammation. J. Exp. Med. 2005, 201, 1113–1123. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Czura, C.J.; Friedman, S.G.; Tracey, K.J. Neural inhibition of inflammation: The cholinergic anti-inflammatory pathway. J. Endotoxin Res. 2003, 9, 409–413. [Google Scholar] [CrossRef] [PubMed]
- Yoshikawa, H.; Kurokawa, M.; Ozaki, N.; Nara, K.; Atou, K.; Takada, E.; Suzuki, N. Nicotine inhibits the production of proinflammatory mediators in human monocytes by suppression of I-kappaB phosphorylation and nuclear factor-kappaB transcriptional activity through nicotinic acetylcholine receptor alpha7. Clin. Exp. Immunol. 2006, 146, 116–123. [Google Scholar] [CrossRef] [PubMed]
- Rosas-Ballina, M.; Goldstein, R.S.; Gallowitsch-Puerta, M.; Yang, L.; Valdés-Ferrer, S.I.; Patel, N.B.; Chavan, S.; Al-Abed, Y.; Yang, H.; Tracey, K.J. The selective alpha7 agonist GTS-21 attenuates cytokine production in human whole blood and human monocytes activated by ligands for TLR2, TLR3, TLR4, TLR9, and RAGE. Mol. Med. 2009, 15, 195–202. [Google Scholar] [CrossRef]
- Reale, M.; De Angelis, F.; Di Nicola, M.; Capello, E.; Di Ioia, M.; De Luca, G.; Lugaresi, A.; Tata, A.M. Relation between pro-inflammatory cytokines and acetylcholine levels in relapsing-remitting multiple sclerosis patients. Int. J. Mol. Sci. 2012, 1310, 12656–12664. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Liu, Z.; Han, B.; Li, P.; Wang, Z.; Fan, Q. Activation of α7nAChR by nicotine reduced the Th17 response in (CD4+)T lymphocytes. Immunol. Investig. 2014, 437, 667–674. [Google Scholar] [CrossRef]
- Di Bari, M.; Reale, M.; Di Nicola, M.; Orlando, V.; Galizia, S.; Porfilio, I.; Costantini, E.; D’Angelo, C.; Ruggieri, S.; Biagioni, S.; et al. Dysregulated homeostasis of acetylcholine levels in immune cells of RR-multiple sclerosis patients. Int. J. Mol. Sci. 2016, 17, 2009. [Google Scholar] [CrossRef] [Green Version]
- Di Bari, M.; Di Pinto, G.; Reale, M.; Mengod, G.; Tata, A.M. Cholinergic system and neuroinflammation: Implication in multiple sclerosis. CNS Agents Med. Chem. 2017, 17, 109–115. [Google Scholar] [CrossRef]
- Gatta, V.; Mengod, G.; Reale, M.; Tata, A.M. Possible Correlation between Cholinergic System Alterations and Neuro/Inflammation in Multiple Sclerosis. Biomedicines 2020, 86, 153. [Google Scholar] [CrossRef]
- Jiang, W.; Li, D.; Han, R.; Zhang, C.; Jin, W.N.; Wood, K.; Liu, Q.; Shi, F.D.; Hao, J. Acetylcholine-producing NK cells attenuate CNS inflammation via modulation of infiltrating monocytes/ macrophages. Proc. Natl. Acad Sci. USA 2017, 114, E6202–E6211. [Google Scholar] [CrossRef] [Green Version]
- Castellani, R.J.; Perry, G. The complexities of the pathology–pathogenesis relationship in Alzheimer disease. Biochemical. Pharmacol. 2014, 88, 671–676. [Google Scholar] [CrossRef]
- De Ferrari, G.V.; Canales, M.A.; Shin, I.; Weiner, L.M.; Silman, I.; Inestrosa, N.C. A structural motif of acetylcholinesterase that promotes amyloid beta-peptide fibril formation. Biochemistry 2001, 4035, 10447–10457. [Google Scholar] [CrossRef]
- Apelt, J.; Kumar, A.; Schliebs, R. Impairment of cholinergic neurotransmission in adult and aged transgenic Tg2576 mouse brain expressing the Swedish mutation of human beta-amyloid precursor protein. Brain Res. 2002; 953, 17–30. [Google Scholar] [CrossRef]
- Wyss-Coray, T.; Rogers, J. Inflammation in Alzheimer disease-a brief review of the basic science and clinical literature. Cold Spring Harb. Perspect. Med. 2012, 21, a006346. [Google Scholar] [CrossRef]
- Reale, M.; D’Angelo, C.; Costantini, E.; Di Nicola, M.; Yarla, N.S.; Kamal, M.A.; Salvador, N.; Perry, G. Expression Profiling of Cytokine, Cholinergic Markers, and Amyloid-β Deposition in the APPSWE/PS1dE9 Mouse Model of Alzheimer’s Disease Pathology. J. Alzheimers Dis. 2018, 621, 467–476. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Holmes, C.; Cunningham, C.; Zotova, E.; Woolford, J.; Dean, C.; Kerr, S.U.; Culliford, D.; Perry, V.H. Systemic inflammation and disease progression in Alzheimer disease. Neurology 2009, 73, 768–774. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Dursun, E.; Gezen-Ak, D.; Hanağası, H.; Bilgiç, B.; Lohmann, E.; Ertan, S.; Atasoy, İ.L.; Alaylıoğlu, M.; Araz, Ö.S.; Önal, B.; et al. The interleukin 1 alpha, interleukin 1 beta, interleukin 6 and alpha-2-macroglobulin serum levels in patients with early or late onset Alzheimer’s disease, mild cognitive impairment or Parkinson’s disease. J. Neuroimmunol. 2015, 283, 50–57. [Google Scholar] [CrossRef] [PubMed]
- Reale, M.; Kamal, M.A.; Velluto, L.; Gambi, D.; Di Nicola, M.; Greig, N.H. Relationship between Inflammatory Mediators, Aβ Levels and ApoE Genotype in Alzheimer Disease. Curr. Alzheimer Res. 2012, 9, 447–457. [Google Scholar] [CrossRef]
- Ferretti, M.T.; Merlini, M.; Spani, C.; Gericke, C.; Schweizer, N.; Enzmann, G.; Engelhardt, B.; Kulic, L.; Suter, T.; Nitsch, R.M. T-cell brain infiltration and immature antigen-presenting cells in transgenic models of Alzheimer’s disease-like cerebral amyloidosis. Brain Behav. Immun. 2016, 54, 211–225. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- McQuillan, K.; Lynch, M.A.; Mills, K.H. Activation of mixed glia by Abeta-specific Th1 and Th17 cells and its regulation by Th2 cells. Brain Behav. Immun. 2010, 24, 598–607. [Google Scholar] [CrossRef]
- Reale, M.; Iarlori, C.; Gambi, F.; Feliciani, C.; Isabella, L.; Gambi, D. The acetylcholinesterase inhibitor, Donepezil, regulates a Th2 bias in Alzheimer’s diseasepatients. Neuropharmacology 2006, 505, 606–613. [Google Scholar] [CrossRef]
- Reale, M.; Di Nicola, M.; Velluto, L.; D’Angelo, C.; Costantini, E.; Lahiri, D.K.; Kamal, M.A.; Yu, Q.-S.; Greig, N.H. Selective Acetyl- and Butyrylcholinesterase Inhibitors Reduce Amyloid-b Ex Vivo Activation of Peripheral Chemo-cytokines From Alzheimer’s Disease Subjects: Exploring the Cholinergic Anti-inflammatory Pathway. Curr. Alzheimer Res. 2014, 11, 608–622. [Google Scholar] [CrossRef] [Green Version]
- Heneka, M.T.; Kummer, M.P.; Latz, E. Innate immune activation in neurodegenerative disease. Nat. Rev. Immunol. 2014, 14, 463–477. [Google Scholar] [CrossRef]
- Bellucci, A.; Bubacco, L.; Longhena, F.; Parrella, E.; Faustini, G.; Porrini, V.; Bono, F.; Missale, C.; Pizzi, M. Nuclear Factor-κB Dysregulation and α-Synuclein Pathology: Critical Interplay in the Pathogenesis of Parkinson’s Disease. Front. Aging Neurosci. 2020, 12, 68. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Alvarez-Erviti, L.; Couch, Y.; Richardson, J.; Cooper, J.M.; Wood, M.J. Alpha-synuclein release by neurons activates the inflammatory response in a microglial cell line. Neurosci. Res. 2011, 694, 337–342. [Google Scholar] [CrossRef] [PubMed]
- Brochard, V.; Combadière, B.; Prigent, A.; Laouar, Y.; Perrin, A.; Beray-Berthat, V.; Bonduelle, O.; Alvarez-Fischer, D.; Callebert, J.; Launay, J.M.; et al. Infiltration of CD4+ lymphocytes into the brain contributes to neurodegeneration in a mouse model of Parkinson disease. J. Clin. Investig. 2009, 1191, 182–192. [Google Scholar] [CrossRef] [PubMed]
- Reale, M.; Iarlori, C.; Thomas, A.; Gambi, D.; Perfetti, B.; Di Nicola, M.; Onofrj, M. Peripheral cytokines profile in Parkinson’s disease. Brain Behav. Immun. 2009, 231, 55–63. [Google Scholar] [CrossRef] [PubMed]
- Harms, A.S.; Ferreira, S.A.; Romero-Ramos, M. Periphery and brain, innate and adaptive immunity in Parkinson’s disease. Acta Neuropathol. 2021. [Google Scholar] [CrossRef] [PubMed]
- Lin, M.T.; Beal, M.F. Mitochondrial dysfunction and oxidative stress in neurodegenerative diseases. Nature. 2006, 443, 787. [Google Scholar] [CrossRef]
- Tejera, D.; Mercan, D.; Sanchez-Caro, J.M.; Hanan, M.; Greenberg, D.; Soreq, H.; Latz, E.; Golenbock, D.; Heneka, M.T. Systemic inflammation impairs microglial Aβ clearance through NLRP3 inflammasome. EMBO J. 2019, 38, e101064. [Google Scholar] [CrossRef] [PubMed]
- Peña, G.; Cai, B.; Liu, J.; Van der Zanden, E.P.; Deitch, E.A.; De Jonge, W.J.; Ulloa, L. Unphosphorylated STAT3 modulates alpha 7 nicotinic receptor signaling and cytokine production in sepsis. Eur. J. Immunol. 2010, 40, 2580–2589. [Google Scholar] [CrossRef] [Green Version]
- Tsoyi, K.; Jang, H.J.; Kim, J.W.; Chang, H.K.; Lee, Y.S.; Pae, H.O.; Kim, H.J.; Seo, H.G.; Lee, J.H.; Chung, H.T.; et al. Stimulation of alpha7 nicotinic acetylcholine receptor by nicotine attenuates inflammatory response in macrophages and improves survival in experimental model of sepsis through heme oxygenase-1 induction. Antioxid Redox Signal. 2011, 14, 2057–2070. [Google Scholar] [CrossRef] [PubMed]
- Maurer, S.V.; Williams, C.L. The Cholinergic System Modulates Memory and Hippocampal Plasticity via Its Interactions with Non-Neuronal Cells. Front. Immunol. 2017, 8, 1489. [Google Scholar] [CrossRef] [Green Version]
- O’Connell, R.M.; Rao, D.S.; Baltimore, D. microRNA regulation of inflammatory responses. Annu. Rev. Immunol. 2012, 30, 295–312. [Google Scholar] [CrossRef]
- Liu, G.; Abraham, E. MicroRNAs in immune response and macrophage polarization. Arterioscler. Thromb. Vasc. Biol. 2013, 33, 170–177. [Google Scholar] [CrossRef] [Green Version]
- Priyadarshini, M.; Arivarasu, N.A.; Shah, A.; Tabrez, S.; Priyamvada, S.; Aatif, M. MicroRNA: Novel modulators of the cholinergic anti-inflammatory pathway. Antiinflamm. Antiallergy Agents Med. Chem. 2013, 12, 136–140. [Google Scholar] [CrossRef]
- Nadorp, B.; Soreq, H. Predicted overlapping microRNA regulators of acetylcholine packaging and degradation in neuroinflammation-related disorders. Front. Mol. Neurosci. 2014, 7, 9. [Google Scholar] [CrossRef] [Green Version]
- Ofek, K.; Soreq, H. Cholinergic involvement and manipulation approaches in multiple system disorders. Chem. Biol. Interact. 2013, 203, 113–119. [Google Scholar] [CrossRef] [PubMed]
- Ponomarev, E.D.; Veremeyko, T.; Barteneva, N.; Krichevsky, A.M.; Weiner, H.L. MicroRNA-124 promotes microglia quiescence and suppresses EAE by deactivating macrophages via the C/EBP-α-PU.1 pathway. Nat. Med. 2011, 17, 64–70. [Google Scholar] [CrossRef] [PubMed]
- Sun, Y.; Li, Q.; Gui, H.; Xu, D.P.; Yang, Y.L.; Su, D.F.; Liu, X. MicroRNA-124 mediates the cholinergic anti-inflammatory action through inhibiting the production of pro-inflammatory cytokines. Cell Res. 2013, 23, 1270–1283. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Zhou, W.; Wang, J.; Li, Z.; Li, J.; Sang, M. MicroRNA-2055b inhibits HMGB1 expression in LPS-induced sepsis. Int. J. Mol. Med. 2016, 38, 312–318. [Google Scholar] [CrossRef] [PubMed]
- Hanieh, H.; Alzahrani, A. MicroRNA-132 suppresses autoimmune encephalomyelitis by inducing cholinergic anti-inflammation: A new Ahr-based exploration. Eur. J. Immunol. 2013, 43, 2771–2782. [Google Scholar]
- Jasiecki, J.; Wasag, B. Butyrylcholinesterase Protein Ends in the Pathogenesis of Alzheimer’s Disease-CouldBCHE Genotyping Be Helpful in Alzheimer’s Therapy. Biomolecules 2019, 9, 592. [Google Scholar] [CrossRef] [Green Version]
- Jasiecki, J.; Limon-Sztencel, A.; Zuk, M.; Chmara, M.; Cysewski, D.; Limon, J.; Wasag, B. Synergy between the alteration in the N-terminal region of butyrylcholinesterase K variant and apolipoprotein E4 in late-onsetAlzheimer’s disease. Sci. Rep. 2019, 9, 5223. [Google Scholar] [CrossRef] [PubMed]
- Reale, M.; Costantini, E.; Di Nicola, M.; D’Angelo, C.; Franchi, S.; D’Aurora, M.; Di Bari, M.; Orlando, V.; Galizia, S.; Ruggieri, S.; et al. Butyrylcholinesterase and Acetylcholinesterase polymorphisms in Multiple Sclerosis patients: Implication in peripheral inflammation. Sci. Rep. 2018, 8, 1319. [Google Scholar] [CrossRef] [PubMed]
- Dong, M.X.; Xu, X.M.; Hu, L.; Liu, Y.; Huang, Y.J.; Wei, Y.D. Serum Butyrylcholinesterase Activity: A Biomarker for Parkinson’s Disease and Related Dementia. BioMed. Res. Int. 2017, 2017, 1524107. [Google Scholar] [CrossRef] [Green Version]
- Simchovitz, A.; Heneka, M.T.; Soreq, H. Personalized genetics of the cholinergic blockade of neuroinflammation. J. Neurochem. 2017, 142 (Suppl. 2), 178–187. [Google Scholar] [CrossRef] [Green Version]
- Hanin, G.; Shenhar-Tsarfaty, S.; Yayon, N.; Yau, Y.H.; Bennett, E.R.; Sklan, E.H.; Rao, D.C.; Rankinen, T.; Bouchard, C.; Geifman-Shochat, S.; et al. Competing targets of microRNA-608 affect anxiety and hypertension. Hum. Mol. Genet. 2014, 23, 4569–4580. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Gong, J.; Zhang, J.P.; Li, B.; Zeng, C.; You, K.; Chen, M.X.; Yuan, Y.; Zhuang, S.M. MicroRNA-125b promotes apoptosis by regulating the expression of Mcl-1, Bcl-w and IL-6R. Oncogene 2013, 32, 3071–3079. [Google Scholar] [CrossRef] [Green Version]
- Russo, P.; Kisialiou, A.; Moroni, R.; Prinzi, G.; Fini, M. Effect of Genetic Polymorphisms (SNPs) in CHRNA7 Gene on Response to Acetylcholinesterase Inhibitors (AChEI) in Patients with Alzheimer’s Disease. Curr. Drug Targets 2017, 8, 1179–1190. [Google Scholar] [CrossRef] [PubMed]
- Chee, L.Y.; Cumming, A. Polymorphisms in the Cholinergic Receptors Muscarinic (CHRM2 and CHRM3) Genes and Alzheimer’s Disease. J. Med. Biotechnol. 2018, 10, 196–199. [Google Scholar]
- Oztürk, A.; DeKosky, S.T.; Kamboh, M.I. Genetic variation in the choline acetyltransferase (CHAT) gene may be associated with the risk of Alzheimer’s disease. Neurobiol. Aging. 2006, 27, 1440–1444. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Cook, L.J.; Ho, L.W.; Wang, L.; Terrenoire, E.; Brayne, C.; Evans, J.G.; Xuereb, J.; Cairns, N.J.; Turic, D.; Hollingworth, P.; et al. Candidate gene association studies of genes involved in neuronal cholinergic transmission in Alzheimer’s disease suggests choline acetyltransferase as a candidate deserving further study. Amer. J. Med. Gen. Part B Neuropsychiatr. Genet. 2005, 132, 5–8. [Google Scholar] [CrossRef] [PubMed]
- Schwarz, S.; Eisele, T.; Diehl, J.; Müller, U.; Förstl, H.; Kurz, A.; Riemenschneider, M. Lack of association between a single nucleotide polymorphism within the choline acetyltransferase gene and patients with Alzheimer’s disease. Neurosci. Lett. 2003, 343, 167–170. [Google Scholar] [CrossRef]
- De Simone, R.; Ajmone-Cat, M.; Carnevale, D.; Minghetti, L. Activation of alpha7 nicotinic acetylcholine receptor by nicotine selectively up-regulates cyclooxygenase-2 and prostaglandin E2 in rat microglial cultures. J. Neuroinflammation. 2005, 2, 4. [Google Scholar] [CrossRef] [Green Version]
- Greig, N.H.; Reale, M.; Tata, A.M. New advances in pharmacological approaches to the cholinergic system: An overview on muscarinic receptor ligands and cholinesterase inhibitors. Recent Pat. CNS Drug Discov. 2013, 8, 123–141. [Google Scholar] [CrossRef]
- Pavlov, V.A.; Tracey, K.J. The cholinergic anti-inflammatory pathway. Brain Behav. Immun. 2005, 19, 493–499. [Google Scholar] [CrossRef]
- Pohanka, M. Inhibitors of Acetylcholinesterase and Butyrylcholinesterase Meet Immunity. Int. J. Mol. Sci. 2014, 15, 9809–9825. [Google Scholar] [CrossRef] [Green Version]



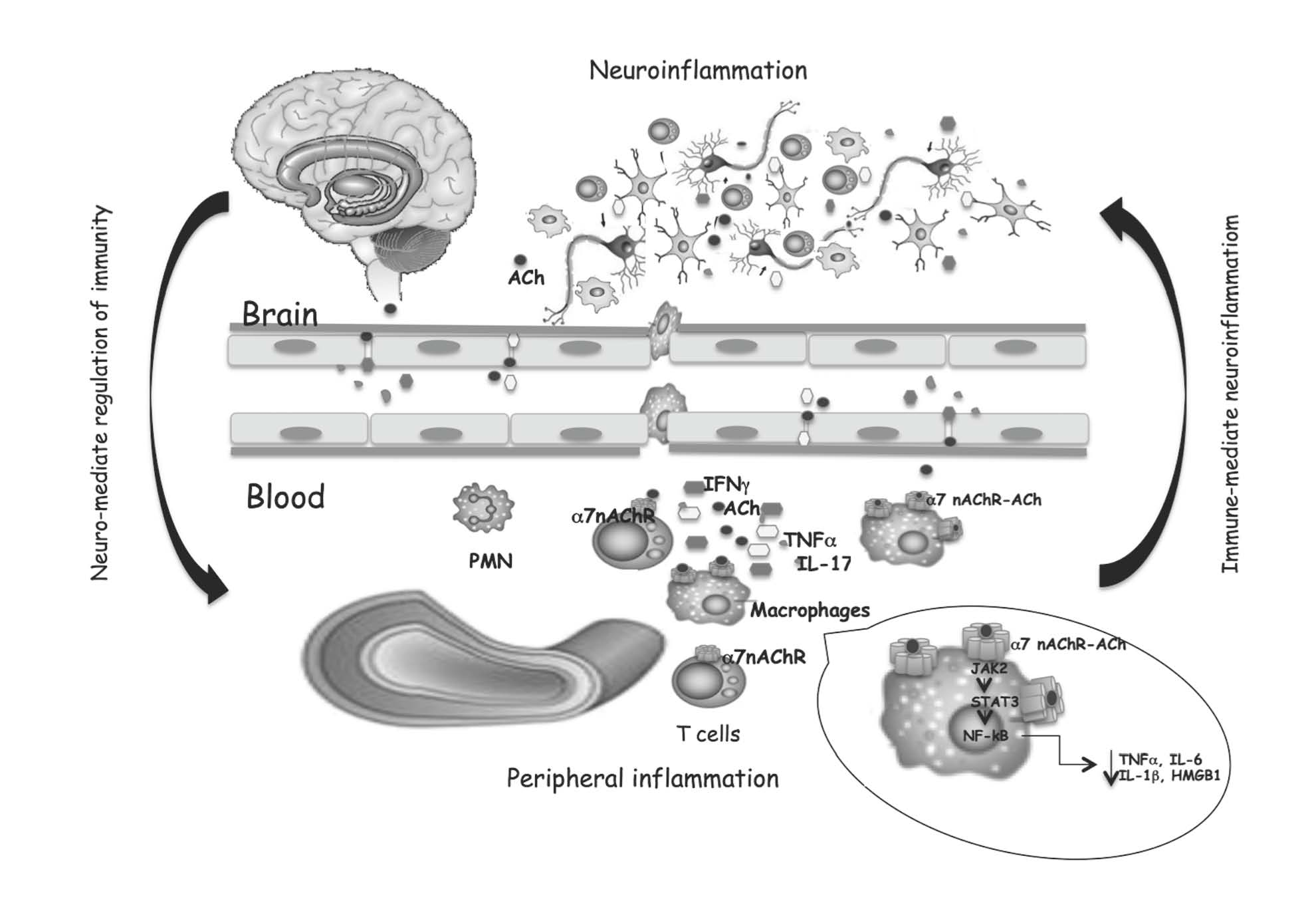{kind=link}
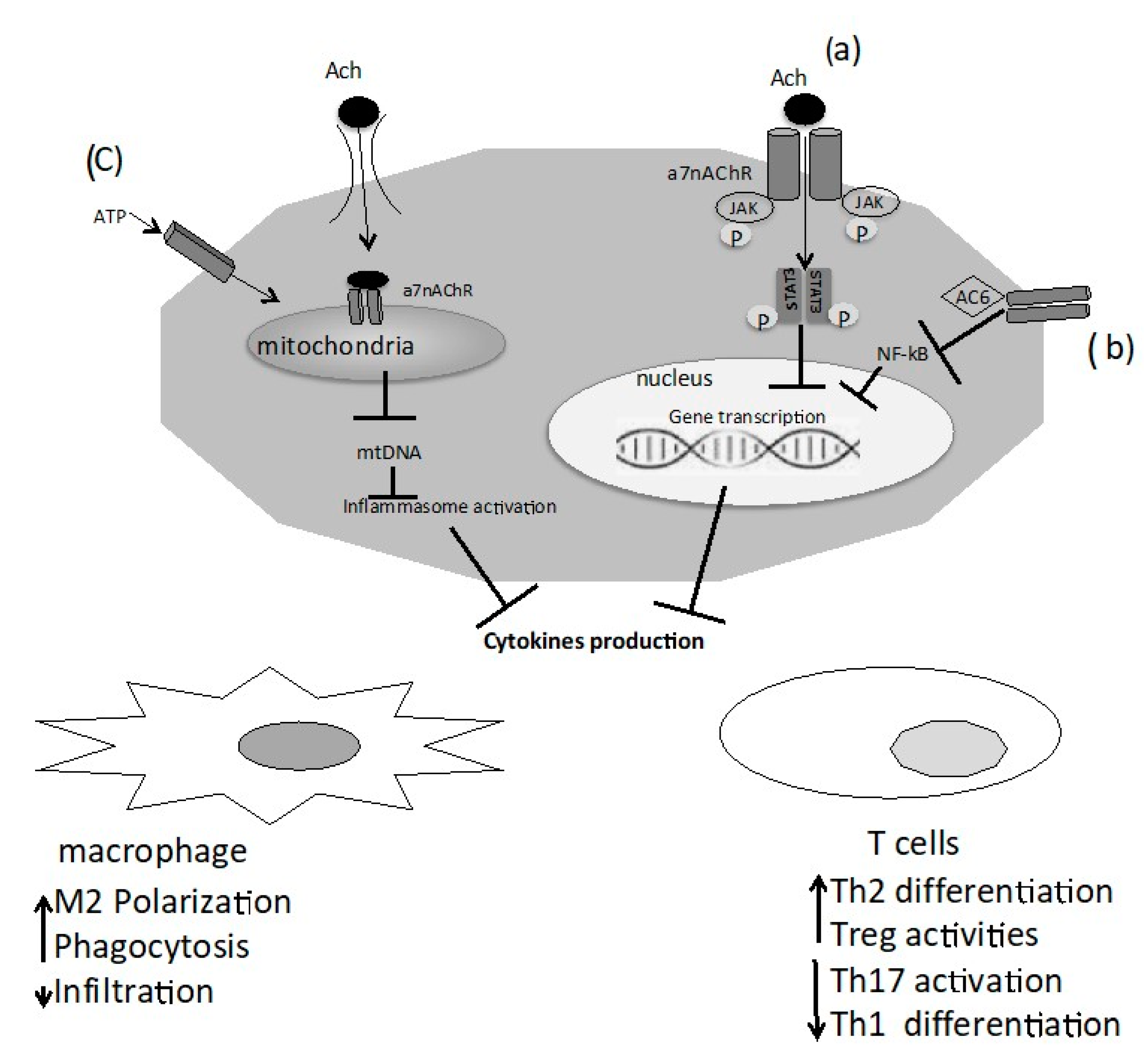{kind=link}
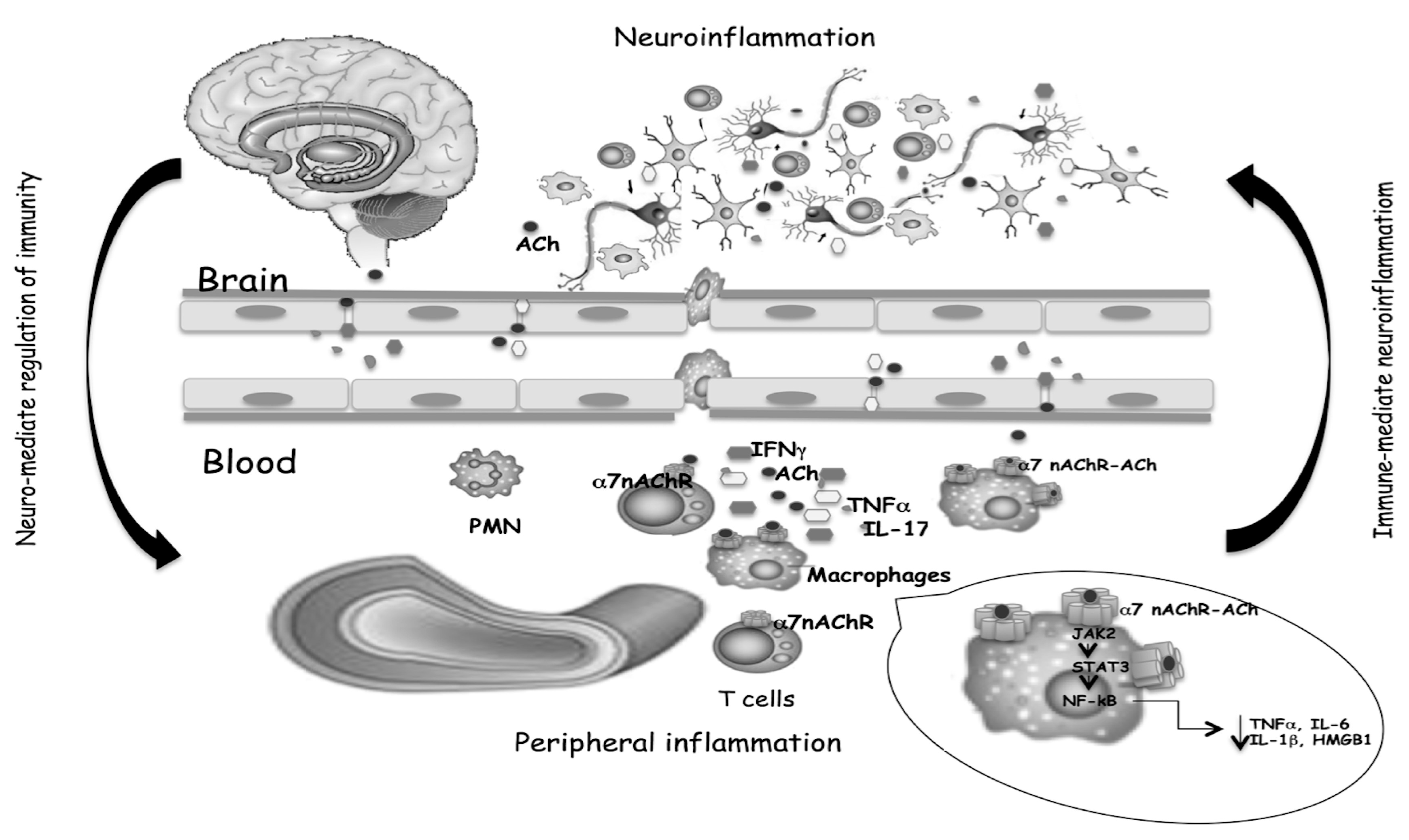{kind=link}
| Cholinergic Components | Lymphocytes | Monocytes | Macrophages |
|---|---|---|---|
| ACh | + [14,19,20] | + [14,19] | + [5] |
| ChAT | + [13] | + [13] | + [13] |
| AChE | + [13] | + [20] | + [20] |
| VAChT | + [13] | NR | NR |
| mAChR | + [21] | + [21] | + [21] |
| nAChR | + [12,20,22] | + [13] | + [13] |
Publisher’s Note: MDPI stays neutral with regard to jurisdictional claims in published maps and institutional affiliations. |
© 2021 by the authors. Licensee MDPI, Basel, Switzerland. This article is an open access article distributed under the terms and conditions of the Creative Commons Attribution (CC BY) license (https://creativecommons.org/licenses/by/4.0/).
Share and Cite
Reale, M.; Costantini, E. Cholinergic Modulation of the Immune System in Neuroinflammatory Diseases. Diseases 2021, 9, 29. https://doi.org/10.3390/diseases9020029
Reale M, Costantini E. Cholinergic Modulation of the Immune System in Neuroinflammatory Diseases. Diseases. 2021; 9(2):29. https://doi.org/10.3390/diseases9020029
Chicago/Turabian StyleReale, Marcella, and Erica Costantini. 2021. "Cholinergic Modulation of the Immune System in Neuroinflammatory Diseases" Diseases 9, no. 2: 29. https://doi.org/10.3390/diseases9020029
APA StyleReale, M., & Costantini, E. (2021). Cholinergic Modulation of the Immune System in Neuroinflammatory Diseases. Diseases, 9(2), 29. https://doi.org/10.3390/diseases9020029