Islet Health, Hormone Secretion, and Insulin Responsivity with Low-Carbohydrate Feeding in Diabetes


Abstract
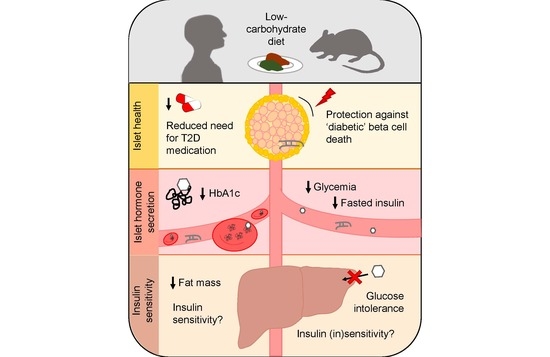
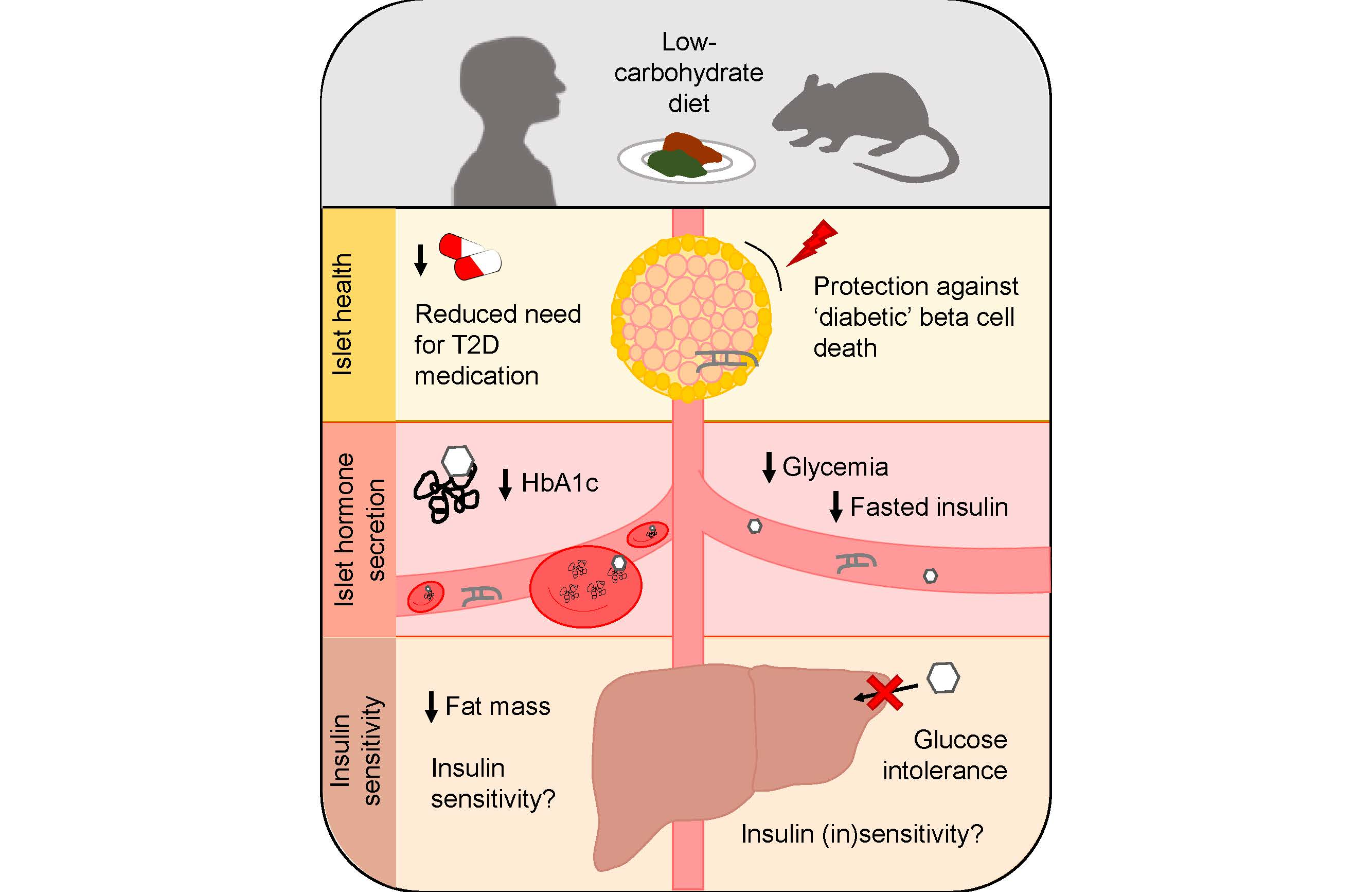
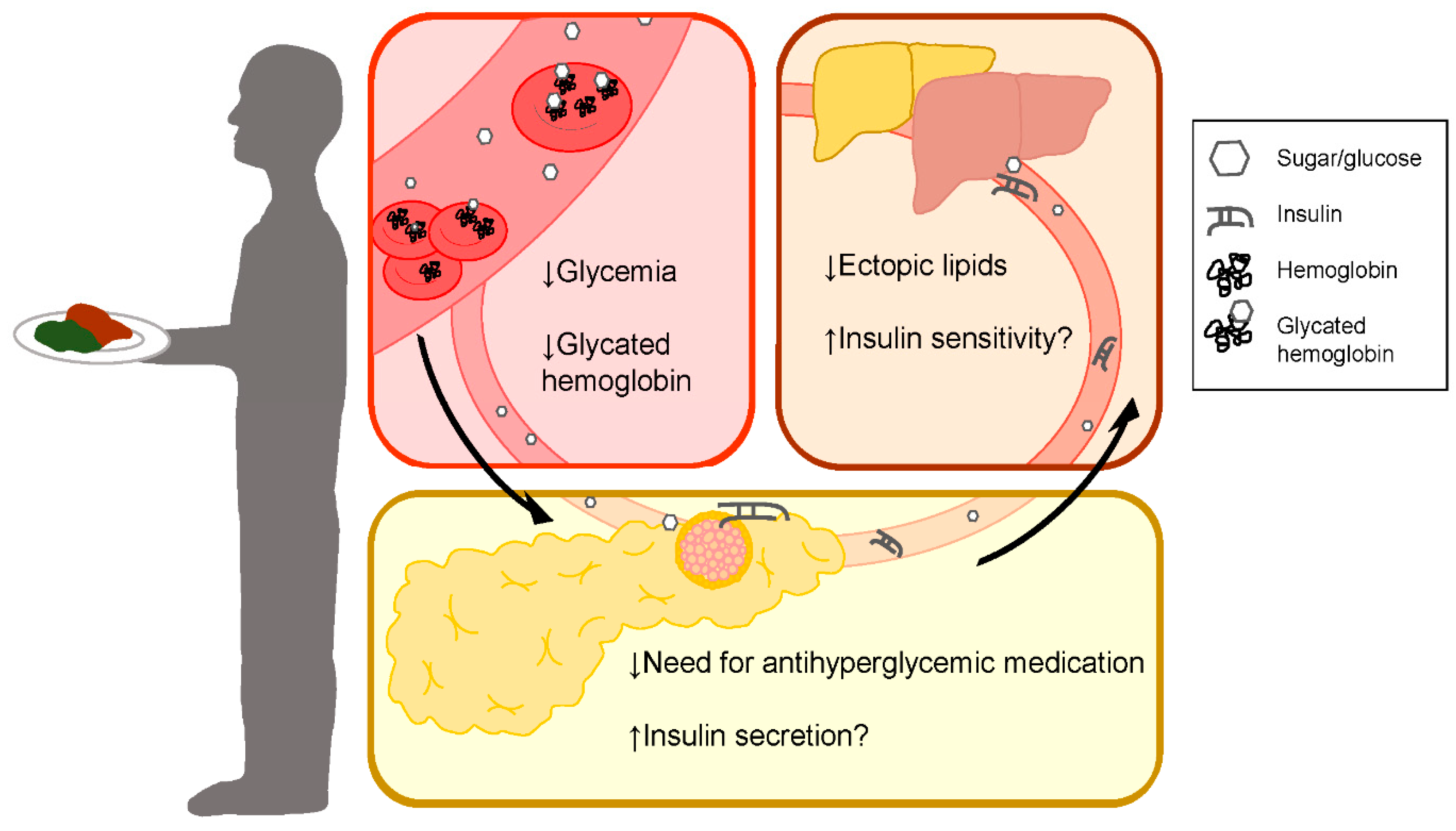
:
1. Introduction
2. Islet Health and Survival
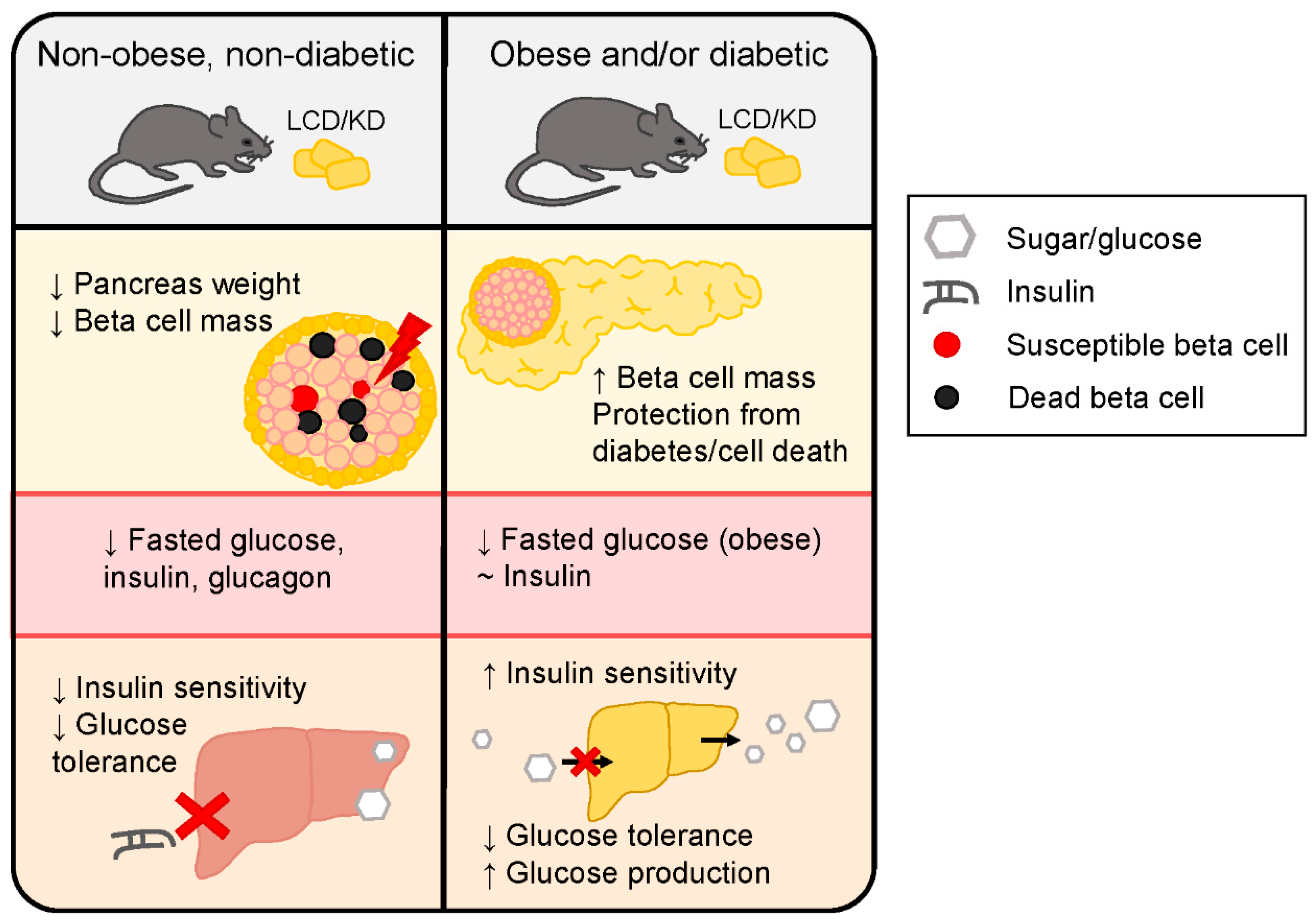
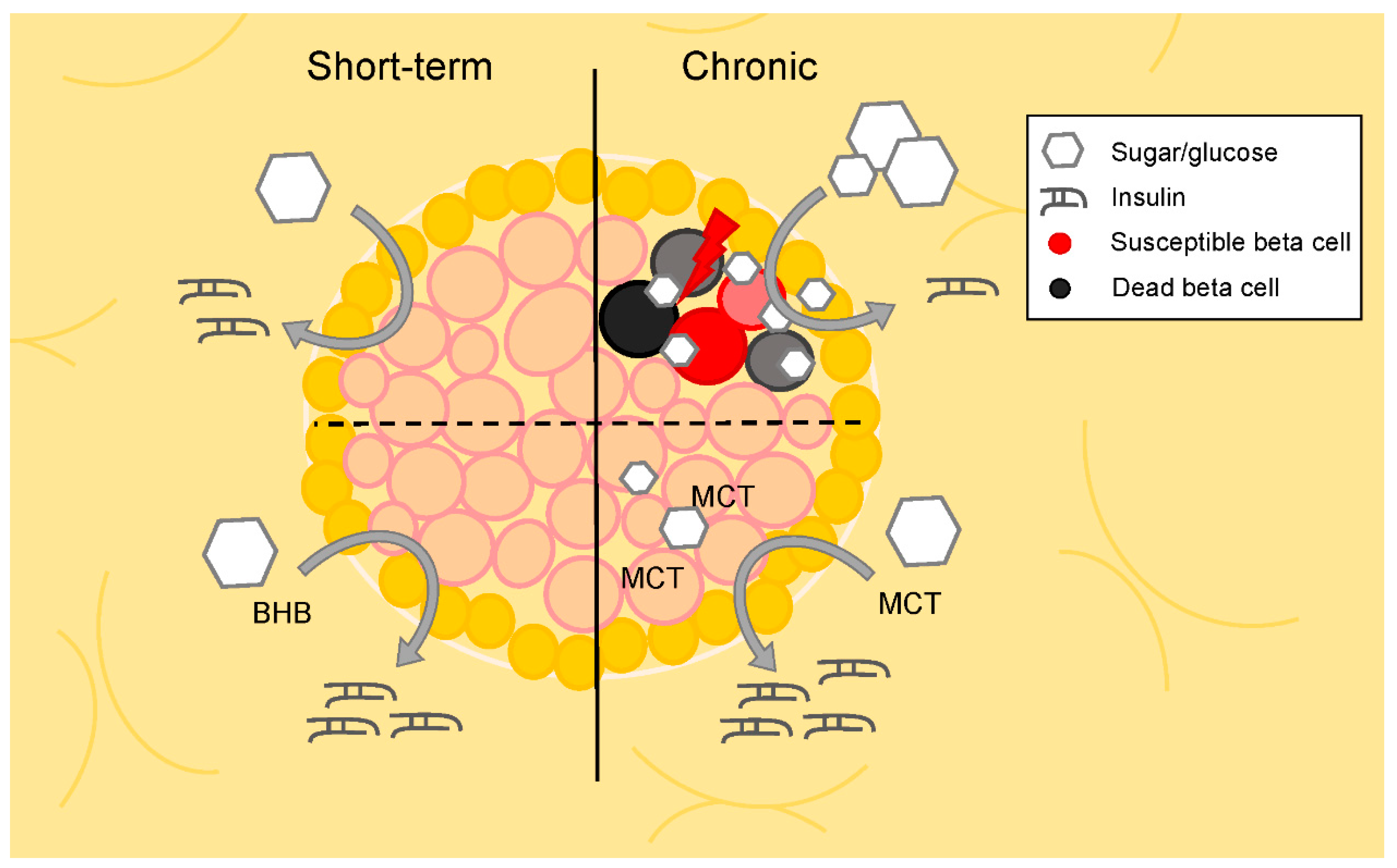
2.1. Non-Obese, Non-Diabetic Animal Studies
2.2. Animal Models of Diabetes and/or Obesity
3. Islet Hormone Secretion
3.1. Cell Studies
3.2. Healthy Animal Models
3.3. Animal Models of Obesity and Diabetes
3.4. Human Studies
4. Insulin Sensitivity
4.1. Non-Obese, Non-Diabetic Mouse Models
4.2. Mouse Models of Obesity and Diabetes
4.3. Human Studies
5. Paracrine Regulation and Metabolic Crosstalk
6. Discussion and Conclusions
Author Contributions
Funding
Acknowledgments
Conflicts of Interest
References
- Esser, N.; Utzschneider, K.M.; Kahn, S.E. Early Beta Cell Dysfunction vs Insulin Hypersecretion as the Primary Event in the Pathogenesis of Dysglycaemia. Diabetologia 2020, 63, 2007–2021. [Google Scholar] [CrossRef] [PubMed]
- Baggio, L.L.; Drucker, D.J. Biology of Incretins: GLP-1 and GIP. Gastroenterology 2007, 132, 2131–2157. [Google Scholar] [CrossRef] [PubMed]
- Rorsman, P.; Eliasson, L.; Renström, E.; Gromada, J.; Barg, S.; Göpel, S. The Cell Physiology of Biphasic Insulin Secretion. News Physiol. Sci. 2000, 15, 72–77. [Google Scholar] [CrossRef] [PubMed]
- Grill, V.; Björklund, A. Overstimulation and β-Cell Function. Diabetes 2001, 50, 48–50. [Google Scholar] [CrossRef] [Green Version]
- Westman, E.C.; Yancy, W.S.; Humphreys, M. Dietary Treatment of Diabetes Mellitus in the Pre-Insulin Era (1914–1922). Perspect. Biol. Med. 2006, 49, 77–83. [Google Scholar] [CrossRef]
- Moran, M. The Evolution of the Nutritional Management of Diabetes. Proc. Nutr. Soc. 2004, 63, 615–620. [Google Scholar] [CrossRef] [Green Version]
- Nuttall, F.Q.; Brunzell, J.D. Principles of Nutrition and Dietary Recommendations for Individuals with Diabetes Mellitus: 1979. Diabetes Care 1979, 28, 520–523. [Google Scholar]
- Article, S. Diabetes Canada Position Statement on Low-Carbohydrate Diets for Adults with Diabetes: A Rapid Review. Can. J. Diabetes 2020, 44, 295–299. [Google Scholar]
- Evert, A.B.; Dennison, M.; Gardner, C.D.; Timothy Garvey, W.; Karen Lau, K.H.; MacLeod, J.; Mitri, J.; Pereira, R.F.; Rawlings, K.; Robinson, S.; et al. Nutrition Therapy for Adults with Diabetes or Prediabetes: A Consensus Report. Diabetes Care 2019, 42, 731–754. [Google Scholar] [CrossRef] [Green Version]
- Managing Your Blood Sugar. Available online: https://www.diabetes.ca/managing-my-diabetes/tools---resources/managing-your-blood-sugar (accessed on 27 September 2020).
- Nathan, D.M. The Diabetes Control and Complications Trial/Epidemiology of Diabetes Interventions and Complications Study at 30 Years: Overview. Diabetes Care 2014, 37, 9–16. [Google Scholar] [CrossRef] [Green Version]
- Johnson, J.D.; Luciani, D.S. Mechanisms of Pancreatic β-Cell Apoptosis in Diabetes and Its Therapies. Adv. Exp. Med. Biol. 2010, 654, 447–462. [Google Scholar]
- Butler, A.E.; Janson, J.; Bonner-Weir, S.; Ritzel, R.; Rizza, R.A.; Butler, P.C. β-Cell Deficit and Increased β-Cell Apoptosis in Humans with Type 2 Diabetes. Diabetes 2003, 52, 102–110. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Rizza, R.A. Pathogenesis of Fasting and Postprandial Hyperglycemia in Type 2 Diabetes: Implications for Therapy. Diabetes 2010, 59, 2697–2707. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Firth, R.G.; Bell, P.M.; Marsh, H.M.; Hansen, I.; Rizza, R.A. Postprandial Hyperglycemia in Patients with Noninsulin-Dependent Diabetes Mellitus. Role of Hepatic and Extrahepatic Tissues. J. Clin. Investig. 1986, 77, 1525–1532. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Heydemann, A. An Overview of Murine High Fat Diet as a Model for Type 2 Diabetes Mellitus. J. Diabetes Res. 2016, 2016. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Paschen, M.; Moede, T.; Valladolid-Acebes, I.; Leibiger, B.; Moruzzi, N.; Jacob, S.; García-Prieto, C.F.; Brismar, K.; Leibiger, I.B.; Berggren, P.O. Diet-Induced b-Cell Insulin Resistance Results in Reversible Loss of Functional b-Cell Mass. FASEB J. 2019, 33, 204–218. [Google Scholar] [CrossRef] [Green Version]
- Matsuda, A.; Makino, N.; Tozawa, T.; Shirahata, N.; Honda, T.; Ikeda, Y.; Sato, H.; Ito, M.; Kakizaki, Y.; Akamatsu, M.; et al. Pancreatic Fat Accumulation, Fibrosis, and Acinar Cell Injury in the Zucker Diabetic Fatty Rat Fed a Chronic High-Fat Diet. Pancreas 2014, 43, 735–743. [Google Scholar] [CrossRef] [Green Version]
- Pinnick, K.E.; Collins, S.C.; Londos, C.; Gauguier, D.; Clark, A.; Fielding, B.A. Pancreatic Ectopic Fat Is Characterized by Adipocyte Infiltration and Altered Lipid Composition. Obesity 2008, 16, 522–530. [Google Scholar] [CrossRef]
- Puchalska, P.; Crawford, P.A. Multi-Dimensional Roles of Ketone Bodies in Fuel Metabolism, Signaling, and Therapeutics. Cell Metab. 2017, 25, 262–284. [Google Scholar] [CrossRef] [Green Version]
- Cooder, H.R. Epilepsy in Children: With Particular Reference to the Ketogenic Diet. Cal. West. Med. 1933, 39, 169–173. [Google Scholar]
- Stubbs, B.J.; Cox, P.J.; Evans, R.D.; Santer, P.; Miller, J.J.; Faull, O.K.; Magor-Elliott, S.; Hiyama, S.; Stirling, M.; Clarke, K. On the Metabolism of Exogenous Ketones in Humans. Front. Physiol. 2017, 8, 1–13. [Google Scholar] [CrossRef] [PubMed]
- Feinman, R.D.; Pogozelski, W.K.; Astrup, A.; Bernstein, R.K.; Fine, E.J.; Westman, E.C.; Accurso, A.; Frassetto, L.; Gower, B.A.; McFarlane, S.I.; et al. Dietary Carbohydrate Restriction as the First Approach in Diabetes Management: Critical Review and Evidence Base. Nutrition 2015, 31, 1–13. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Johnstone, A.M.; Horgan, G.W.; Murison, S.D.; Bremner, D.M.; Lobley, G.E. Effects of a High-Protein Ketogenic Diet on Hunger, Appetite, and Weight Loss in Obese Men Feeding Ad Libitum. Am. J. Clin. Nutr. 2008, 87, 44–55. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Dyson, P.A.; Beatty, S.; Matthews, D.R. A Low-Carbohydrate Diet Is More Effective in Reducing Body Weight than Healthy Eating in Both Diabetic and Non-Diabetic Subjects. Diabet. Med. 2007, 24, 1430–1435. [Google Scholar] [CrossRef]
- Weber, D.D.; Aminzadeh-Gohari, S.; Tulipan, J.; Catalano, L.; Feichtinger, R.G.; Kofler, B. Ketogenic Diet in the Treatment of Cancer—Where Do We Stand? Mol. Metab. 2020, 33, 102–121. [Google Scholar] [CrossRef]
- Tan-Shalaby, J. Ketogenic Diets and Cancer: Emerging Evidence. Fed. Pract. 2017, 34, 37S–42S. [Google Scholar]
- Murai, N.; Saito, N.; Kodama, E.; Iida, T.; Mikura, K.; Imai, H.; Kaji, M.; Hashizume, M.; Kigawa, Y.; Koizumi, G.; et al. Insulin and Proinsulin Dynamics Progressively Deteriorate From Within the Normal Range Toward Impaired Glucose Tolerance. J. Endocr. Soc. 2020, 4. [Google Scholar] [CrossRef]
- Quan, W.; Jo, E.K.; Lee, M.S. Role of Pancreatic β-Cell Death and Inflammation in Diabetes. Diabetes Obes. Metab. 2013, 15, 141–151. [Google Scholar] [CrossRef]
- Van Vliet, S.; Koh, H.-C.; Patterson, B.; Yoshino, M.; LaForest, R.; Gropler, R.; Klein, S.; Mittendorfer, B. Obesity Is Associated with Increased Basal and Postprandial β-Cell Insulin Secretion Even in the Absence of Insulin Resistance. Diabetes 2020, 69, 2112–2119. [Google Scholar] [CrossRef]
- Rojas, J.; Bermudez, V.; Palmar, J.; Martínez, M.S.; Olivar, L.C.; Nava, M.; Tomey, D.; Rojas, M.; Salazar, J.; Garicano, C.; et al. Pancreatic Beta Cell Death: Novel Potential Mechanisms in Diabetes Therapy. J. Diabetes Res. 2018. [Google Scholar] [CrossRef] [Green Version]
- Weir, G.C. Glucolipotoxicity, β-Cells, and Diabetes: The Emperor Has No Clothes. Diabetes 2020, 69, 273–278. [Google Scholar] [CrossRef] [PubMed]
- Ellenbroek, J.H.; Van Dijck, L.; Töns, H.A.; Rabelink, T.J.; Carlotti, F.; Ballieux, B.E.P.B.; De Koning, E.J.P. Long-Term Ketogenic Diet Causes Glucose Intolerance and Reduced β- and α-Cell Mass but No Weight Loss in Mice. Am. J. Physiol. Endocrinol. Metab. 2014, 306, 552–558. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Bielohuby, M.; Sisley, S.; Sandoval, D.; Herbach, N.; Zengin, A.; Fischereder, M.; Menhofer, D.; Stoehr, B.J.M.; Stemmer, K.; Wanke, R.; et al. Impaired Glucose Tolerance in Rats Fed Low-Carbohydrate, High-Fat Diets. Am. J. Physiol. Endocrinol. Metab. 2013, 305, E1059–E1070. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Her, T.K.; Lagakos, W.S.; Brown, M.R.; LeBrasseur, N.K.; Rakshit, K.; Matveyenko, A.V. Dietary Carbohydrates Modulate Metabolic and β-Cell Adaptation to High-Fat Diet-Induced Obesity. Am. J. Physiol. Endocrinol. Metab. 2020, 318, E856–E865. [Google Scholar] [CrossRef] [PubMed]
- Al-Khalifa, A.; Mathew, T.C.; Al-Zaid, N.S.; Mathew, E.; Dashti, H. Low Carbohydrate Ketogenic Diet Prevents the Induction of Diabetes Using Streptozotocin in Rats. Exp. Toxicol. Pathol. 2011, 63, 663–669. [Google Scholar] [CrossRef]
- Al-Khalifa, A.; Mathew, T.C.; Al-Zaid, N.S.; Mathew, E.; Dashti, H.M. Therapeutic Role of Low-Carbohydrate Ketogenic Diet in Diabetes. Nutrition 2009, 25, 1177–1185. [Google Scholar] [CrossRef]
- Tattikota, S.G.; Rathjen, T.; McAnulty, S.J.; Wessels, H.H.; Akerman, I.; Van De Bunt, M.; Hausser, J.; Esguerra, J.L.S.; Musahl, A.; Pandey, A.K.; et al. Argonaute2 Mediates Compensatory Expansion of the Pancreatic β Cell. Cell Metab. 2014, 19, 122–134. [Google Scholar] [CrossRef] [Green Version]
- Mirhashemi, F.; Kluth, O.; Scherneck, S.; Vogel, H.; Kluge, R.; Schürmann, A.; Joost, H.G.; Neschen, S. High-Fat, Carbohydrate-Free Diet Markedly Aggravates Obesity but Prevents β-Cell Loss and Diabetes in the Obese, Diabetes-Susceptible Db/Db Strain. Obes. Facts 2008, 1, 292–297. [Google Scholar] [CrossRef]
- Leiter, E.H.; Coleman, D.L.; Ingram, D.K.; Reynolds, M.A. Influence of Dietary Carbohydrate on the Induction of Diabetes in C57BL/KsJ-Db/Db Diabetes Mice. J. Nutr. 1983, 113, 184–195. [Google Scholar] [CrossRef]
- Jürgens, H.S.; Neschen, S.; Ortmann, S.; Scherneck, S.; Schmolz, K.; Schüler, G.; Schmidt, S.; Blüher, M.; Klaus, S.; Perez-Tilve, D.; et al. Development of Diabetes in Obese, Insulin-Resistant Mice: Essential Role of Dietary Carbohydrate in Beta Cell Destruction. Diabetologia 2007, 50, 1481–1489. [Google Scholar] [CrossRef] [Green Version]
- Lamont, B.J.; Waters, M.F.; Andrikopoulos, S. A Low-Carbohydrate High-Fat Diet Increases Weight Gain and Does Not Improve Glucose Tolerance, Insulin Secretion or β-Cell Mass in NZO Mice. Nutr. Diabetes 2016, 6, e194. [Google Scholar] [CrossRef] [PubMed]
- Kluth, O.; Mirhashemi, F.; Scherneck, S.; Kaiser, D.; Kluge, R.; Neschen, S.; Joost, H.G.; Schürmann, A. Dissociation of Lipotoxicity and Glucotoxicity in a Mouse Model of Obesity Associated Diabetes: Role of Forkhead Box O1 (FOXO1) in Glucose-Induced Beta Cell Failure. Diabetologia 2011, 54, 605–616. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Honors, M.A.; Davenport, B.M.; Kinzig, K.P. Effects of Consuming a High Carbohydrate Diet after Eight Weeks of Exposure to a Ketogenic Diet. Nutr. Metab. 2009, 6, 1–9. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Biden, T.J.; Taylor, K.W. Effects of Ketone Bodies on Insulin Release and Islet-Cell Metabolism in the Rat. Biochem. J. 1983, 212, 371–377. [Google Scholar] [CrossRef] [Green Version]
- Rhodes, C.J.; Campbell, I.L.; Szopa, T.M.; Biden, T.J.; Reynolds, P.D.; Fernando, O.N.; Taylor, K.W. Effects of Glucose and D-3-Hydroxybutyrate on Human Pancreatic Islet Cell Function. Clin. Sci. 1985, 68, 567–572. [Google Scholar] [CrossRef]
- Malaisse, W.J.; Lebrun, P.; Yaylali, B.; Camara, J.; Valverde, I.; Sener, A. Ketone Bodies and Islet Function: 45Ca Handling, Insulin Synthesis, and Release. Am. J. Physiol. Endocrinol. Metab. 1990, 259, E117–E122. [Google Scholar] [CrossRef]
- MacDonald, M.J.; Longacre, M.J.; Stoker, S.W.; Brown, L.J.; Hasan, N.M.; Kendrick, M.A. Acetoacetate and β-Hydroxybutyrate in Combination with Other Metabolites Release Insulin from INS-1 Cells and Provide Clues about Pathways in Insulin Secretion. Am. J. Physiol. Cell Physiol. 2008, 294, C442–C450. [Google Scholar] [CrossRef] [Green Version]
- Robinson, A.M.; Williamson, D.H. Physiological Roles of Ketone Bodies as Substrates and Signals in Mammalian Tissues. Physiol. Rev. 1980, 60, 143–187. [Google Scholar] [CrossRef]
- Zhou, Y.-P.; Grill, V. Long Term Exposure to Fatty Acids and Ketones Inhibits B-Cell Functions in Human Pancreatic Islets of Langerhans. J. Clin. Endocrinol. Metab. 1995, 80, 1584–1590. [Google Scholar]
- Pujol, J.B.; Christinat, N.; Ratinaud, Y.; Savoia, C.; Mitchell, S.E.; Dioum, E.H.M. Coordination of GPR40 and Ketogenesis Signaling by Medium Chain Fatty Acids Regulates Beta Cell Function. Nutrients 2018, 10, 473. [Google Scholar] [CrossRef] [Green Version]
- Berne, C. The Effect of Fatty Acids and Ketone Bodies on the Biosynthesis of Insulin in Isolated Pancreatic Islets of Obese Hyperglycemic Mice. Horm. Metab. Res. 1975, 7, 385–389. [Google Scholar] [CrossRef] [PubMed]
- Zhao, C.; Wilson, M.C.; Schuit, F.; Halestrap, A.P.; Rutter, G.A. Expression and Distribution of Lactate/Monocarboxylate Transporter Isoforms in Pancreatic Islets and the Exocrine Pancreas. Diabetes 2001, 50, 361–366. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Hasan, N.M.; Longacre, M.J.; Seed Ahmed, M.; Kendrick, M.A.; Gu, H.; Ostenson, C.G.; Fukao, T.; MacDonald, M.J. Lower Succinyl-CoA:3-Ketoacid-CoA Transferase (SCOT) and ATP Citrate Lyase in Pancreatic Islets of a Rat Model of Type 2 Diabetes: Knockdown of SCOT Inhibits Insulin Release in Rat Insulinoma Cells. Arch. Biochem. Biophys. 2010, 499, 62–68. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Berne, C. Determination of D 3 Hydroxybutyrate Dehydrogenase in Mouse Pancreatic Islets with a Photokinetic Technique Using Bacterial Luciferase. Enzyme 1976, 21, 127–136. [Google Scholar] [CrossRef] [PubMed]
- Berne, C. The Metabolism of Lipids in Mouse Pancreatic Islets. The Oxidation of Fatty Acids and Ketone Bodies. Biochem. J. 1975, 152, 661–666. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Kennedy, A.R.; Pissios, P.; Otu, H.; Xue, B.; Asakura, K.; Furukawa, N.; Marino, F.E.; Liu, F.F.; Kahn, B.B.; Libermann, T.A.; et al. A High-Fat, Ketogenic Diet Induces a Unique Metabolic State in Mice. Am. J. Physiol. Endocrinol. Metab. 2007, 292, 1724–1739. [Google Scholar] [CrossRef]
- Grandl, G.; Straub, L.; Rudigier, C.; Arnold, M.; Wueest, S.; Konrad, D.; Wolfrum, C. Short-Term Feeding of a Ketogenic Diet Induces More Severe Hepatic Insulin Resistance than an Obesogenic High-Fat Diet. J. Physiol. 2018, 596, 4597–4609. [Google Scholar] [CrossRef]
- Jornayvaz, F.R.; Jurczak, M.J.; Lee, H.Y.; Birkenfeld, A.L.; Frederick, D.W.; Zhang, D.; Zhang, X.M.; Samuel, V.T.; Shulman, G.I. A High-Fat, Ketogenic Diet Causes Hepatic Insulin Resistance in Mice, despite Increasing Energy Expenditure and Preventing Weight Gain. Am. J. Physiol. Endocrinol. Metab. 2010, 299, 808–815. [Google Scholar] [CrossRef]
- Morrison, C.D.; Hill, C.M.; DuVall, M.A.; Coulter, C.E.; Gosey, J.L.; Herrera, M.J.; Maisano, L.E.; Sikaffy, H.X.; McDougal, D.H. Consuming a Ketogenic Diet Leads to Altered Hypoglycemiccounter-Regulation in Mice. J. Diabetes Complicat. 2020, 34, 107557. [Google Scholar] [CrossRef]
- Badman, M.K.; Kennedy, A.R.; Adams, A.C.; Pissios, P.; Maratos-Flier, E. A Very Low Carbohydrate Ketogenic Diet Improves Glucose Tolerance in Ob/Ob Mice Independently of Weight Loss. Am. J. Physiol. Endocrinol. Metab. 2009, 297, E1197–E1204. [Google Scholar] [CrossRef] [Green Version]
- Garbow, J.R.; Doherty, J.M.; Schugar, R.C.; Travers, S.; Weber, M.L.; Wentz, A.E.; Ezenwajiaku, N.; Cotter, D.G.; Brunt, E.M.; Crawford, P.A. Hepatic Steatosis, Inflammation, and ER Stress in Mice Maintained Long Term on a Very Low-Carbohydrate Ketogenic Diet. Am. J. Physiol. Gastrointest. Liver Physiol. 2011, 300, 956–967. [Google Scholar] [CrossRef] [PubMed]
- Morens, C.; Sirot, V.; Scheurink, A.J.W.; Van Dijk, G. Low-Carbohydrate Diets Affect Energy Balance and Fuel Homeostasis Differentially in Lean and Obese Rats. Am. J. Physiol. Regul. Integr. Comp. Physiol. 2006, 291, 1622–1629. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Kinzig, K.P.; Honors, M.A.; Hargrave, S.L. Insulin Sensitivity and Glucose Tolerance Are Altered by Maintenance on a Ketogenic Diet. Endocrinology 2010, 151, 3105–3114. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Fabbrini, E.; Higgins, P.B.; Magkos, F.; Bastarrachea, R.A.; Saroja Voruganti, V.; Comuzzie, A.G.; Shade, R.E.; Gastaldelli, A.; Horton, J.D.; Omodei, D.; et al. Metabolic Response to High-Carbohydrate and Low-Carbohydrate Meals in a Nonhuman Primate Model. Am. J. Physiol. Endocrinol. Metab. 2013, 304, 444–451. [Google Scholar] [CrossRef] [Green Version]
- Park, S.; Kim, D.S.; Kang, S.; Daily, J.W. A Ketogenic Diet Impairs Energy and Glucose Homeostasis by the Attenuation of Hypothalamic Leptin Signaling and Hepatic Insulin Signaling in a Rat Model of Non-Obese Type 2 Diabetes. Exp. Biol. Med. 2011, 236, 194–204. [Google Scholar] [CrossRef]
- Zhang, X.; Qin, J.; Zhao, Y.; Shi, J.; Lan, R.; Gan, Y.; Ren, H.; Zhu, B.; Qian, M.; Du, B. Long-Term Ketogenic Diet Contributes to Glycemic Control but Promotes Lipid Accumulation and Hepatic Steatosis in Type 2 Diabetic Mice. Nutr. Res. 2016, 36, 349–358. [Google Scholar] [CrossRef]
- Okuda, T.; Morita, N. A Very Low Carbohydrate Ketogenic Diet Increases Hepatic Glycosphingolipids Related to Regulation of Insulin Signalling. J. Funct. Foods 2016, 21, 70–74. [Google Scholar] [CrossRef]
- Westman, E.C.; Yancy, W.S.; Mavropoulos, J.C.; Marquart, M.; McDuffie, J.R. The Effect of a Low-Carbohydrate, Ketogenic Diet versus a Low-Glycemic Index Diet on Glycemic Control in Type 2 Diabetes Mellitus. Nutr. Metab. 2008, 5, 1–9. [Google Scholar] [CrossRef] [Green Version]
- Hall, K.D.; Chen, K.Y.; Guo, J.; Lam, Y.Y.; Leibel, R.L.; Mayer, L.E.S.; Reitman, M.L.; Rosenbaum, M.; Smith, S.R.; Walsh, B.T.; et al. Energy Expenditure and Body Composition Changes after an Isocaloric Ketogenic Diet in Overweight and Obese Men. Am. J. Clin. Nutr. 2016, 104, 324–333. [Google Scholar] [CrossRef] [Green Version]
- Boden, G.; Sargrad, K.; Homko, C.; Mozzoli, M.; Stein, T.P. Effect of a Low-Carbohydrate Diet on Appetite, Blood Glucose Levels, and Insulin Resistance in Obese Patients with Type 2 Diabetes. Ann. Intern. Med. 2005, 142, 403–411. [Google Scholar] [CrossRef]
- Gannon, M.C.; Nuttall, F.Q. Effect of a High-Protein, Low-Carbohydrate Diet on Blood Glucose Control in People with Type 2 Diabetes. Diabetes 2004, 53, 2375–2382. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Goss, A.M.; Gower, B.A.; Soleymani, T.; Stewart, M.C.; Pendergrass, M.; Lockhart, M.; Krantz, O.; Dowla, S.; Bush, N.; Garr Barry, V.; et al. Effects of Weight Loss during a Very Low Carbohydrate Diet on Specific Adipose Tissue Depots and Insulin Sensitivity in Older Adults with Obesity: A Randomized Clinical Trial. Metabolism 2020, 17, 1–12. [Google Scholar] [CrossRef] [PubMed]
- Rosenbaum, M.; Hall, K.D.; Guo, J.; Ravussin, E.; Mayer, L.S.; Reitman, M.L.; Smith, S.R.; Walsh, B.T.; Leibel, R.L. Glucose and Lipid Homeostasis and Inflammation in Humans Following an Isocaloric Ketogenic Diet. Obesity 2019, 27, 971–981. [Google Scholar] [CrossRef] [PubMed]
- Samaha, F.F.; Iqbal, N.; Seshadri, P.; Chicano, K.L.; Daily, D.A.; McGrory, J.; Williams, T.; Williams, M.; Gracely, E.J.; Stern, L. A Low-Carbohydrate as Compared with a Low-Fat Diet in Severe Obesity. N. Engl. J. Med. 2003, 348, 2074–2081. [Google Scholar] [CrossRef] [Green Version]
- Tay, J.; Luscombe-Marsh, N.D.; Thompson, C.H.; Noakes, M.; Buckley, J.D.; Wittert, G.A.; Yancy Jr, W.S.; Brinkworth, G.D. Comparison of Low- and High-Carbohydrate Diets for Type 2 Diabetes Management: A Randomized Trial. Am. J. Clin. Nutr. 2015, 102, 780–790. [Google Scholar] [CrossRef]
- Allick, G.; Bisschop, P.H.; Ackermans, M.T.; Endert, E.; Meijer, A.J.; Kuipers, F.; Sauerwein, H.P.; Romijn, J.A. A Low-Carbohydrate/High-Fat Diet Improves Glucoregulation in Type 2 Diabetes Mellitus by Reducing Postabsorptive Glycogenolysis. J. Clin. Endocrinol. Metab. 2004, 89, 6193–6197. [Google Scholar] [CrossRef] [Green Version]
- Vernon, M.C.; Mavropoulos, J.; Transue, M.; Yancy, W.S.; Al, V.E.T. Clinical Experience of a Carbohydrate-Restricted Diet: Effect on Diabetes Mellitus. Metab. Syndr. Relat. Disord. 2003, 1, 233–237. [Google Scholar] [CrossRef]
- Hussain, T.A.; Mathew, T.C.; Dashti, A.A.; Asfar, S.; Al-Zaid, N.; Dashti, H.M. Effect of Low-Calorie versus Low-Carbohydrate Ketogenic Diet in Type 2 Diabetes. Nutrition 2012, 28, 1016–1021. [Google Scholar] [CrossRef]
- Yancy, W.S.; Foy, M.; Chalecki, A.M.; Vernon, M.C.; Westman, E.C. A Low-Carbohydrate, Ketogenic Diet to Treat Type 2 Diabetes. Nutr. Metab. 2005, 2, 34. [Google Scholar] [CrossRef]
- Nielsen, J.V.; Joensson, E.A. Low-Carbohydrate Diet in Type 2 Diabetes: Stable Improvement of Bodyweight and Glycemic Control during 44 Months Follow-Up. Nutr. Metab. 2008, 5, 14. [Google Scholar] [CrossRef] [Green Version]
- Müller, J.E.; Sträter-Müller, D.; Marks, H.J.; Gläsner, M.; Kneppe, P.; Clemens-Harmening, B.; Menker, H. Carbohydrate Restricted Diet in Conjunction with Metformin and Liraglutide Is an Effective Treatment in Patients with Deteriorated Type 2 Diabetes Mellitus: Proof-of-Concept Study. Nutr. Metab. 2011, 8, 92. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Verkoelen, H.; Govers, E.; Maas, H.; Gh, K. Low Carbohydrate Lifestyle Reduces Significantly Insulin Need in Type 2 Diabetes Patients. Interv. Obes. Diabetes 2020, 4, 411–424. [Google Scholar]
- Chen, C.; Id, W.H.; Chen, H.; Chang, C.; Lee, T.; Chen, H.; Kang, Y.; Chie, W.; Id, C.J.; Wang, D.; et al. Effect of a 90 g/Day Low-Carbohydrate Diet on Glycaemic Control, Small, Dense Low-Density Lipoprotein and Carotid Intima-Media Thickness in Type 2 Diabetic Patients: An 18-Month Randomised Controlled Trial. PLoS ONE 2020, 15, e0240158. [Google Scholar] [CrossRef] [PubMed]
- Walton, C.M.; Perry, K.; Hart, R.H.; Berry, S.L.; Bikman, B.T. Improvement in Glycemic and Lipid Profiles in Type 2 Diabetics with a 90-Day Ketogenic Diet. J. Diabetes Res. 2019, 2019. [Google Scholar] [CrossRef] [Green Version]
- McKenzie, A.L.; Hallberg, S.J.; Creighton, B.C.; Volk, B.M.; Link, T.M.; Abner, M.K.; Glon, R.M.; McCarter, J.P.; Volek, J.S.; Phinney, S.D. A Novel Intervention Including Individualized Nutritional Recommendations Reduces Hemoglobin A1c Level, Medication Use, and Weight in Type 2 Diabetes. JMIR Diabetes 2017, 2, e5. [Google Scholar] [CrossRef]
- o’Neill, D.F.; Westman, E.C.; Bernstein, R.K. The Effects of a Low-Carbohydrate Regimen on Glycemic Control and Serum Lipids in Diabetes Mellitus. Metab. Syndr. Relat. Disord. 2003, 1, 291–298. [Google Scholar] [CrossRef] [Green Version]
- Athinarayanan, S.J.; Adams, R.N.; Hallberg, S.J.; McKenzie, A.L.; Bhanpuri, N.H.; Campbell, W.W.; Volek, J.S.; Phinney, S.D.; McCarter, J.P. Long-Term Effects of a Novel Continuous Remote Care Intervention Including Nutritional Ketosis for the Management of Type 2 Diabetes: A 2-Year Nonrandomized Clinical Trial. Front. Endocrinol. 2019, 10, 348. [Google Scholar] [CrossRef] [Green Version]
- Hallberg, S.J.; McKenzie, A.L.; Williams, P.T.; Bhanpuri, N.H.; Peters, A.L.; Campbell, W.W.; Hazbun, T.L.; Volk, B.M.; McCarter, J.P.; Phinney, S.D.; et al. Effectiveness and Safety of a Novel Care Model for the Management of Type 2 Diabetes at 1 Year: An Open-Label, Non-Randomized, Controlled Study. Diabetes Ther. 2018, 9, 583–612. [Google Scholar] [CrossRef] [Green Version]
- Daly, M.E.; Paisey, R.; Paisey, R.; Millward, B.A.; Eccles, C.; Williams, K.; Hammersley, S.; MacLeod, K.M.; Gale, T.J. Short-Term Effects of Severe Dietary Carbohydrate-Restriction Advice in Type 2 Diabetes-A Randomized Controlled Trial. Diabet. Med. 2006, 23, 15–20. [Google Scholar] [CrossRef]
- Muniyappa, R.; Lee, S.; Chen, H.; Quon, M.J. Current Approaches for Assessing Insulin Sensitivity and Resistance in Vivo: Advantages, Limitations, and Appropriate Usage. Am. J. Physiol. Endocrinol. Metab. 2008, 294, E15–E26. [Google Scholar] [CrossRef] [Green Version]
- Lee, S.; Muniyappa, R.; Yan, X.; Chen, H.; Yue, L.Q.; Hong, E.G.; Kim, J.K.; Quon, M.J. Comparison between Surrogate Indexes of Insulin Sensitivity and Resistance and Hyperinsulinemic Euglycemic Clamp Estimates in Mice. Am. J. Physiol. Endocrinol. Metab. 2008, 294, E261–E270. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Muniyappa, R.; Chen, H.; Muzumdar, R.H.; Einstein, F.H.; Yan, X.; Yue, L.Q.; Barzilai, N.; Quon, M.J. Comparison between Surrogate Indexes of Insulin Sensitivity/Resistance and Hyperinsulinemic Euglycemic Clamp Estimates in Rats. Am. J. Physiol. Endocrinol. Metab. 2009, 297, E1023–E1029. [Google Scholar] [CrossRef] [PubMed]
- Roberts, M.N.; Wallace, M.A.; Tomilov, A.A.; Zhou, Z.; Marcotte, G.R.; Tran, D.; Perez, G.; Gutierrez-Casado, E.; Koike, S.; Knotts, T.A.; et al. A Ketogenic Diet Extends Longevity and Healthspan in Adult Mice. Cell Metab. 2017, 26, 539–546.e5. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Ribeiro, L.C.; Chittó, A.L.; Müller, A.P.; Rocha, J.K.; Da Silva, M.C.; Quincozes-Santos, A.; Nardin, P.; Rotta, L.N.; Ziegler, D.R.; Gonçalves, C.A.; et al. Ketogenic Diet-Fed Rats Have Increased Fat Mass and Phosphoenolpyruvate Carboxykinase Activity. Mol. Nutr. Food Res. 2008, 52, 1365–1371. [Google Scholar] [CrossRef]
- Hu, S.; Togo, J.; Wang, L.; Wu, Y.; Yang, D.; Xu, Y.; Li, L.; Li, B.; Li, M.; Li, J.; et al. Effects of Dietary Macronutrients and Body Composition on Glucose Homeostasis in Mice. Natl. Sci. Rev. 2020. [Google Scholar] [CrossRef]
- Bisschop, P.H.; De Metz, J.; Ackermans, M.T.; Endert, E.; Pijl, H.; Kuipers, F.; Meijer, A.J.; Sauerwein, H.P.; Romijn, J.A. Dietary Fat Content Alters Insulin-Mediated Glucose Metabolism in Healthy Men. Am. J. Clin. Nutr. 2001, 73, 554–559. [Google Scholar] [CrossRef] [Green Version]
- Mulvihill, E.E.; Varin, E.M.; Gladanac, B.; Campbell, J.E.; Ussher, J.R.; Baggio, L.L.; Yusta, B.; Ayala, J.; Burmeister, M.A.; Matthews, D.; et al. Cellular Sites and Mechanisms Linking Reduction of Dipeptidyl Peptidase-4 Activity to Control of Incretin Hormone Action and Glucose Homeostasis. Cell Metab. 2017, 25, 152–165. [Google Scholar] [CrossRef] [Green Version]
- Mulvihill, E.E. Dipeptidyl Peptidase Inhibitor Therapy in Type 2 Diabetes: Control of the Incretin Axis and Regulation of Postprandial Glucose and Lipid Metabolism. Peptides 2018, 100, 158–164. [Google Scholar] [CrossRef]
- Campbell, J.E.; Drucker, D.J. Pharmacology, Physiology, and Mechanisms of Incretin Hormone Action. Cell Metab. 2013, 17, 819–837. [Google Scholar] [CrossRef] [Green Version]
- Stubbs, B.J.; Cox, P.J.; Evans, R.D.; Cyranka, M.; Clarke, K.; De Wet, H. A Ketone Ester Drink Lowers Human Ghrelin and Appetite. Obesity 2018, 26, 269–273. [Google Scholar] [CrossRef]
- Wallenius, V.; Elias, E.; Elebring, E.; Haisma, B.; Casselbrant, A.; Larraufie, P.; Spak, E.; Reimann, F.; Le Roux, C.W.; Docherty, N.G.; et al. Suppression of Enteroendocrine Cell Glucagon-like Peptide (GLP)-1 Release by Fat-Induced Small Intestinal Ketogenesis: A Mechanism Targeted by Roux-En-Y Gastric Bypass Surgery but Not by Preoperative Very-Low-Calorie Diet. Gut 2020, 69, 1423–1431. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Hirasawa, A.; Tsumaya, K.; Awaji, T.; Katsuma, S.; Adachi, T.; Yamada, M.; Sugimoto, Y.; Miyazaki, S.; Tsujimoto, G. Free Fatty Acids Regulate Gut Incretin Glucagon-like Peptide-1 Secretion through GPR120. Nat. Med. 2005, 11, 90–94. [Google Scholar] [CrossRef] [PubMed]
- Hara, T.; Kashihara, D.; Ichimura, A.; Kimura, I.; Tsujimoto, G.; Hirasawa, A. Role of Free Fatty Acid Receptors in the Regulation of Energy Metabolism. Biochim. Biophys. Acta Mol. Cell Biol. Lipids 2014, 1841, 1292–1300. [Google Scholar] [CrossRef] [PubMed]
- Edfalk, S.; Steneberg, P.; Edlund, H. Gpr40 Is Expressed in Enteroendocrine Cells and Mediates Free Fatty Acid Stimulation of Incretin Secretion. Diabetes 2008, 57, 2280–2287. [Google Scholar] [CrossRef] [Green Version]
- Supale, S.; Li, N.; Brun, T.; Maechler, P. Mitochondrial Dysfunction in Pancreatic β Cells. Trends Endocrinol. Metab. 2012, 23, 477–487. [Google Scholar] [CrossRef]
- Rutter, G.A.; Georgiadou, E.; Martinez-Sanchez, A.; Pullen, T.J. Metabolic and Functional Specialisations of the Pancreatic Beta Cell: Gene Disallowance, Mitochondrial Metabolism and Intercellular Connectivity. Diabetologia 2020, 63, 1990–1998. [Google Scholar] [CrossRef]
- Hu, S.; Wang, L.; Yang, D.; Li, L.; Togo, J.; Wu, Y.; Liu, Q.; Li, B.; Li, M.; Wang, G.; et al. Dietary Fat, but Not Protein or Carbohydrate, Regulates Energy Intake and Causes Adiposity in Mice. Cell Metab. 2018, 28, 415–431.e4. [Google Scholar] [CrossRef] [Green Version]
- Colberg, S.R.; Sigal, R.J.; Fernhall, B.; Regensteiner, J.G.; Blissmer, B.J.; Rubin, R.R.; Chasan-Taber, L.; Albright, A.L.; Braun, B. Exercise and Type 2 Diabetes: The American College of Sports Medicine and the American Diabetes Association: Joint Position Statement. Diabetes Care 2010, 33, e147–e167. [Google Scholar] [CrossRef] [Green Version]
- Monda, V.; Sessa, F.; Ruberto, M.; Carotenuto, M.; Marsala, G.; Monda, M.; Cambria, M.T.; Astuto, M.; Distefano, A.; Messina, G. Aerobic Exercise and Metabolic Syndrome: The Role of Sympathetic Activity and the Redox System. Diabetes Metab. Syndr. Obes. Targets Ther. 2020, 13, 2433–2442. [Google Scholar] [CrossRef]
- Polito, R.; Francavilla, V.C.; Ambrosi, A.; Tartaglia, N.; Tafuri, D.; Monda, M.; Messina, A.; Sessa, F.; Di Maio, G.; Ametta, A.; et al. The Orexin-A Serum Levels Are Strongly Modulated by Physical Activity Intervention in Diabetes Mellitus Patients. J. Hum. Sport Exerc. 2020, 15, S244–S251. [Google Scholar]
- Wroble, K.A.; Trott, M.N.; Schweitzer, G.G.; Rahman, R.S.; Kelly, P.V.; Weiss, E.P. Low-Carbohydrate, Ketogenic Diet Impairs Anaerobic Exercise Performance in Exercise-Trained Women and Men: A Randomized-Sequence Crossover Trial. J. Sports Med. Phys. Fit. 2019, 59, 600–607. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Pilis, K.; Pilis, A.; Stec, K.; Pilis, W.; Langfort, J.; Letkiewicz, S.; Michalski, C.; Czuba, M.; Zych, M.; Chalimoniuk, M. Three-Year Chronic Consumption of Low-Carbohydrate Diet Impairs Exercise Performance and Has a Small Unfavorable Effect on Lipid Profile in Middle-Aged Men. Nutrients 2018, 10, 1914. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Ferreira, G.A.; Felippe, L.C.; Silva, R.L.S.; Bertuzzi, R.; De Oliveira, F.R.; Pires, F.O.; Lima-Silva, A.E. Effect of Pre-Exercise Carbohydrate Availability on Fat Oxidation and Energy Expenditure after a High-Intensity Exercise. Brazilian J. Med. Biol. Res. 2018, 51, 1–8. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Burke, L.M.; Ross, M.L.; Garvican-Lewis, L.A.; Welvaert, M.; Heikura, I.A.; Forbes, S.G.; Mirtschin, J.G.; Cato, L.E.; Strobel, N.; Sharma, A.P.; et al. Low Carbohydrate, High Fat Diet Impairs Exercise Economy and Negates the Performance Benefit from Intensified Training in Elite Race Walkers. J. Physiol. 2017, 595, 2785–2807. [Google Scholar] [CrossRef] [Green Version]
- Greene, D.A.; Varley, B.J.; Hartwig, T.B.; Chapman, P.; Rigney, M. A Low-Carbohydrate Ketogenic Diet Reduces Body Mass without Compromising Performance in Powerlifting and Olympic Weightlifting Athletes. J. Strength Cond. Res. 2018, 32, 3373–3382. [Google Scholar] [CrossRef]
- Paoli, A.; Grimaldi, K.; D’Agostino, D.; Cenci, L.; Moro, T.; Bianco, A.; Palma, A. Ketogenic Diet Does Not Affect Strength Performance in Elite Artistic Gymnasts. J. Int. Soc. Sports Nutr. 2012, 9, 34. [Google Scholar] [CrossRef] [Green Version]
- McSwiney, F.T.; Wardrop, B.; Hyde, P.N.; Lafountain, R.A.; Volek, J.S.; Doyle, L. Keto-Adaptation Enhances Exercise Performance and Body Composition Responses to Training in Endurance Athletes. Metabolism 2018, 81, 25–34. [Google Scholar] [CrossRef]
- Golden, S.H.; Brown, A.; Cauley, J.A.; Chin, M.H.; Gary-Webb, T.L.; Kim, C.; Sosa, J.A.; Sumner, A.E.; Anton, B. Health Disparities in Endocrine Disorders: Biological, Clinical, and Nonclinical Factors—An Endocrine Society Scientific Statement. J. Clin. Endocrinol. Metab. 2012, 97, E1579–E1639. [Google Scholar] [CrossRef] [Green Version]
- O’Neill, B.; Raggi, P. The Ketogenic Diet: Pros and Cons. Atherosclerosis 2020, 292, 119–126. [Google Scholar] [CrossRef] [Green Version]
- Kirkpatrick, C.F.; Bolick, J.P.; Kris-Etherton, P.M.; Sikand, G.; Aspry, K.E.; Soffer, D.E.; Willard, K.E.; Maki, K.C. Review of Current Evidence and Clinical Recommendations on the Effects of Low-Carbohydrate and Very-Low-Carbohydrate (Including Ketogenic) Diets for the Management of Body Weight and Other Cardiometabolic Risk Factors: A Scientific Statement from the Nati. J. Clin. Lipidol. 2019, 13, 689–711. [Google Scholar] [CrossRef] [Green Version]
- Fung, T.T.; Van Dam, R.M.; Hankinson, S.E.; Stampfer, M.; Willett, W.C.; Hu, F.B. Low-Carbohydrate Diets and All-Cause and Cause-Specific Mortality: Two Cohort Studies. Ann. Intern. Med. 2010, 153, 289–298. [Google Scholar] [CrossRef] [PubMed]
- Stern, L.; Iqbal, N.; Seshadri, P.; Chicano, K.L.; Daily, D.A.; McGrory, J.; Williams, M.; Gracely, E.J.; Samaha, F.F. The Effects of Low-Carbohydrate versus Conventional Weight Loss Diets in Severely Obese Adults: One-Year Follow-up of a Randomized Trial. Ann. Intern. Med. 2004, 140, 778–785. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Tzur, A.; Nijholt, R.; Sparagna, V.; Ritson, A. Adhering to the Ketogenic Diet—Is it Easy or Hard? (Research Review). Available online: https://sci-fit.net/adhere-ketogenic-diet/ (accessed on 18 September 2020).




{kind=link}
{kind=link}
{kind=link}
{kind=link}
| Diet | Carbohydrate | Fat | Protein | ||
|---|---|---|---|---|---|
| CRD | Carbohydrate-restricted diet | A diet which intends to decrease carbohydrate consumption | <40% | >30% | 4–60% |
| LCD | Low-carbohydrate diet | A CRD with less than 30% of kcal from carbohydrates without evidence of elevated ketone bodies | <30% | 30–95% | 4–60% |
| KD | Ketogenic diet | An LCD with elevated ketone bodies but some dietary carbohydrate and typically low protein | <10% | >70% | 4–20% |
| CFD | Carbohydrate-free diet | A diet containing no carbohydrates (preclinical) | 0% | 8–88% | 12–83% |
Publisher’s Note: MDPI stays neutral with regard to jurisdictional claims in published maps and institutional affiliations. |
© 2020 by the authors. Licensee MDPI, Basel, Switzerland. This article is an open access article distributed under the terms and conditions of the Creative Commons Attribution (CC BY) license (http://creativecommons.org/licenses/by/4.0/).
Share and Cite
Locatelli, C.A.A.; Mulvihill, E.E. Islet Health, Hormone Secretion, and Insulin Responsivity with Low-Carbohydrate Feeding in Diabetes. Metabolites 2020, 10, 455. https://doi.org/10.3390/metabo10110455
Locatelli CAA, Mulvihill EE. Islet Health, Hormone Secretion, and Insulin Responsivity with Low-Carbohydrate Feeding in Diabetes. Metabolites. 2020; 10(11):455. https://doi.org/10.3390/metabo10110455
Chicago/Turabian StyleLocatelli, Cassandra A. A., and Erin E. Mulvihill. 2020. "Islet Health, Hormone Secretion, and Insulin Responsivity with Low-Carbohydrate Feeding in Diabetes" Metabolites 10, no. 11: 455. https://doi.org/10.3390/metabo10110455
APA StyleLocatelli, C. A. A., & Mulvihill, E. E. (2020). Islet Health, Hormone Secretion, and Insulin Responsivity with Low-Carbohydrate Feeding in Diabetes. Metabolites, 10(11), 455. https://doi.org/10.3390/metabo10110455