Oxidative Stress in Cytokine-Induced Dysfunction of the Pancreatic Beta Cell: Known Knowns and Known Unknowns

Abstract
:1. Introduction
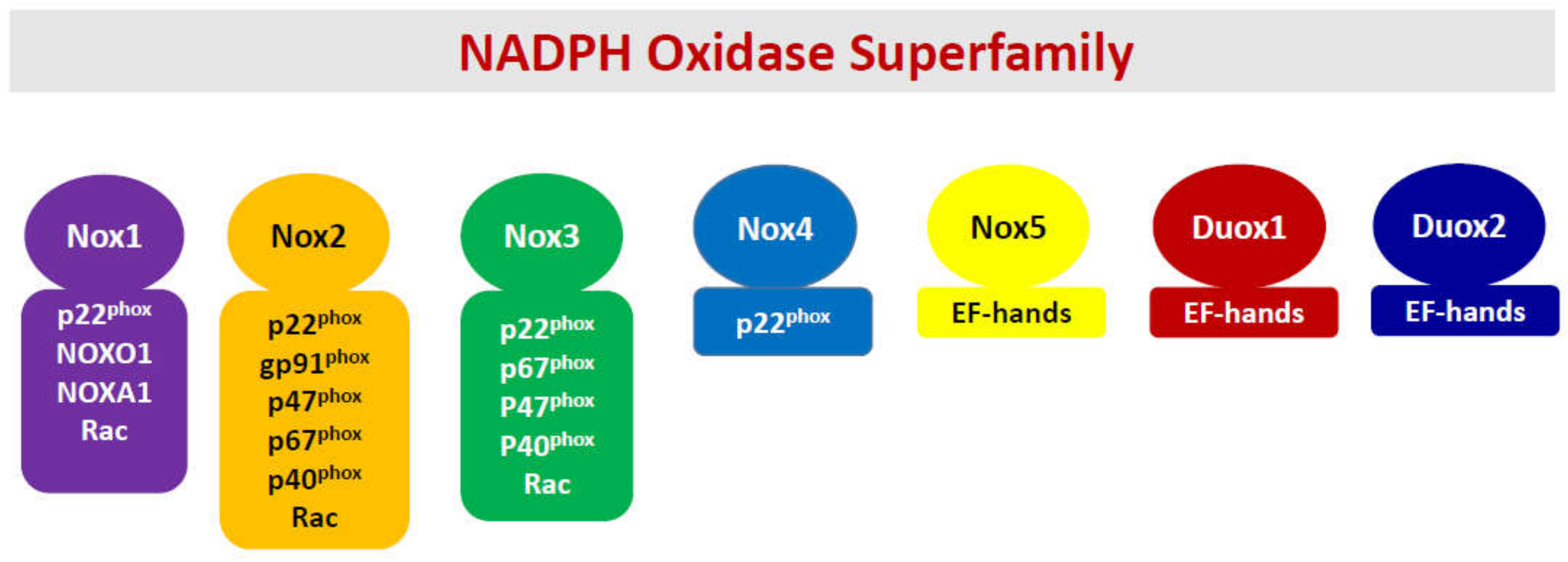
2. NADPH Oxidases Play Key Roles in Islet Function in Health and under Stress Induced by Cytokines
3. Regulatory Roles of Noxs in Physiological Insulin Secretion
4. Regulatory Roles of Nox1 in Cytokine-Induced Dysfunction of the Beta Cell
5. Regulatory Roles of Nox2 in Cytokine-Induced Dysfunction of the Beta Cell
6. Roles of Nox3, Nox4, and Nox5 in Cytokine-Induced Dysfunction of the Beta Cell
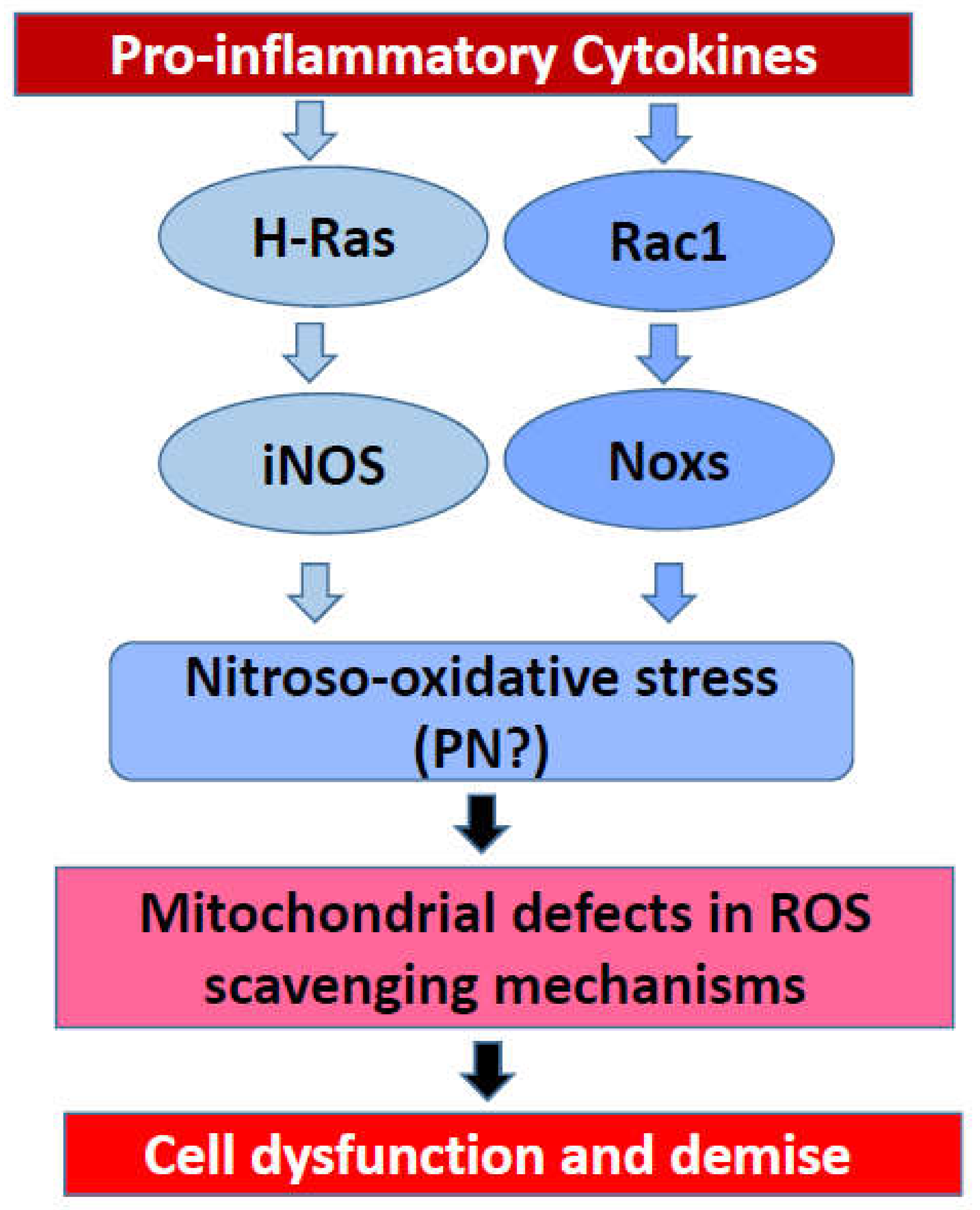
7. Potential Crosstalk between iNOS and Nox2 Signaling Pathways in the Onset of Cytokine-Induced Metabolic Dysregulation of the Islet Beta Cell
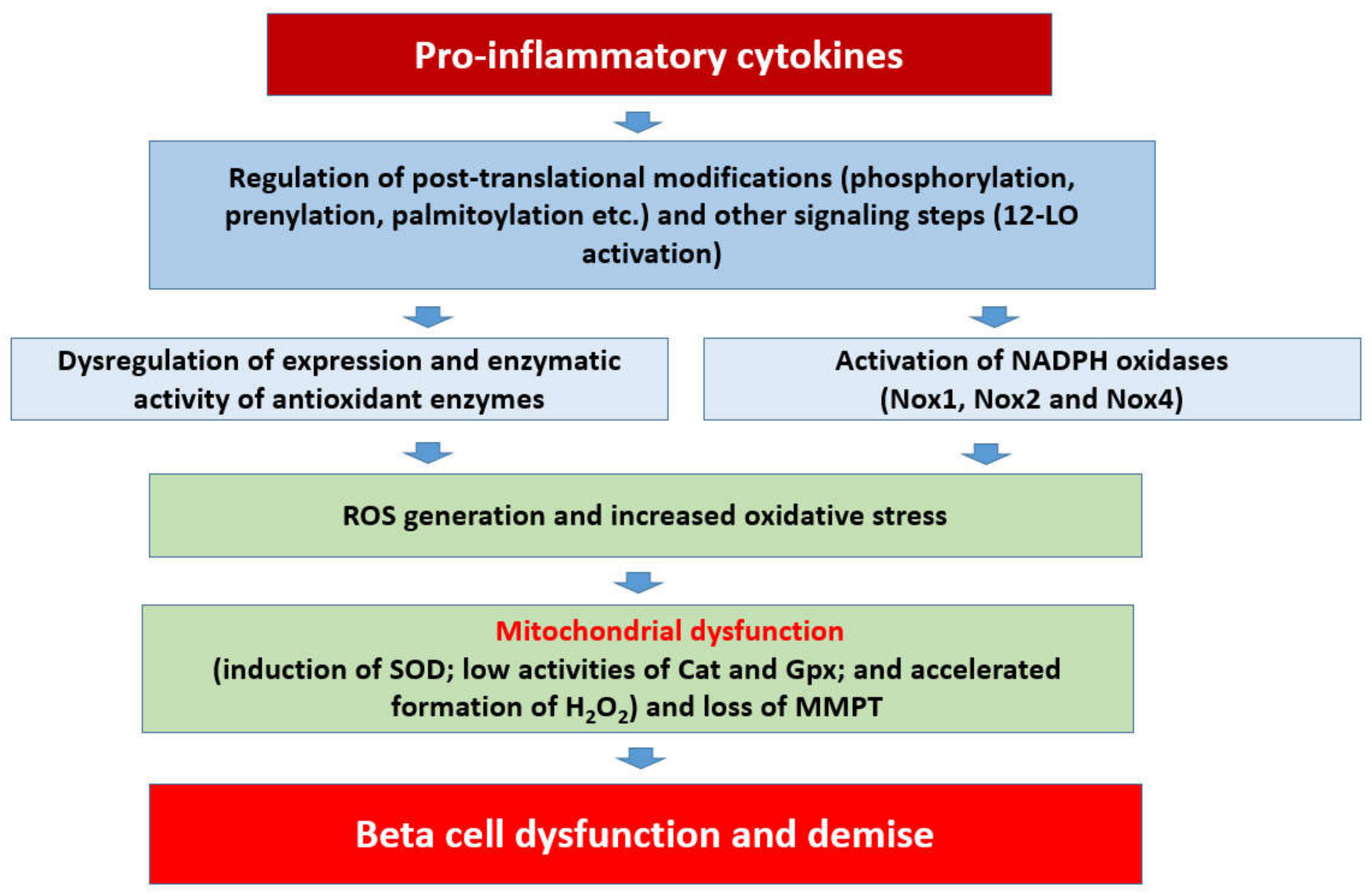
8. Restoration of Intracellular Redox Environment Prevents Cytokine-Induced Metabolic Defects in the Beta Cell
9. Conclusions
Funding
Acknowledgments
Conflicts of Interest
Abbreviations
| DPI | diphenyleneiodonium |
| Duox1 | dual oxidase1 |
| Duox2 | dual oxidase2 |
| EF hands | calcium binding domains or motifs |
| ER stress | endoplasmic reticulum stress |
| FTase | farnesyltransferase |
| GEFs | guanine nucleotide exchange factors |
| GSH | reduced glutathione |
| GSIS | glucose-stimulated insulin secretion |
| 12-HETE | 12-hydroxyeicosatetranoic acid |
| IFNγ | interferon γ |
| IL-1β | interleukin 1β |
| iNOS | inducible nitric oxide synthase |
| 12-LO | 12-lipoxygenase |
| MCP-1 | monocyte chemoattractant protein-1 |
| MMPT | mitochondrial membrane pore transition |
| Nrf2 | nuclear factor erythroid 2-related factor 2 |
| NO | nitric oxide |
| Nox1 | NADPH oxidase 1 |
| Nox2 | NADPH oxidase 2 |
| Nox3 | NADPH oxidase 3 |
| Nox4 | NADPH oxidase 4 |
| Nox5 | NADPH oxidase 5 |
| PKC | protein kinase C |
| PN | peroxynitrite |
| Prdx1 | peroxyredoxin1 |
| PrdxIII | peroxyredoxin III |
| Prdx6 | peroxyredoxin 6 |
| siRNA | small interfering RNA |
| SOD | superoxide dismutase |
| TNFα | tumor necrosis factorα |
| NAC | N-acetylcysteine |
| NSC23766 | N6-[2-[4-(Diethylamino)-1-methylbu-tyl]amino]-6-methyl-4-pyrimidinyl]-2-methyl- 4,6-qu-inolinediamine trihydrochloride |
| NOD | non-obese diabetic |
| Rac1 | Ras-related C3 botulinum toxin substrate 1 |
| RNS | reactive nitrogen species |
| ROS | reactive oxygen species |
| T1DM | type 1 diabetes mellitus |
| Tiam1 | T-cell lymphoma invasion and metastasis-inducing protein 1 |
References
- Nerup, J.; Mandrup-Poulsen, T.; Mølvig, J.; Helqvist, S.; Wogensen, L.; Egeberg, J. Mechanisms of pancreatic beta-cell destruction in type I diabetes. Diabetes Care 1988, 11 (Suppl. 1), 16–23. [Google Scholar]
- Kaminitz, A.; Stein, J.; Yaniv, I.; Askenasy, N. The vicious cycle of apoptotic beta-cell death in type 1 diabetes. Immunol. Cell Biol. 2007, 85, 582–589. [Google Scholar] [CrossRef]
- Mehmeti, I.; Gurgul-Convey, E.; Lenzen, S.; Lortz, S. Induction of the intrinsic apoptosis pathway in insulin-secreting cells is dependent on oxidative damage of mitochondria but independent of caspase-12 activation. Biochim. Biophys. Acta 2011, 1813, 1827–1835. [Google Scholar] [CrossRef] [Green Version]
- Zhong, L.; Tran, T.; Baguley, T.D.; Lee, S.J.; Henke, A.; To, A.; Li, S.; Yu, S.; Grieco, F.A.; Roland, J.; et al. A novel inhibitor of inducible NOS dimerization protects against cytokine-induced rat beta cell dysfunction. Br. J. Pharmacol. 2018, 175, 3470–3485. [Google Scholar] [CrossRef]
- Berchtold, L.A.; Prause, M.; Størling, J.; Mandrup-Poulsen, T. Cytokines and pancreatic β-Cell apoptosis. Adv. Clin. Chem. 2016, 75, 99–158. [Google Scholar] [CrossRef]
- Brun, T.; Maechler, P. Beta-cell mitochondrial carriers and the diabetogenic stress response. Biochim. Biophys. Acta 2016, 1863, 2540–2549. [Google Scholar] [CrossRef]
- Eizirik, D.L.; Colli, M.L.; Ortis, F. The role of inflammation in insulitis and beta-cell loss in type 1 diabetes. Nat. Rev. Endocrinol. 2009, 5, 219–226. [Google Scholar] [CrossRef]
- Rabinovitch, A.; Suarez-Pinzon, W.L. Cytokines and their roles in pancreatic islet beta-cell destruction and insulin-dependent diabetes mellitus. Biochem. Pharmacol. 1998, 55, 1139–1149. [Google Scholar] [CrossRef]
- Padgett, L.E.; Broniowska, K.A.; Hansen, P.A.; Corbett, J.A.; Tse, H.M. The role of reactive oxygen species and proinflammatory cytokines in type 1 diabetes pathogenesis. Ann. N. Y. Acad. Sci. 2013, 1281, 16–35. [Google Scholar] [CrossRef] [Green Version]
- Veluthakal, R.; Arora, D.K.; Goalstone, M.L.; Kowluru, R.A.; Kowluru, A. Metabolic stress induces Caspase-3 mediated degradation and inactivation of farnesyl and geranylgeranyl transferase activities in pancreatic β-Cells. Cell. Physiol. Biochem. 2016, 39, 2110–2120. [Google Scholar] [CrossRef]
- Kowluru, A. Role of G-proteins in islet function in health and diabetes. Diabetes Obes. Metab. 2017, 19 (Suppl. 1), 63–75. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Eguchi, K.; Manabe, I. Macrophages and islet inflammation in type 2 diabetes. Diabetes Obes. Metab. 2013, 15 (Suppl. 3), 152–158. [Google Scholar] [CrossRef] [PubMed]
- Hasnain, S.Z.; Prins, J.B.; McGuckin, M.A. Oxidative and endoplasmic reticulum stress in β-cell dysfunction in diabetes. J. Mol. Endocrinol. 2016, 56, R33–R54. [Google Scholar] [CrossRef] [Green Version]
- Ogihara, T.; Mirmira, R.G. An islet in distress: β cell failure in type 2 diabetes. J. Diabetes Investig. 2010, 1, 123–133. [Google Scholar] [CrossRef] [Green Version]
- Cardozo, A.K.; Ortis, F.; Storling, J.; Feng, Y.M.; Rasschaert, J.; Tonnesen, M.; van Eylen, F.; Mandrup-Poulsen, T.; Herchuelz, A.; Eizirik, D.L. Cytokines downregulate the sarcoendoplasmic reticulum pump Ca2+ ATPase 2b and deplete endoplasmic reticulum Ca2+, leading to induction of endoplasmic reticulum stress in pancreatic beta-cells. Diabetes 2005, 54, 452–461. [Google Scholar] [CrossRef] [Green Version]
- Lenzen, S.; Drinkgern, J.; Tiedge, M. Low antioxidant enzyme gene expression in pancreatic islets compared with various other mouse tissues. Free Radic. Biol. Med. 1996, 20, 463–466. [Google Scholar] [CrossRef]
- Gurgul-Convey, E.; Mehmeti, I.; Plötz, T.; Jörns, A.; Lenzen, S. Sensitivity profile of the human EndoC-βH1 beta cell line to proinflammatory cytokines. Diabetologia 2016, 59, 2125–2133. [Google Scholar] [CrossRef] [Green Version]
- Miki, A.; Ricordi, C.; Sakuma, Y.; Yamamoto, T.; Misawa, R.; Mita, A.; Molano, R.D.; Vaziri, N.D.; Pileggi, A.; Ichii, H. Divergent antioxidant capacity of human islet cell subsets: A potential cause of beta-cell vulnerability in diabetes and islet transplantation. PLoS ONE 2018, 13, e0196570. [Google Scholar] [CrossRef] [Green Version]
- Gurgul-Convey, E.; Mehmeti, I.; Lortz, S.; Lenzen, S. Cytokine toxicity in insulin-producing cells is mediated by nitro-oxidative stress-induced hydroxyl radical formation in mitochondria. J. Mol. Med. 2011, 89, 785–798. [Google Scholar] [CrossRef]
- Lenzen, S. Chemistry and biology of reactive species with special reference to the antioxidative defence status in pancreatic β-cells. Biochim. Biophys. Acta Gen. Subj. 2017, 1861, 1929–1942. [Google Scholar] [CrossRef]
- Brozzi, F.; Nardelli, T.R.; Lopes, M.; Millard, I.; Barthson, J.; Igoillo-Esteve, M.; Grieco, F.A.; Villate, O.; Oliveira, J.M.; Casimir, M.; et al. Cytokines induce endoplasmic reticulum stress in human, rat and mouse beta cells via different mechanisms. Diabetologia 2015, 58, 2307–2316. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Eizirik, D.L.; Sandler, S.; Welsh, N.; Cetkovic-Cvrlje, M.; Nieman, A.; Geller, D.A.; Pipeleers, D.G.; Bendtzen, K.; Hellerström, C. Cytokines suppress human islet function irrespective of their effects on nitric oxide generation. J. Clin. Investig. 1994, 93, 1968–1974. [Google Scholar] [CrossRef] [Green Version]
- Welsh, N.; Eizirik, D.L.; Sandler, S. Nitric oxide and pancreatic beta-cell destruction in insulin dependent diabetes mellitus: Don’t take NO for an answer. Autoimmunity 1994, 18, 285–290. [Google Scholar] [CrossRef] [PubMed]
- Acharya, J.D.; Ghaskadbi, S.S. Islets and their antioxidant defense. Islets 2010, 2, 225–235. [Google Scholar] [CrossRef]
- Newsholme, P.; Morgan, D.; Rebelato, E.; Oliveira-Emilio, H.C.; Procopio, J.; Curi, R.; Carpinelli, A. Insights into the critical role of NADPH oxidase(s) in the normal and dysregulated pancreatic beta cell. Diabetologia 2009, 52, 2489–2498. [Google Scholar] [CrossRef] [Green Version]
- Kowluru, A.; Kowluru, R.A. Phagocyte-like NADPH oxidase [Nox2] in cellular dysfunction in models of glucolipotoxicity and diabetes. Biochem. Pharmacol. 2014, 88, 275–283. [Google Scholar] [CrossRef]
- Buvelot, H.; Jaquet, V.; Krause, K.H. Mammalian NADPH Oxidases. Methods Mol. Biol. 2019, 1982, 17–36. [Google Scholar] [CrossRef]
- Laddha, A.P.; Kulkarni, Y.A. NADPH oxidase: A membrane-bound enzyme and its inhibitors in diabetic complications. Eur. J. Pharmacol. 2020, 881, 173206. [Google Scholar] [CrossRef]
- Schröder, K. NADPH oxidases: Current aspects and tools. Redox Biol. 2020, 34, 101512. [Google Scholar] [CrossRef]
- Guichard, C.; Moreau, R.; Pessayre, D.; Epperson, T.K.; Krause, K.H. NOX family NADPH oxidases in liver and in pancreatic islets: A role in the metabolic syndrome and diabetes? Biochem. Soc. Trans. 2008, 36, 920–929. [Google Scholar] [CrossRef] [Green Version]
- Grandvaux, N.; Soucy-Faulkner, A.; Fink, K. Innate host defense: Nox and Duox on phox’s tail. Biochimie 2007, 89, 1113–1122. [Google Scholar] [CrossRef]
- Bedard, K.; Jaquet, V.; Krause, K.H. NOX5: From basic biology to signaling and disease. Free Radic. Biol. Med. 2012, 52, 725–734. [Google Scholar] [CrossRef]
- Leto, T.L.; Morand, S.; Hurt, D.; Ueyama, T. Targeting and regulation of reactive oxygen species generation by Nox family NADPH oxidases. Antioxid. Redox Signal. 2009, 11, 2607–2619. [Google Scholar] [CrossRef]
- Kato, K.; Hecker, L. NADPH oxidases: Pathophysiology and therapeutic potential in age-associated pulmonary fibrosis. Redox Biol. 2020, 33, 101541. [Google Scholar] [CrossRef]
- Vignais, P.V. The superoxide-generating NADPH oxidase: Structural aspects and activation mechanism. Cell. Mol. Life Sci. 2002, 59, 1428–1459. [Google Scholar] [CrossRef]
- Leusen, J.H.; Verhoeven, A.J.; Roos, D. Interactions between the components of the human NADPH oxidase: A review about the intrigues in the phox family. Front. Biosci. 1996, 1, d72–d90. [Google Scholar] [CrossRef] [Green Version]
- Weaver, J.R.; Holman, T.R.; Imai, Y.; Jadhav, A.; Kenyon, V.; Maloney, D.J.; Nadler, J.L.; Rai, G.; Simeonov, A.; Taylor-Fishwick, D.A. Integration of pro-inflammatory cytokines, 12-lipoxygenase and NOX-1 in pancreatic islet beta cell dysfunction. Mol. Cell. Endocrinol. 2012, 358, 88–95. [Google Scholar] [CrossRef]
- Weaver, J.R.; Grzesik, W.; Taylor-Fishwick, D.A. Inhibition of NADPH oxidase-1 preserves beta cell function. Diabetologia 2015, 58, 113–121. [Google Scholar] [CrossRef]
- Imai, Y.; Dobrian, A.D.; Weaver, J.R.; Butcher, M.J.; Cole, B.K.; Galkina, E.V.; Morris, M.A.; Taylor-Fishwick, D.A.; Nadler, J.L. Interaction between cytokines and inflammatory cells in islet dysfunction, insulin resistance and vascular disease. Diabetes Obes. Metab. 2013, 15 (Suppl. 3), 117–129. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Rebelato, E.; Mares-Guia, T.R.; Graciano, M.F.; Labriola, L.; Britto, L.R.; Garay-Malpartida, H.M.; Curi, R.; Sogayar, M.C.; Carpinelli, A.R. Expression of NADPH oxidase in human pancreatic islets. Life Sci. 2012, 91, 244–249. [Google Scholar] [CrossRef]
- Wang, X.; Elksnis, A.; Wikström, P.; Walum, E.; Welsh, N.; Carlsson, P.O. The novel NADPH oxidase 4 selective inhibitor GLX7013114 counteracts human islet cell death in vitro. PLoS ONE 2018, 13, e0204271. [Google Scholar] [CrossRef] [PubMed]
- Syed, I.; Kyathanahalli, C.N.; Jayaram, B.; Govind, S.; Rhodes, C.J.; Kowluru, R.A.; Kowluru, A. Increased phagocyte-like NADPH oxidase and ROS generation in type 2 diabetic ZDF rat and human islets: Role of Rac1-JNK1/2 signaling pathway in mitochondrial dysregulation in the diabetic islet. Diabetes 2011, 60, 2843–2852. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Li, N.; Li, B.; Brun, T.; Deffert-Delbouille, C.; Mahiout, Z.; Daali, Y.; Ma, X.J.; Krause, K.H.; Maechler, P. NADPH oxidase NOX2 defines a new antagonistic role for reactive oxygen species and cAMP/PKA in the regulation of insulin secretion. Diabetes 2012, 61, 2842–2850. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Anvari, E.; Wikström, P.; Walum, E.; Welsh, N. The novel NADPH oxidase 4 inhibitor GLX351322 counteracts glucose intolerance in high-fat diet-treated C57BL/6 mice. Free Radic. Res. 2015, 49, 1308–1318. [Google Scholar] [CrossRef] [Green Version]
- Bouzakri, K.; Veyrat-Durebex, C.; Holterman, C.; Arous, C.; Barbieux, C.; Bosco, D.; Altirriba, J.; Alibashe, M.; Tournier, B.B.; Gunton, J.E.; et al. Beta-cell-specific expression of nicotinamide adenine dinucleotide phosphate oxidase 5 aggravates high-fat diet-induced impairment of islet insulin secretion in mice. Antioxid. Redox Signal. 2020, 32, 618–635. [Google Scholar] [CrossRef]
- Uchizono, Y.; Takeya, R.; Iwase, M.; Sasaki, N.; Oku, M.; Imoto, H.; Iida, M.; Sumimoto, H. Expression of isoforms of NADPH oxidase components in rat pancreatic islets. Life Sci. 2006, 80, 133–139. [Google Scholar] [CrossRef]
- Oliveira, H.R.; Verlengia, R.; Carvalho, C.R.; Britto, L.R.; Curi, R.; Carpinelli, A.R. Pancreatic beta-cells express phagocyte-like NAD(P)H oxidase. Diabetes 2003, 52, 1457–1463. [Google Scholar] [CrossRef] [Green Version]
- Weaver, J.; Taylor-Fishwick, D.A. Relationship of NADPH Oxidase-1 expression to beta cell dysfunction induced by inflammatory cytokines. Biochem. Biophys. Res. Commun. 2017, 485, 290–294. [Google Scholar] [CrossRef]
- Bleich, D.; Chen, S.; Gu, J.L.; Nadler, J.L. The role of 12-lipoxygenase in pancreatic -cells (Review). Int. J. Mol. Med. 1998, 1, 265–272. [Google Scholar] [CrossRef]
- Prasad, K.M.; Thimmalapura, P.R.; Woode, E.A.; Nadler, J.L. Evidence that increased 12-lipoxygenase expression impairs pancreatic beta cell function and viability. Biochem. Biophys. Res. Commun. 2003, 308, 427–432. [Google Scholar] [CrossRef]
- Kowluru, A. GPCRs, G Proteins, and their impact on β-cell function. Compr. Physiol. 2020, 10, 453–490. [Google Scholar] [CrossRef] [PubMed]
- Michalska, M.; Wolf, G.; Walther, R.; Newsholme, P. Effects of pharmacological inhibition of NADPH oxidase or iNOS on pro-inflammatory cytokine, palmitic acid or H2O2-induced mouse islet or clonal pancreatic β-cell dysfunction. Biosci. Rep. 2010, 30, 445–453. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Subasinghe, W.; Syed, I.; Kowluru, A. Phagocyte-like NADPH oxidase promotes cytokine-induced mitochondrial dysfunction in pancreatic β-cells: Evidence for regulation by Rac1. Am. J. Physiol. Regul. Integr. Comp. Physiol. 2011, 300, R12–R20. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Mohammed, A.M.; Syeda, K.; Hadden, T.; Kowluru, A. Upregulation of phagocyte-like NADPH oxidase by cytokines in pancreatic beta-cells: Attenuation of oxidative and nitrosative stress by 2-bromopalmitate. Biochem. Pharmacol. 2013, 85, 109–114. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Belambri, S.A.; Rolas, L.; Raad, H.; Hurtado-Nedelec, M.; Dang, P.M.; El-Benna, J. NADPH oxidase activation in neutrophils: Role of the phosphorylation of its subunits. Eur. J. Clin. Investig. 2018, 48 (Suppl. 2), e12951. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- El-Benna, J.; Dang, P.M.; Gougerot-Pocidalo, M.A.; Marie, J.C.; Braut-Boucher, F. p47phox, the phagocyte NADPH oxidase/NOX2 organizer: Structure, phosphorylation and implication in diseases. Exp. Mol. Med. 2009, 41, 217–225. [Google Scholar] [CrossRef] [Green Version]
- Rastogi, R.; Geng, X.; Li, F.; Ding, Y. NOX activation by subunit interaction and underlying mechanisms in disease. Front. Cell. Neurosci. 2016, 10, 301. [Google Scholar] [CrossRef] [Green Version]
- Morgan, D.; Oliveira-Emilio, H.R.; Keane, D.; Hirata, A.E.; Santos da Rocha, M.; Bordin, S.; Curi, R.; Newsholme, P.; Carpinelli, A.R. Glucose, palmitate and pro-inflammatory cytokines modulate production and activity of a phagocyte-like NADPH oxidase in rat pancreatic islets and a clonal beta cell line. Diabetologia 2007, 50, 359–369. [Google Scholar] [CrossRef] [Green Version]
- Xiang, F.L.; Lu, X.; Strutt, B.; Hill, D.J.; Feng, Q. NOX2 deficiency protects against streptozotocin-induced beta-cell destruction and development of diabetes in mice. Diabetes 2010, 59, 2603–2611. [Google Scholar] [CrossRef] [Green Version]
- Veluthakal, R.; Sidarala, V.; Kowluru, A. NSC23766, a known inhibitor of Tiam1-Rac1 signaling module, prevents the onset of type 1 diabetes in the NOD mouse model. Cell. Physiol. Biochem. 2016, 39, 760–767. [Google Scholar] [CrossRef] [Green Version]
- Pacher, P.; Beckman, J.S.; Liaudet, L. Nitric oxide and peroxynitrite in health and disease. Physiol. Rev. 2007, 87, 315–424. [Google Scholar] [CrossRef] [Green Version]
- Ghasemi, M.; Mayasi, Y.; Hannoun, A.; Eslami, S.M.; Carandang, R. Nitric oxide and mitochondrial function in neurological diseases. Neuroscience 2018, 376, 48–71. [Google Scholar] [CrossRef] [PubMed]
- Islam, B.U.; Habib, S.; Ahmad, P.; Allarakha, S.; Moinuddin; Ali, A. Pathophysiological role of peroxynitrite induced DNA damage in human diseases: A special focus on poly(ADP-ribose) polymerase (PARP). Indian J. Clin. Biochem. 2015, 30, 368–385. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Kanaan, G.N.; Harper, M.E. Cellular redox dysfunction in the development of cardiovascular diseases. Biochim. Biophys. Acta Gen. Subj. 2017, 1861, 2822–2829. [Google Scholar] [CrossRef]
- Poitout, V.; Robertson, R.P. Glucolipotoxicity: Fuel excess and beta-cell dysfunction. Endocr. Rev. 2008, 29, 351–366. [Google Scholar] [CrossRef]
- Delaney, C.A.; Tyrberg, B.; Bouwens, L.; Vaghef, H.; Hellman, B.; Eizirik, D.L. Sensitivity of human pancreatic islets to peroxynitrite-induced cell dysfunction and death. FEBS Lett. 1996, 394, 300–306. [Google Scholar] [CrossRef] [Green Version]
- Lakey, J.R.; Suarez-Pinzon, W.L.; Strynadka, K.; Korbutt, G.S.; Rajotte, R.V.; Mabley, J.G.; Szabó, C.; Rabinovitch, A. Peroxynitrite is a mediator of cytokine-induced destruction of human pancreatic islet beta cells. Lab. Investig. 2001, 81, 1683–1692. [Google Scholar] [CrossRef]
- Suarez-Pinzon, W.L.; Mabley, J.G.; Strynadka, K.; Power, R.F.; Szabó, C.; Rabinovitch, A. An inhibitor of inducible nitric oxide synthase and scavenger of peroxynitrite prevents diabetes development in NOD mice. J. Autoimmun. 2001, 16, 449–455. [Google Scholar] [CrossRef]
- Mabley, J.G.; Southan, G.J.; Salzman, A.L.; Szabó, C. The combined inducible nitric oxide synthase inhibitor and free radical scavenger guanidinoethyldisulfide prevents multiple low-dose streptozotocin-induced diabetes in vivo and interleukin-1beta-induced suppression of islet insulin secretion in vitro. Pancreas 2004, 28, E39–E44. [Google Scholar] [CrossRef]
- Stancill, J.S.; Happ, J.T.; Broniowska, K.A.; Hogg, N.; Corbett, J.A. Peroxiredoxin 1 plays a primary role in protecting pancreatic β-cells from hydrogen peroxide and peroxynitrite. Am. J. Physiol. Regul. Integr. Comp. Physiol. 2020, 318, R1004–R1013. [Google Scholar] [CrossRef]
- Tran, P.O.; Parker, S.M.; LeRoy, E.; Franklin, C.C.; Kavanagh, T.J.; Zhang, T.; Zhou, H.; Vliet, P.; Oseid, E.; Harmon, J.S.; et al. Adenoviral overexpression of the glutamylcysteine ligase catalytic subunit protects pancreatic islets against oxidative stress. J. Biol. Chem. 2004, 279, 53988–53993. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Gurgul, E.; Lortz, S.; Tiedge, M.; Jörns, A.; Lenzen, S. Mitochondrial catalase overexpression protects insulin-producing cells against toxicity of reactive oxygen species and proinflammatory cytokines. Diabetes 2004, 53, 2271–2280. [Google Scholar] [CrossRef] [Green Version]
- Lortz, S.; Gurgul-Convey, E.; Lenzen, S.; Tiedge, M. Importance of mitochondrial superoxide dismutase expression in insulin-producing cells for the toxicity of reactive oxygen species and proinflammatory cytokines. Diabetologia 2005, 48, 1541–1548. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Lortz, S.; Lenzen, S.; Mehmeti, I. Impact of scavenging hydrogen peroxide in the endoplasmic reticulum for β cell function. J. Mol. Endocrinol. 2015, 55, 21–29. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Mehmeti, I.; Lenzen, S.; Lortz, S. Modulation of Bcl-2-related protein expression in pancreatic beta cells by pro-inflammatory cytokines and its dependence on the antioxidative defense status. Mol. Cell. Endocrinol. 2011, 332, 88–96. [Google Scholar] [CrossRef] [Green Version]
- Wolf, G.; Aumann, N.; Michalska, M.; Bast, A.; Sonnemann, J.; Beck, J.F.; Lendeckel, U.; Newsholme, P.; Walther, R. Peroxiredoxin III protects pancreatic ß cells from apoptosis. J. Endocrinol. 2010, 207, 163–175. [Google Scholar] [CrossRef] [PubMed]
- Jain, D.; Weber, G.; Eberhard, D.; Mehana, A.E.; Eglinger, J.; Welters, A.; Bartosinska, B.; Jeruschke, K.; Weiss, J.; Päth, G.; et al. DJ-1 protects pancreatic beta cells from cytokine- and streptozotocin-mediated cell death. PLoS ONE 2015, 10, e0138535. [Google Scholar] [CrossRef]
- Eberhard, D.; Lammert, E. The role of the antioxidant protein DJ-1 in type 2 diabetes mellitus. Adv. Exp. Med. Biol. 2017, 1037, 173–186. [Google Scholar] [CrossRef]
- Chen, H.Q.; Tannous, M.; Veluthakal, R.; Amin, R.; Kowluru, A. Novel roles for palmitoylation of Ras in IL-1 beta-induced nitric oxide release and caspase 3 activation in insulin-secreting beta cells. Biochem. Pharmacol. 2003, 66, 1681–1694. [Google Scholar] [CrossRef]
- Tannous, M.; Amin, R.; Popoff, M.R.; Fiorentini, C.; Kowluru, A. Positive modulation by Ras of interleukin-1beta-mediated nitric oxide generation in insulin-secreting clonal beta (HIT-T15) cells. Biochem. Pharmacol. 2001, 62, 1459–1468. [Google Scholar] [CrossRef]
- Tannous, M.; Veluthakal, R.; Amin, R.; Kowluru, A. IL-1beta-induced nitric oxide release from insulin-secreting beta-cells: Further evidence for the involvement of GTP-binding proteins. Diabetes Metab. 2002, 28, 3S78–3S84, discussion 73S108–73S112. [Google Scholar] [PubMed]
- Sidarala, V.; Veluthakal, R.; Syeda, K.; Vlaar, C.; Newsholme, P.; Kowluru, A. Phagocyte-like NADPH oxidase (Nox2) promotes activation of p38MAPK in pancreatic β-cells under glucotoxic conditions: Evidence for a requisite role of Ras-related C3 botulinum toxin substrate 1 (Rac1). Biochem. Pharmacol. 2015, 95, 301–310. [Google Scholar] [CrossRef] [PubMed]
- Hernandez-Perez, M.; Chopra, G.; Fine, J.; Conteh, A.M.; Anderson, R.M.; Linnemann, A.K.; Benjamin, C.; Nelson, J.B.; Benninger, K.S.; Nadler, J.L.; et al. Inhibition of 12/15-Lipoxygenase protects against β-Cell oxidative stress and glycemic deterioration in mouse models of type 1 diabetes. Diabetes 2017, 66, 2875–2887. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Ma, K.; Xiao, A.; Park, S.H.; Glenn, L.; Jackson, L.; Barot, T.; Weaver, J.R.; Taylor-Fishwick, D.A.; Luci, D.K.; Maloney, D.J.; et al. 12-Lipoxygenase inhibitor improves functions of cytokine-treated human islets and type 2 diabetic islets. J. Clin. Endocrinol. Metab. 2017, 102, 2789–2797. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Jastroch, M. Unraveling the molecular machinery that promotes pancreatic β-cell dysfunction during oxidative stress: Focus on “Phagocyte-like NADPH oxidase promotes cytokine-induced mitochondrial dysfunction in pancreatic β-cells: Evidence for regulation by Rac1”. Am. J. Physiol. Regul. Integr. Comp. Physiol. 2011, 300, R9–R11. [Google Scholar] [CrossRef] [Green Version]




{kind=link}
{kind=link}
{kind=link}
| Inhibitor | Mechanism(s) of Action | Reference |
|---|---|---|
| GGTI-2147 | Inhibitor of prenylation of G proteins (Rac1) | [53] |
| NSC23766 | Inhibitor of Tiam1-Rac1 signaling pathway | [53] |
| Manumycin | Pan inhibitor of farnesylation of G proteins (Ras) | [79,80,81] |
| Damnacanthal | Pan inhibitor of farnesylation of G proteins (Ras) | [80,81] |
| Cerulenin | Inhibitor of palmitoylation of G proteins | [79] |
| 2-bromopalmitate | Inhibitor of palmitoylation of G proteins | [54,79] |
| DPI | Pan inhibitor of Noxs | [37,41,58] |
| Apocynin | Pan inhibitor of Noxs2 | [37,53,54] |
| Gp91ds-tat | Inhibitor of Nox2 | [82] |
| Guanidinoethylsulphide | Inhibitor of iNOS and scavenger of PN | [67,68] |
| GLX7013114 | Specific inhibitor of Nox4 | [41] |
| GLX351322 | Selective inhibitor of Nox4 (inhibits other Noxs) | [41] |
| GLX481372 | Selective inhibitor of Nox4 (inhibits other Noxs) | [41] |
| Dapsone | Inhibits expression/activity of Nox4 and DUOX1 | [41] |
| ML171 | Inhibitor of Nox1 | [38,41] |
| Phox-I2 | Inhibitor of Nox2 | [41] |
| ML351 | Inhibitor of 12/15-LO | [83] |
| NCTT-956 | Inhibitor of 12-LO | [37,84] |
| GF109203X | Inhibitor of PKC | [58] |
Publisher’s Note: MDPI stays neutral with regard to jurisdictional claims in published maps and institutional affiliations. |
© 2020 by the author. Licensee MDPI, Basel, Switzerland. This article is an open access article distributed under the terms and conditions of the Creative Commons Attribution (CC BY) license (http://creativecommons.org/licenses/by/4.0/).
Share and Cite
Kowluru, A. Oxidative Stress in Cytokine-Induced Dysfunction of the Pancreatic Beta Cell: Known Knowns and Known Unknowns. Metabolites 2020, 10, 480. https://doi.org/10.3390/metabo10120480
Kowluru A. Oxidative Stress in Cytokine-Induced Dysfunction of the Pancreatic Beta Cell: Known Knowns and Known Unknowns. Metabolites. 2020; 10(12):480. https://doi.org/10.3390/metabo10120480
Chicago/Turabian StyleKowluru, Anjaneyulu. 2020. "Oxidative Stress in Cytokine-Induced Dysfunction of the Pancreatic Beta Cell: Known Knowns and Known Unknowns" Metabolites 10, no. 12: 480. https://doi.org/10.3390/metabo10120480
APA StyleKowluru, A. (2020). Oxidative Stress in Cytokine-Induced Dysfunction of the Pancreatic Beta Cell: Known Knowns and Known Unknowns. Metabolites, 10(12), 480. https://doi.org/10.3390/metabo10120480