Linking Gut Microbiome and Lipid Metabolism: Moving beyond Associations


 ,
, 
 ,
,  and
and 
Abstract
:1. Lipids and Gut Microbes
2. Lipid Pool of the Human Gut
3. Synthesis of Lipids by Gut Microbes
3.1. Sphingolipids
3.2. Sterol
3.2.1. Bile Acids and Derivatives
3.2.2. Cholesterol
3.3. Fatty Acyls and Conjugates
3.4. Endocannabinoids
3.5. Carnitine and Acyl Carnitines
4. Functional Profiling and Metabolic Modeling of Human Gut Microbiome for Understanding Microbial-Host-Lipid Co-Metabolism
5. Conclusions and Future Perspectives
Author Contributions
Funding
Acknowledgments
Conflicts of Interest
References
- Tran, T.T.; Postal, B.G.; Demignot, S.; Ribeiro, A.; Osinski, C.; Pais de Barros, J.P.; Blachnio-Zabielska, A.; Leturque, A.; Rousset, M.; Ferre, P.; et al. Short Term Palmitate Supply Impairs Intestinal Insulin Signaling via Ceramide Production. J. Biol. Chem. 2016, 291, 16328–16338. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Iqbal, J.; Hussain, M.M. Intestinal lipid absorption. Am. J. Physiol. Endocrinol. Metab. 2009, 296, E1183–E1194. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Wei, F.; Lamichhane, S.; Orešič, M.; Hyötyläinen, T. Lipidomes in health and disease: Analytical strategies and considerations. TrAC Trends Anal. Chem. 2019, 120, 115664. [Google Scholar] [CrossRef]
- VanHook, A.M. Microbial metabolites shape lipid metabolism. Sci. Signal. 2020, 13, eabc1552. [Google Scholar] [CrossRef]
- Visconti, A.; Le Roy, C.I.; Rosa, F.; Rossi, N.; Martin, T.C.; Mohney, R.P.; Li, W.; de Rinaldis, E.; Bell, J.T.; Venter, J.C.; et al. Interplay between the human gut microbiome and host metabolism. Nat. Commun. 2019, 10, 4505. [Google Scholar] [CrossRef] [Green Version]
- Bar, N.; Korem, T.; Weissbrod, O.; Zeevi, D.; Rothschild, D.; Leviatan, S.; Kosower, N.; Lotan-Pompan, M.; Weinberger, A.; Le Roy, C.I.; et al. A reference map of potential determinants for the human serum metabolome. Nature 2020. [Google Scholar] [CrossRef]
- Kostic, A.D.; Gevers, D.; Siljander, H.; Vatanen, T.; Hyötyläinen, T.; Hämäläinen, A.-M.; Peet, A.; Tillmann, V.; Pöhö, P.; Mattila, I. The dynamics of the human infant gut microbiome in development and in progression toward type 1 diabetes. Cell Host Microbe 2015, 17, 260–273. [Google Scholar] [CrossRef] [Green Version]
- Pedersen, H.K.; Gudmundsdottir, V.; Nielsen, H.B.; Hyotylainen, T.; Nielsen, T.; Jensen, B.A.; Forslund, K.; Hildebrand, F.; Prifti, E.; Falony, G.; et al. Human gut microbes impact host serum metabolome and insulin sensitivity. Nature 2016, 535, 376–381. [Google Scholar] [CrossRef]
- Fu, J.; Bonder, M.J.; Cenit, M.C.; Tigchelaar, E.F.; Maatman, A.; Dekens, J.A.; Brandsma, E.; Marczynska, J.; Imhann, F.; Weersma, R.K.; et al. The Gut Microbiome Contributes to a Substantial Proportion of the Variation in Blood Lipids. Circ. Res. 2015, 117, 817–824. [Google Scholar] [CrossRef]
- Liu, H.; Pan, L.L.; Lv, S.; Yang, Q.; Zhang, H.; Chen, W.; Lv, Z.; Sun, J. Alterations of Gut Microbiota and Blood Lipidome in Gestational Diabetes Mellitus With Hyperlipidemia. Front. Physiol. 2019, 10, 1015. [Google Scholar] [CrossRef] [Green Version]
- Benítez-Páez, A.; Kjølbæk, L.; Gómez Del Pulgar, E.M.; Brahe, L.K.; Astrup, A.; Matysik, S.; Schött, H.F.; Krautbauer, S.; Liebisch, G.; Boberska, J.; et al. A Multi-omics Approach to Unraveling the Microbiome-Mediated Effects of Arabinoxylan Oligosaccharides in Overweight Humans. mSystems 2019, 4. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Albouery, M.; Buteau, B.; Gregoire, S.; Cherbuy, C.; Pais de Barros, J.P.; Martine, L.; Chain, F.; Cabaret, S.; Berdeaux, O.; Bron, A.M.; et al. Age-Related Changes in the Gut Microbiota Modify Brain Lipid Composition. Front. Cell. Infect. Microbiol. 2019, 9, 444. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Just, S.; Mondot, S.; Ecker, J.; Wegner, K.; Rath, E.; Gau, L.; Streidl, T.; Hery-Arnaud, G.; Schmidt, S.; Lesker, T.R.; et al. The gut microbiota drives the impact of bile acids and fat source in diet on mouse metabolism. Microbiome 2018, 6, 134. [Google Scholar] [CrossRef] [PubMed]
- Lamichhane, S.; Sen, P.; Dickens, A.M.; Orešič, M.; Bertram, H.C. Gut metabolome meets microbiome: A methodological perspective to understand the relationship between host and microbe. Methods 2018, 149, 3–12. [Google Scholar] [CrossRef]
- Karu, N.; Deng, L.; Slae, M.; Guo, A.C.; Sajed, T.; Huynh, H.; Wine, E.; Wishart, D.S. A review on human fecal metabolomics: Methods, applications and the human fecal metabolome database. Anal. Chim. Acta 2018, 1030, 1–24. [Google Scholar] [CrossRef]
- Gregory, K.E.; Bird, S.S.; Gross, V.S.; Marur, V.R.; Lazarev, A.V.; Walker, W.A.; Kristal, B.S. Method development for fecal lipidomics profiling. Anal. Chem. 2013, 85, 1114–1123. [Google Scholar] [CrossRef] [Green Version]
- Van Meulebroek, L.; De Paepe, E.; Vercruysse, V.; Pomian, B.; Bos, S.; Lapauw, B.; Vanhaecke, L. Holistic Lipidomics of the Human Gut Phenotype Using Validated Ultra-High-Performance Liquid Chromatography Coupled to Hybrid Orbitrap Mass Spectrometry. Anal. Chem. 2017, 89, 12502–12510. [Google Scholar] [CrossRef] [Green Version]
- Trost, K.; Ahonen, L.; Suvitaival, T.; Christiansen, N.; Nielsen, T.; Thiele, M.; Jacobsen, S.; Krag, A.; Rossing, P.; Hansen, T.; et al. Describing the fecal metabolome in cryogenically collected samples from healthy participants. Sci. Rep. 2020, 10, 885. [Google Scholar] [CrossRef] [Green Version]
- Bowden, J.A.; Heckert, A.; Ulmer, C.Z.; Jones, C.M.; Koelmel, J.P.; Abdullah, L.; Ahonen, L.; Alnouti, Y.; Armando, A.M.; Asara, J.M.; et al. Harmonizing lipidomics: NIST interlaboratory comparison exercise for lipidomics using SRM 1950-Metabolites in Frozen Human Plasma. J. Lipid Res. 2017, 58, 2275–2288. [Google Scholar] [CrossRef] [Green Version]
- Lamichhane, S.; Sen, P.; Dickens, A.M.; Hyötyläinen, T.; Orešič, M. An overview of metabolomics data analysis: Current tools and future perspectives. In Comprehensive Analytical Chemistry; Elsevier: Amsterdam, The Netherlands, 2018; Volume 82, pp. 387–2413. [Google Scholar]
- Zullig, T.; Trotzmuller, M.; Kofeler, H.C. Lipidomics from sample preparation to data analysis: A primer. Anal. Bioanal. Chem. 2020, 412, 2191–2209. [Google Scholar] [CrossRef] [Green Version]
- O’Shea, K.; Misra, B.B. Software tools, databases and resources in metabolomics: Updates from 2018 to 2019. Metabolomics 2020, 16, 36. [Google Scholar] [CrossRef] [PubMed]
- Gao, X.; Pujos-Guillot, E.; Sebedio, J.L. Development of a quantitative metabolomic approach to study clinical human fecal water metabolome based on trimethylsilylation derivatization and GC/MS analysis. Anal. Chem. 2010, 82, 6447–6456. [Google Scholar] [CrossRef] [PubMed]
- Phua, L.C.; Koh, P.K.; Cheah, P.Y.; Ho, H.K.; Chan, E.C. Global gas chromatography/time-of-flight mass spectrometry (GC/TOFMS)-based metabonomic profiling of lyophilized human feces. J. Chromatogr. B Anal. Technol. Biomed. Life Sci. 2013, 937, 103–113. [Google Scholar] [CrossRef] [PubMed]
- Kind, T.; Wohlgemuth, G.; Lee, D.Y.; Lu, Y.; Palazoglu, M.; Shahbaz, S.; Fiehn, O. FiehnLib: Mass spectral and retention index libraries for metabolomics based on quadrupole and time-of-flight gas chromatography/mass spectrometry. Anal. Chem. 2009, 81, 10038–10048. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Kopka, J.; Schauer, N.; Krueger, S.; Birkemeyer, C.; Usadel, B.; Bergmuller, E.; Dormann, P.; Weckwerth, W.; Gibon, Y.; Stitt, M.; et al. [email protected]: The Golm Metabolome Database. Bioinformatics 2005, 21, 1635–1638. [Google Scholar] [CrossRef] [Green Version]
- Wishart, D.S.; Feunang, Y.D.; Marcu, A.; Guo, A.C.; Liang, K.; Vazquez-Fresno, R.; Sajed, T.; Johnson, D.; Li, C.; Karu, N.; et al. HMDB 4.0: The human metabolome database for 2018. Nucleic Acids Res. 2018, 46, D608–D617. [Google Scholar] [CrossRef]
- Fahy, E.; Sud, M.; Cotter, D.; Subramaniam, S. LIPID MAPS online tools for lipid research. Nucleic Acids Res. 2007, 35, W606–W612. [Google Scholar] [CrossRef] [Green Version]
- Guijas, C.; Montenegro-Burke, J.R.; Domingo-Almenara, X.; Palermo, A.; Warth, B.; Hermann, G.; Koellensperger, G.; Huan, T.; Uritboonthai, W.; Aisporna, A.E.; et al. METLIN: A Technology Platform for Identifying Knowns and Unknowns. Anal. Chem. 2018, 90, 3156–3164. [Google Scholar] [CrossRef] [Green Version]
- Gil-de-la-Fuente, A.; Godzien, J.; Saugar, S.; Garcia-Carmona, R.; Badran, H.; Wishart, D.S.; Barbas, C.; Otero, A. CEU Mass Mediator 3.0: A Metabolite Annotation Tool. J. Proteome Res. 2019, 18, 797–802. [Google Scholar] [CrossRef]
- Zierer, J.; Jackson, M.A.; Kastenmüller, G.; Mangino, M.; Long, T.; Telenti, A.; Mohney, R.P.; Small, K.S.; Bell, J.T.; Steves, C.J.; et al. The fecal metabolome as a functional readout of the gut microbiome. Nat. Genet. 2018, 50, 790–795. [Google Scholar] [CrossRef]
- Bradley, P.H.; Pollard, K.S. Building a chemical blueprint for human blood. Nature 2020, 588, 36–37. [Google Scholar] [CrossRef] [PubMed]
- Brown, E.M.; Ke, X.; Hitchcock, D.; Jeanfavre, S.; Avila-Pacheco, J.; Nakata, T.; Arthur, T.D.; Fornelos, N.; Heim, C.; Franzosa, E.A.; et al. Bacteroides-Derived Sphingolipids Are Critical for Maintaining Intestinal Homeostasis and Symbiosis. Cell Host Microbe 2019, 25, 668–680. [Google Scholar] [CrossRef] [PubMed]
- Oliphant, K.; Allen-Vercoe, E. Macronutrient metabolism by the human gut microbiome: Major fermentation by-products and their impact on host health. Microbiome 2019, 7, 91. [Google Scholar] [CrossRef] [PubMed]
- Cani, P.D.; Van Hul, M.; Lefort, C.; Depommier, C.; Rastelli, M.; Everard, A. Microbial regulation of organismal energy homeostasis. Nat. Metab. 2019, 1, 34–46. [Google Scholar] [CrossRef] [PubMed]
- Hannun, Y.A.; Obeid, L.M. Author Correction: Sphingolipids and their metabolism in physiology and disease. Nat. Rev. Mol. Cell Biol. 2018, 19, 673. [Google Scholar] [CrossRef] [Green Version]
- Kolter, T. A view on sphingolipids and disease. Chem. Phys. Lipids 2011, 164, 590–606. [Google Scholar] [CrossRef]
- Heaver, S.L.; Johnson, E.L.; Ley, R.E. Sphingolipids in host-microbial interactions. Curr. Opin. Microbiol. 2018, 43, 92–99. [Google Scholar] [CrossRef]
- Johnson, E.L.; Heaver, S.L.; Waters, J.L.; Kim, B.I.; Bretin, A.; Goodman, A.L.; Gewirtz, A.T.; Worgall, T.S.; Ley, R.E. Sphingolipids produced by gut bacteria enter host metabolic pathways impacting ceramide levels. Nat. Commun. 2020, 11, 2471. [Google Scholar] [CrossRef]
- Lee, M.T.; Le, H.H.; Johnson, E.L. Dietary sphinganine is selectively assimilated by members of the mammalian gut microbiome. J. Lipid Res. 2020. [Google Scholar] [CrossRef]
- Devlin, A.S.; Fischbach, M.A. A biosynthetic pathway for a prominent class of microbiota-derived bile acids. Nat. Chem. Biol. 2015, 11, 685–690. [Google Scholar] [CrossRef] [Green Version]
- Ridlon, J.M.; Kang, D.J.; Hylemon, P.B. Bile salt biotransformations by human intestinal bacteria. J. Lipid Res. 2006, 47, 241–259. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Fiorucci, S.; Biagioli, M.; Zampella, A.; Distrutti, E. Bile Acids Activated Receptors Regulate Innate Immunity. Front. Immunol. 2018, 9, 1853. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Ramírez-Pérez, O.; Cruz-Ramón, V.; Chinchilla-López, P.; Méndez-Sánchez, N. The Role of the Gut Microbiota in Bile Acid Metabolism. Ann. Hepatol. 2017, 16, s15–s20. [Google Scholar] [CrossRef] [PubMed]
- Ridlon, J.M.; Kang, D.J.; Hylemon, P.B.; Bajaj, J.S. Bile acids and the gut microbiome. Curr. Opin. Gastroenterol. 2014, 30, 332–338. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Jia, X.; Lu, S.; Zeng, Z.; Liu, Q.; Dong, Z.; Chen, Y.; Zhu, Z.; Hong, Z.; Zhang, T.; Du, G.; et al. Characterization of Gut Microbiota, Bile Acid Metabolism, and Cytokines in Intrahepatic Cholangiocarcinoma. Hepatology 2020, 71, 893–906. [Google Scholar] [CrossRef]
- Humbert, L.; Maubert, M.A.; Wolf, C.; Duboc, H.; Mahé, M.; Farabos, D.; Seksik, P.; Mallet, J.M.; Trugnan, G.; Masliah, J.; et al. Bile acid profiling in human biological samples: Comparison of extraction procedures and application to normal and cholestatic patients. J. Chromatogr. B Anal. Technol. Biomed. Life Sci. 2012, 899, 135–145. [Google Scholar] [CrossRef]
- Winston, J.A.; Theriot, C.M. Diversification of host bile acids by members of the gut microbiota. Gut Microbes 2020, 11, 158–171. [Google Scholar] [CrossRef]
- Fukiya, S.; Arata, M.; Kawashima, H.; Yoshida, D.; Kaneko, M.; Minamida, K.; Watanabe, J.; Ogura, Y.; Uchida, K.; Itoh, K.; et al. Conversion of cholic acid and chenodeoxycholic acid into their 7-oxo derivatives by Bacteroides intestinalis AM-1 isolated from human feces. FEMS Microbiol. Lett. 2009, 293, 263–270. [Google Scholar] [CrossRef] [Green Version]
- Ridlon, J.M.; Bajaj, J.S. The human gut sterolbiome: Bile acid-microbiome endocrine aspects and therapeutics. Acta Pharm. Sin. B 2015, 5, 99–105. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Le Roy, T.; Lécuyer, E.; Chassaing, B.; Rhimi, M.; Lhomme, M.; Boudebbouze, S.; Ichou, F.; Haro Barceló, J.; Huby, T.; Guerin, M.; et al. The intestinal microbiota regulates host cholesterol homeostasis. BMC Biol. 2019, 17, 94. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Kenny, D.J.; Plichta, D.R.; Shungin, D.; Koppel, N.; Hall, A.B.; Fu, B.; Vasan, R.S.; Shaw, S.Y.; Vlamakis, H.; Balskus, E.P.; et al. Cholesterol Metabolism by Uncultured Human Gut Bacteria Influences Host Cholesterol Level. Cell Host Microbe 2020, 28, 245–257. [Google Scholar] [CrossRef] [PubMed]
- Kishino, S.; Takeuchi, M.; Park, S.B.; Hirata, A.; Kitamura, N.; Kunisawa, J.; Kiyono, H.; Iwamoto, R.; Isobe, Y.; Arita, M.; et al. Polyunsaturated fatty acid saturation by gut lactic acid bacteria affecting host lipid composition. Proc. Natl. Acad. Sci. USA 2013, 110, 17808–17813. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Nanthirudjanar, T.; Furumoto, H.; Zheng, J.; Kim, Y.I.; Goto, T.; Takahashi, N.; Kawada, T.; Park, S.B.; Hirata, A.; Kitamura, N.; et al. Gut Microbial Fatty Acid Metabolites Reduce Triacylglycerol Levels in Hepatocytes. Lipids 2015, 50, 1093–1102. [Google Scholar] [CrossRef] [PubMed]
- Miyamoto, J.; Igarashi, M.; Watanabe, K.; Karaki, S.I.; Mukouyama, H.; Kishino, S.; Li, X.; Ichimura, A.; Irie, J.; Sugimoto, Y.; et al. Gut microbiota confers host resistance to obesity by metabolizing dietary polyunsaturated fatty acids. Nat. Commun. 2019, 10, 4007. [Google Scholar] [CrossRef] [Green Version]
- Druart, C.; Bindels, L.B.; Schmaltz, R.; Neyrinck, A.M.; Cani, P.D.; Walter, J.; Ramer-Tait, A.E.; Delzenne, N.M. Ability of the gut microbiota to produce PUFA-derived bacterial metabolites: Proof of concept in germ-free versus conventionalized mice. Mol. Nutr. Food Res. 2015, 59, 1603–1613. [Google Scholar] [CrossRef]
- Druart, C.; Dewulf, E.M.; Cani, P.D.; Neyrinck, A.M.; Thissen, J.P.; Delzenne, N.M. Gut microbial metabolites of polyunsaturated fatty acids correlate with specific fecal bacteria and serum markers of metabolic syndrome in obese women. Lipids 2014, 49, 397–402. [Google Scholar] [CrossRef]
- Cani, P.D.; Plovier, H.; Van Hul, M.; Geurts, L.; Delzenne, N.M.; Druart, C.; Everard, A. Endocannabinoids--at the crossroads between the gut microbiota and host metabolism. Nat. Rev. Endocrinol. 2016, 12, 133–143. [Google Scholar] [CrossRef]
- DiPatrizio, N.V. Endocannabinoids in the Gut. Cannabis Cannabinoid Res. 2016, 1, 67–77. [Google Scholar] [CrossRef]
- Forte, N.; Fernández-Rilo, A.C.; Palomba, L.; Di Marzo, V.; Cristino, L. Obesity Affects the Microbiota-Gut-Brain Axis and the Regulation Thereof by Endocannabinoids and Related Mediators. Int. J. Mol. Sci. 2020, 21, 1554. [Google Scholar] [CrossRef] [Green Version]
- Jansma, J.; Brinkman, F.; van Hemert, S.; El Aidy, S. Targeting the endocannabinoid system with microbial interventions to improve gut integrity. Prog. Neuropsychopharmacol. Biol. Psychiatry 2020. [Google Scholar] [CrossRef]
- Lacroix, S.; Pechereau, F.; Leblanc, N.; Boubertakh, B.; Houde, A.; Martin, C.; Flamand, N.; Silvestri, C.; Raymond, F.; Di Marzo, V.; et al. Rapid and Concomitant Gut Microbiota and Endocannabinoidome Response to Diet-Induced Obesity in Mice. mSystems 2019, 4. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Yang, S.T.; Kreutzberger, A.J.B.; Lee, J.; Kiessling, V.; Tamm, L.K. The role of cholesterol in membrane fusion. Chem. Phys. Lipids 2016, 199, 136–143. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Kriaa, A.; Bourgin, M.; Potiron, A.; Mkaouar, H.; Jablaoui, A.; Gérard, P.; Maguin, E.; Rhimi, M. Microbial impact on cholesterol and bile acid metabolism: Current status and future prospects. J. Lipid Res. 2019, 60, 323–332. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Koppel, N.; Maini Rekdal, V.; Balskus, E.P. Chemical transformation of xenobiotics by the human gut microbiota. Science 2017, 356. [Google Scholar] [CrossRef] [PubMed]
- Kindt, A.; Liebisch, G.; Clavel, T.; Haller, D.; Hörmannsperger, G.; Yoon, H.; Kolmeder, D.; Sigruener, A.; Krautbauer, S.; Seeliger, C.; et al. The gut microbiota promotes hepatic fatty acid desaturation and elongation in mice. Nat. Commun. 2018, 9, 3760. [Google Scholar] [CrossRef] [Green Version]
- Reigstad, C.S.; Salmonson, C.E.; Rainey, J.F., 3rd; Szurszewski, J.H.; Linden, D.R.; Sonnenburg, J.L.; Farrugia, G.; Kashyap, P.C. Gut microbes promote colonic serotonin production through an effect of short-chain fatty acids on enterochromaffin cells. FASEB J. 2015, 29, 1395–1403. [Google Scholar] [CrossRef] [Green Version]
- Berger, M.; Gray, J.A.; Roth, B.L. The expanded biology of serotonin. Annu. Rev. Med. 2009, 60, 355–366. [Google Scholar] [CrossRef] [Green Version]
- Verhoeckx, K.C.; Voortman, T.; Balvers, M.G.; Hendriks, H.F.; Wortelboer, H.M.; Witkamp, R.F. Presence, formation and putative biological activities of N-acyl serotonins, a novel class of fatty-acid derived mediators, in the intestinal tract. Biochim. Biophys. Acta 2011, 1811, 578–586. [Google Scholar] [CrossRef]
- Sharon, G.; Garg, N.; Debelius, J.; Knight, R.; Dorrestein, P.C.; Mazmanian, S.K. Specialized metabolites from the microbiome in health and disease. Cell Metab. 2014, 20, 719–730. [Google Scholar] [CrossRef] [Green Version]
- Rousseaux, C.; Thuru, X.; Gelot, A.; Barnich, N.; Neut, C.; Dubuquoy, L.; Dubuquoy, C.; Merour, E.; Geboes, K.; Chamaillard, M.; et al. Lactobacillus acidophilus modulates intestinal pain and induces opioid and cannabinoid receptors. Nat. Med. 2007, 13, 35–37. [Google Scholar] [CrossRef]
- Muccioli, G.G.; Naslain, D.; Bäckhed, F.; Reigstad, C.S.; Lambert, D.M.; Delzenne, N.M.; Cani, P.D. The endocannabinoid system links gut microbiota to adipogenesis. Mol. Syst. Biol. 2010, 6, 392. [Google Scholar] [CrossRef] [PubMed]
- Everard, A.; Belzer, C.; Geurts, L.; Ouwerkerk, J.P.; Druart, C.; Bindels, L.B.; Guiot, Y.; Derrien, M.; Muccioli, G.G.; Delzenne, N.M.; et al. Cross-talk between Akkermansia muciniphila and intestinal epithelium controls diet-induced obesity. Proc. Natl. Acad. Sci. USA 2013, 110, 9066–9071. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Lynch, A.; Crowley, E.; Casey, E.; Cano, R.; Shanahan, R.; McGlacken, G.; Marchesi, J.R.; Clarke, D.J. The Bacteroidales produce an N-acylated derivative of glycine with both cholesterol-solubilising and hemolytic activity. Sci. Rep. 2017, 7, 13270. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Cohen, L.J.; Kang, H.S.; Chu, J.; Huang, Y.H.; Gordon, E.A.; Reddy, B.V.; Ternei, M.A.; Craig, J.W.; Brady, S.F. Functional metagenomic discovery of bacterial effectors in the human microbiome and isolation of commendamide, a GPCR G2A/132 agonist. Proc. Natl. Acad. Sci. USA 2015, 112, E4825–E4834. [Google Scholar] [CrossRef] [Green Version]
- Meadows, J.A.; Wargo, M.J. Carnitine in bacterial physiology and metabolism. Microbiology 2015, 161, 1161–1174. [Google Scholar] [CrossRef]
- Ghonimy, A.; Zhang, D.M.; Farouk, M.H.; Wang, Q. The Impact of Carnitine on Dietary Fiber and Gut Bacteria Metabolism and Their Mutual Interaction in Monogastrics. Int. J. Mol. Sci. 2018, 19, 1008. [Google Scholar] [CrossRef] [Green Version]
- Tang, W.H.W.; Li, D.Y.; Hazen, S.L. Dietary metabolism, the gut microbiome, and heart failure. Nat. Rev. Cardiol. 2019, 16, 137–154. [Google Scholar] [CrossRef]
- Sitaraman, R. Phospholipid catabolism by gut microbiota and the risk of cardiovascular disease. J. Med. Microbiol. 2013, 62, 948–950. [Google Scholar] [CrossRef]
- Hulme, H.; Meikle, L.M.; Strittmatter, N.; van der Hooft, J.J.J.; Swales, J.; Bragg, R.A.; Villar, V.H.; Ormsby, M.J.; Barnes, S.; Brown, S.L.; et al. Microbiome-derived carnitine mimics as previously unknown mediators of gut-brain axis communication. Sci. Adv. 2020, 6, eaax6328. [Google Scholar] [CrossRef] [Green Version]
- Turroni, S.; Fiori, J.; Rampelli, S.; Schnorr, S.L.; Consolandi, C.; Barone, M.; Biagi, E.; Fanelli, F.; Mezzullo, M.; Crittenden, A.N.; et al. Fecal metabolome of the Hadza hunter-gatherers: A host-microbiome integrative view. Sci. Rep. 2016, 6, 32826. [Google Scholar] [CrossRef]
- Huang, H.J.; Zhang, A.Y.; Cao, H.C.; Lu, H.F.; Wang, B.H.; Xie, Q.; Xu, W.; Li, L.J. Metabolomic analyses of faeces reveals malabsorption in cirrhotic patients. Dig. Liver Dis. 2013, 45, 677–682. [Google Scholar] [CrossRef] [PubMed]
- Quince, C.; Walker, A.W.; Simpson, J.T.; Loman, N.J.; Segata, N. Corrigendum: Shotgun metagenomics, from sampling to analysis. Nat. Biotechnol. 2017, 35, 1211. [Google Scholar] [CrossRef] [PubMed]
- Conlan, S.; Kong, H.H.; Segre, J.A. Species-level analysis of DNA sequence data from the NIH Human Microbiome Project. PLoS ONE 2012, 7, e47075. [Google Scholar] [CrossRef] [PubMed]
- Franzosa, E.A.; Sirota-Madi, A.; Avila-Pacheco, J.; Fornelos, N.; Haiser, H.J.; Reinker, S.; Vatanen, T.; Hall, A.B.; Mallick, H.; McIver, L.J.; et al. Gut microbiome structure and metabolic activity in inflammatory bowel disease. Nat. Microbiol. 2019, 4, 293–305. [Google Scholar] [CrossRef] [PubMed]
- Hudson, L.E.; Anderson, S.E.; Corbett, A.H.; Lamb, T.J. Gleaning Insights from Fecal Microbiota Transplantation and Probiotic Studies for the Rational Design of Combination Microbial Therapies. Clin. Microbiol. Rev. 2017, 30, 191–231. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Kultima, J.R.; Coelho, L.P.; Forslund, K.; Huerta-Cepas, J.; Li, S.S.; Driessen, M.; Voigt, A.Y.; Zeller, G.; Sunagawa, S.; Bork, P. MOCAT2: A metagenomic assembly, annotation and profiling framework. Bioinformatics 2016, 32, 2520–2523. [Google Scholar] [CrossRef] [PubMed]
- Glass, E.M.; Wilkening, J.; Wilke, A.; Antonopoulos, D.; Meyer, F. Using the metagenomics RAST server (MG-RAST) for analyzing shotgun metagenomes. Cold Spring Harb. Protoc. 2010, 2010, pdb.prot5368. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Franzosa, E.A.; McIver, L.J.; Rahnavard, G.; Thompson, L.R.; Schirmer, M.; Weingart, G.; Lipson, K.S.; Knight, R.; Caporaso, J.G.; Segata, N.; et al. Species-level functional profiling of metagenomes and metatranscriptomes. Nat. Methods 2018, 15, 962–968. [Google Scholar] [CrossRef] [PubMed]
- Huson, D.H.; Auch, A.F.; Qi, J.; Schuster, S.C. MEGAN analysis of metagenomic data. Genome Res. 2007, 17, 377–386. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Seshadri, R.; Kravitz, S.A.; Smarr, L.; Gilna, P.; Frazier, M. CAMERA: A community resource for metagenomics. PLoS Biol. 2007, 5, e75. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Breitwieser, F.P.; Lu, J.; Salzberg, S.L. A review of methods and databases for metagenomic classification and assembly. Brief. Bioinform. 2017. [Google Scholar] [CrossRef] [PubMed]
- Zhang, S.W.; Jin, X.Y.; Zhang, T. Gene Prediction in Metagenomic Fragments with Deep Learning. Biomed. Res. Int. 2017, 2017, 4740354. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Liang, Q.; Bible, P.W.; Liu, Y.; Zou, B.; Wei, L. DeepMicrobes: Taxonomic classification for metagenomics with deep learning. NAR Genom. Bioinform. 2020, 2. [Google Scholar] [CrossRef] [Green Version]
- Rojas-Carulla, M.; Tolstikhin, I.; Luque, G.; Youngblut, N.; Ley, R.; Schölkopf, B. Genet: Deep representations for metagenomics. arXiv 2019, arXiv:1901.11015. [Google Scholar]
- Janda, J.M.; Abbott, S.L. 16S rRNA gene sequencing for bacterial identification in the diagnostic laboratory: Pluses, perils, and pitfalls. J. Clin. Microbiol. 2007, 45, 2761–2764. [Google Scholar] [CrossRef] [Green Version]
- Lozupone, C.A.; Knight, R. Species divergence and the measurement of microbial diversity. FEMS Microbiol. Rev. 2008, 32, 557–578. [Google Scholar] [CrossRef]
- Chen, W.; Zhang, C.K.; Cheng, Y.; Zhang, S.; Zhao, H. A comparison of methods for clustering 16S rRNA sequences into OTUs. PLoS ONE 2013, 8, e70837. [Google Scholar] [CrossRef]
- DeSantis, T.Z.; Hugenholtz, P.; Larsen, N.; Rojas, M.; Brodie, E.L.; Keller, K.; Huber, T.; Dalevi, D.; Hu, P.; Andersen, G.L. Greengenes, a chimera-checked 16S rRNA gene database and workbench compatible with ARB. Appl. Environ. Microbiol. 2006, 72, 5069–5072. [Google Scholar] [CrossRef] [Green Version]
- Quast, C.; Pruesse, E.; Yilmaz, P.; Gerken, J.; Schweer, T.; Yarza, P.; Peplies, J.; Glockner, F.O. The SILVA ribosomal RNA gene database project: Improved data processing and web-based tools. Nucleic Acids Res. 2013, 41, D590–D596. [Google Scholar] [CrossRef]
- Nilsson, R.H.; Larsson, K.H.; Taylor, A.F.S.; Bengtsson-Palme, J.; Jeppesen, T.S.; Schigel, D.; Kennedy, P.; Picard, K.; Glockner, F.O.; Tedersoo, L.; et al. The UNITE database for molecular identification of fungi: Handling dark taxa and parallel taxonomic classifications. Nucleic Acids Res. 2019, 47, D259–D264. [Google Scholar] [CrossRef]
- Bolyen, E.; Rideout, J.R.; Dillon, M.R.; Bokulich, N.A.; Abnet, C.C.; Al-Ghalith, G.A.; Alexander, H.; Alm, E.J.; Arumugam, M.; Asnicar, F.; et al. Reproducible, interactive, scalable and extensible microbiome data science using QIIME 2. Nat. Biotechnol. 2019, 37, 852–857. [Google Scholar] [CrossRef]
- Hugerth, L.W.; Andersson, A.F. Analysing microbial community composition through amplicon sequencing: From sampling to hypothesis testing. Front. Microbiol. 2017, 8, 1561. [Google Scholar] [CrossRef] [PubMed]
- Carlos, N.; Tang, Y.W.; Pei, Z. Pearls and pitfalls of genomics-based microbiome analysis. Emerg. Microbes Infect. 2012, 1, e45. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Sharma, V.K.; Kumar, N.; Prakash, T.; Taylor, T.D. MetaBioME: A database to explore commercially useful enzymes in metagenomic datasets. Nucleic Acids Res. 2010, 38, D468–D472. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Poretsky, R.; Rodriguez, R.L.; Luo, C.; Tsementzi, D.; Konstantinidis, K.T. Strengths and limitations of 16S rRNA gene amplicon sequencing in revealing temporal microbial community dynamics. PLoS ONE 2014, 9, e93827. [Google Scholar] [CrossRef] [Green Version]
- Singer, E.; Bushnell, B.; Coleman-Derr, D.; Bowman, B.; Bowers, R.M.; Levy, A.; Gies, E.A.; Cheng, J.F.; Copeland, A.; Klenk, H.P.; et al. High-resolution phylogenetic microbial community profiling. ISME J. 2016, 10, 2020–2032. [Google Scholar] [CrossRef]
- Shoaie, S.; Nielsen, J. Elucidating the interactions between the human gut microbiota and its host through metabolic modeling. Front. Genet. 2014, 5, 86. [Google Scholar] [CrossRef] [Green Version]
- Sen, P.; Oresic, M. Metabolic Modeling of Human Gut Microbiota on a Genome Scale: An Overview. Metabolites 2019, 9, 22. [Google Scholar] [CrossRef] [Green Version]
- Bauer, E.; Thiele, I. From Network Analysis to Functional Metabolic Modeling of the Human Gut Microbiota. mSystems 2018, 3. [Google Scholar] [CrossRef] [Green Version]
- Ji, B.; Nielsen, J. New insight into the gut microbiome through metagenomics. Adv. Genom. Genet. 2015, 5, 77–91. [Google Scholar]
- Baig, S. Reviewing personal bacteria—The human microbiome project. J. Coll. Physicians Surg. Pak. 2012, 22, 3–4. [Google Scholar] [PubMed]
- Shoaie, S.; Ghaffari, P.; Kovatcheva-Datchary, P.; Mardinoglu, A.; Sen, P.; Pujos-Guillot, E.; de Wouters, T.; Juste, C.; Rizkalla, S.; Chilloux, J. Quantifying diet-induced metabolic changes of the human gut microbiome. Cell Metab. 2015, 22, 320–331. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Heinken, A.; Ravcheev, D.A.; Baldini, F.; Heirendt, L.; Fleming, R.M.; Thiele, I. Personalized modeling of the human gut microbiome reveals distinct bile acid deconjugation and biotransformation potential in healthy and IBD individuals. BioRxiv 2017. [Google Scholar] [CrossRef] [Green Version]
- Arkin, A.P.; Cottingham, R.W.; Henry, C.S.; Harris, N.L.; Stevens, R.L.; Maslov, S.; Dehal, P.; Ware, D.; Perez, F.; Canon, S.; et al. KBase: The United States Department of Energy Systems Biology Knowledgebase. Nat. Biotechnol. 2018, 36, 566–569. [Google Scholar] [CrossRef] [Green Version]
- Seaver, S.M.D.; Liu, F.; Zhang, Q.; Jeffryes, J.; Faria, J.P.; Edirisinghe, J.N.; Mundy, M.; Chia, N.; Noor, E.; Beber, M.E.; et al. The ModelSEED Biochemistry Database for the integration of metabolic annotations and the reconstruction, comparison and analysis of metabolic models for plants, fungi and microbes. Nucleic Acids Res. 2020. [Google Scholar] [CrossRef]
- Heirendt, L.; Arreckx, S.; Pfau, T.; Mendoza, S.N.; Richelle, A.; Heinken, A.; Haraldsdóttir, H.S.; Wachowiak, J.; Keating, S.M.; Vlasov, V. Creation and analysis of biochemical constraint-based models using the COBRA Toolbox v. 3.0. Nat. Protoc. 2019, 14, 639–702. [Google Scholar] [CrossRef] [Green Version]
- Agren, R.; Liu, L.; Shoaie, S.; Vongsangnak, W.; Nookaew, I.; Nielsen, J. The RAVEN toolbox and its use for generating a genome-scale metabolic model for Penicillium chrysogenum. PLoS Comput. Biol. 2013, 9, e1002980. [Google Scholar] [CrossRef] [Green Version]
- Kumar, M.; Ji, B.; Babaei, P.; Das, P.; Lappa, D.; Ramakrishnan, G.; Fox, T.E.; Haque, R.; Petri, W.A., Jr.; Bäckhed, F. Gut microbiota dysbiosis is associated with malnutrition and reduced plasma amino acid levels: Lessons from genome-scale metabolic modeling. Metab. Eng. 2018, 49, 128–142. [Google Scholar] [CrossRef] [Green Version]
- Magnúsdóttir, S.; Heinken, A.; Kutt, L.; Ravcheev, D.A.; Bauer, E.; Noronha, A.; Greenhalgh, K.; Jäger, C.; Baginska, J.; Wilmes, P. Generation of genome-scale metabolic reconstructions for 773 members of the human gut microbiota. Nat. Biotechnol. 2016. [Google Scholar] [CrossRef]
- Noronha, A.; Modamio, J.; Jarosz, Y.; Guerard, E.; Sompairac, N.; Preciat, G.; Danielsdottir, A.D.; Krecke, M.; Merten, D.; Haraldsdottir, H.S.; et al. The Virtual Metabolic Human database: Integrating human and gut microbiome metabolism with nutrition and disease. Nucleic Acids Res. 2019, 47, D614–D624. [Google Scholar] [CrossRef]
- King, Z.A.; Lu, J.; Dräger, A.; Miller, P.; Federowicz, S.; Lerman, J.A.; Ebrahim, A.; Palsson, B.O.; Lewis, N.E. BiGG Models: A platform for integrating, standardizing and sharing genome-scale models. Nucleic Acids Res. 2015, 44, D515–D522. [Google Scholar] [CrossRef] [PubMed]
- Poupin, N.; Vinson, F.; Moreau, A.; Batut, A.; Chazalviel, M.; Colsch, B.; Fouillen, L.; Guez, S.; Khoury, S.; Dalloux-Chioccioli, J.; et al. Improving lipid mapping in Genome Scale Metabolic Networks using ontologies. Metab. Off. J. Metab. Soc. 2020, 16, 44. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Molenaar, M.R.; Jeucken, A.; Wassenaar, T.A.; van de Lest, C.H.A.; Brouwers, J.F.; Helms, J.B. LION/web: A web-based ontology enrichment tool for lipidomic data analysis. Gigascience 2019, 8. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Ni, Z.; Fedorova, M. LipidLynxX: A data transfer hub to support integration of large scale lipidomics datasets. bioRxiv 2020. [Google Scholar] [CrossRef] [Green Version]
- Fahy, E.; Subramaniam, S. RefMet: A reference nomenclature for metabolomics. Nat. Methods 2020, 17, 1173–1174. [Google Scholar] [CrossRef]
- Liebisch, G.; Fahy, E.; Aoki, J.; Dennis, E.A.; Durand, T.; Ejsing, C.S.; Fedorova, M.; Feussner, I.; Griffiths, W.J.; Kofeler, H.; et al. Update on LIPID MAPS classification, nomenclature, and shorthand notation for MS-derived lipid structures. J. Lipid Res. 2020, 61, 1539–1555. [Google Scholar] [CrossRef]
- Sanchez, B.J.; Li, F.; Kerkhoven, E.J.; Nielsen, J. SLIMEr: Probing flexibility of lipid metabolism in yeast with an improved constraint-based modeling framework. BMC Syst. Biol. 2019, 13, 4. [Google Scholar] [CrossRef] [Green Version]



{kind=link}
{kind=link}
| Lipid Category | Lipid Sub Class | Example/Related Lipids | Microbes | References |
|---|---|---|---|---|
| Sphingolipids | Ceramide phosphoinositols | PI-Cer(d18:1/22:0) | Bacteroidetes (genera Bacteroides, Prevotella, Porphyromonas, Bacteroides theta, thetaiotaomicron, ovatus and fragilis | [33] |
| Ceramide phosphoethanolamines | N-Acyl ceramide phosphoethanolamine | |||
| Sphinganines | 3-ketosphinganine sphinganine | |||
| N-acylsphinganines | dihydroceramide | |||
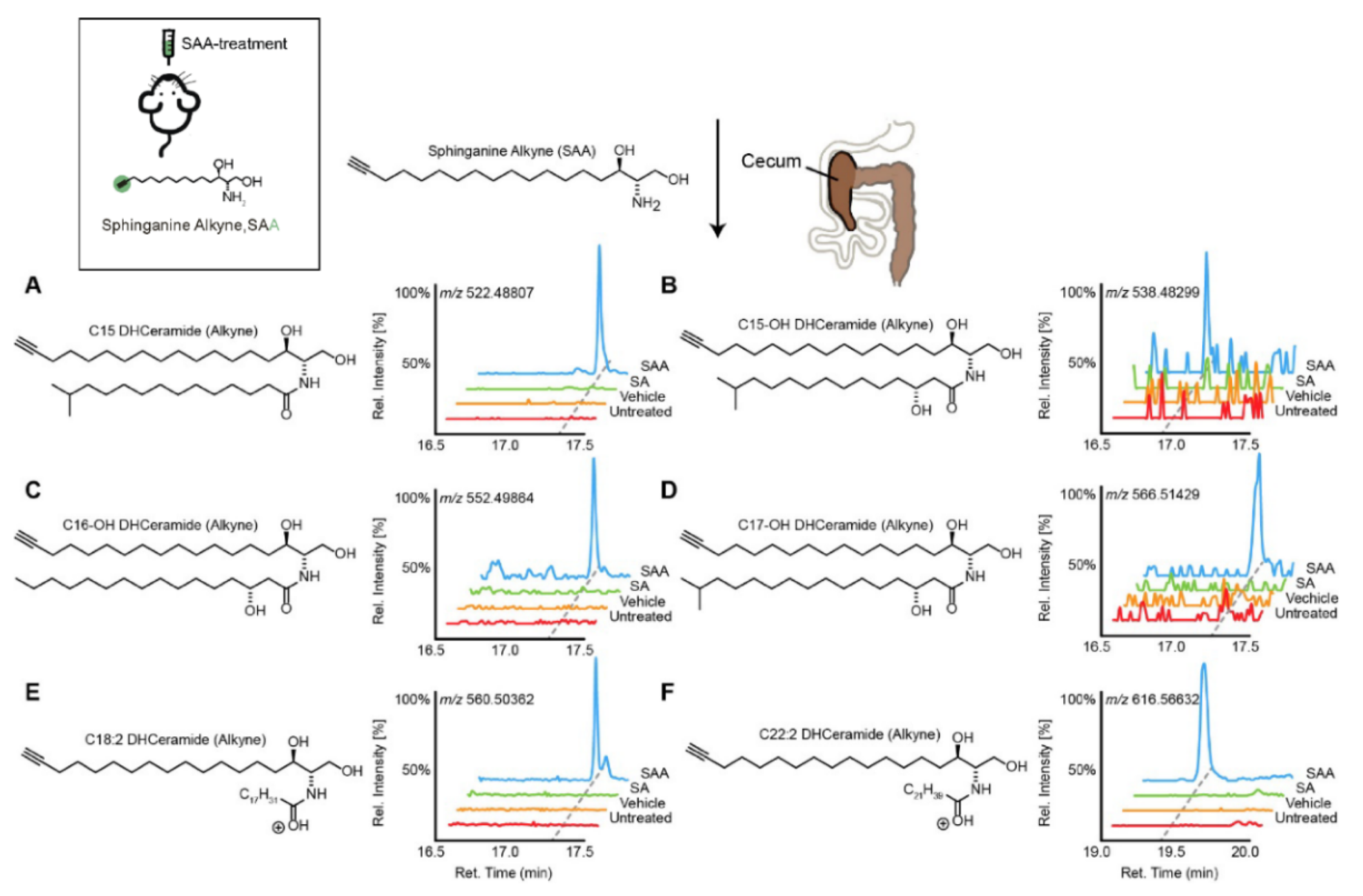
| C15-, C15OH-, C16OH-, C17OH-, C18:2-, C22:2- dihydrocermide | [40] | |||
| Sphingoid base 1-phosphates | sphinganine-1-phosphate (d17:0) sphinganine-1-phosphate (d18:0) | [39] | ||
| Sterol | C24 bile acids, alcohols, and derivatives | deoxycholic acid lithocholic acid ursodeoxycholate iso-deoxycholic acid iso-lithocholic acid 7-oxo-lithocholic Acid | Bacteroides, Clostridium, Lactobacillus, and Bifidobacteria and Alloscardovia sp. | [15,46,47,48,49] [41,50] |
| Taurine conjugates | tauroursodeoxycholic acid | |||
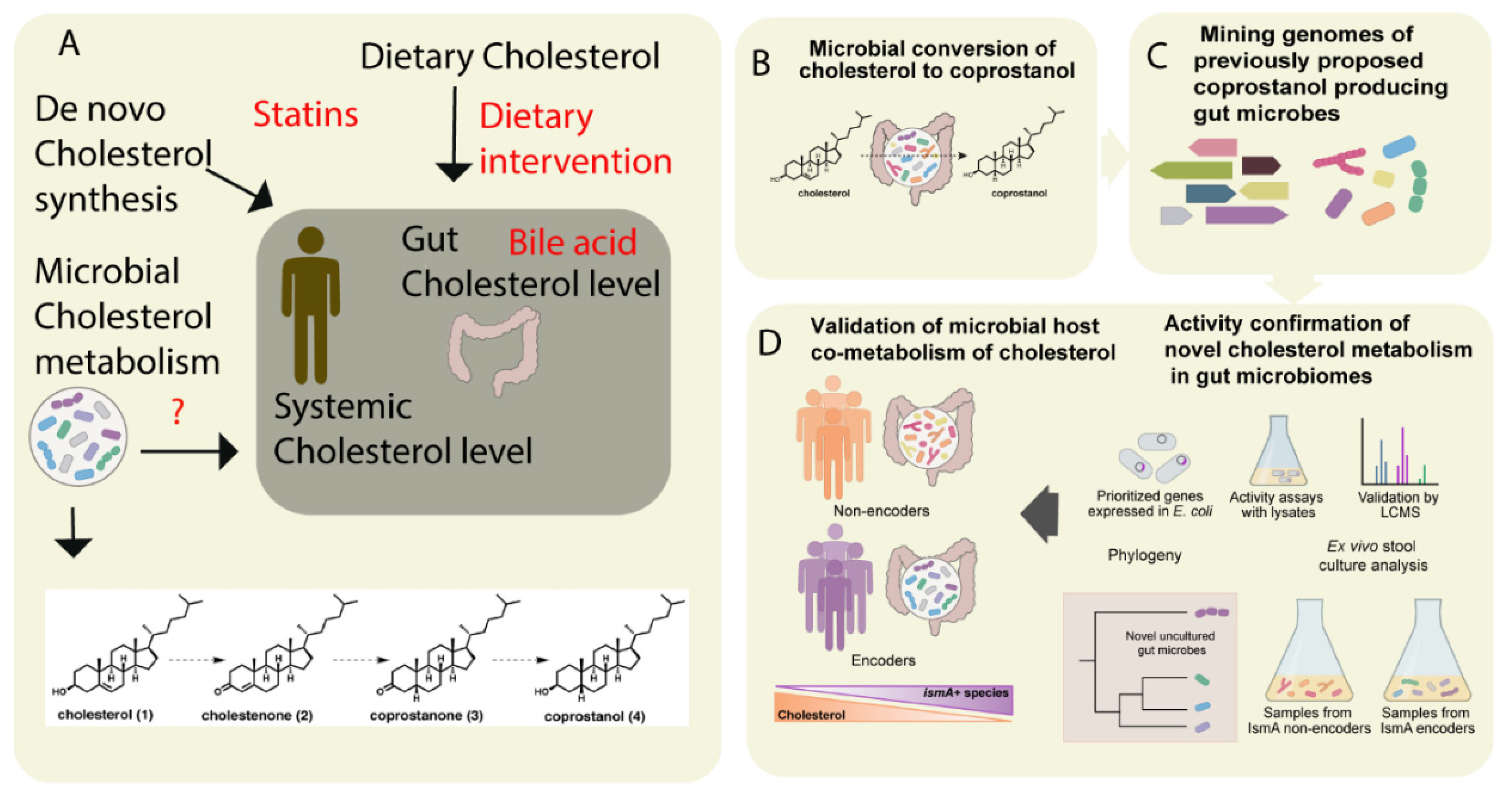
| Cholesterol and derivatives | cholestenone coprostanone coprostanol | Eubacterium coprostanoligenes, Bacteroides intestinalis, Faecalibacterium prausnitzii | [51,52] | |
| Fatty Acyls | Other Octadecanoids | 10-hydroxy-12 (Z)-octadecenoic acid (18:1) (HYA), 10-hydroxy-12,15(Z,Z) octadecenoic acid (18:2) (αHYA), 10-hydroxy-6,12(Z,Z)-octadecadienoic acid (18:2) (γHYA), 10-hydroxyoctadecanoic acid (HYB), 10-hydoroxy-trans-11-octade-cenoic acid (HYC), 10-oxo-12(Z)-octadecenoic acid (18:1) (KetoA), 10-oxo-12,15(Z,Z) (18:2) octadecenoic acid (αKetoA), 10-oxo-6,12(Z,Z)-octadecenoic acid (18:2) (γKetoA), 10-oxo-octadecanoic acid, 10-oxo-trans-11-octadecenoic acid | Lactobacillus genus (Lactobacillus plantarum, Lactobacillus salivarius, Lactobacillus gasseri, Lactobacillus acidophilus and Lactobacillus johnsonii) Bifidobacterium spp., Eubacterium ventriosum and Lactobacillus spp. | [53,54,55,56,57] |
| Unsaturated fatty acids | oleic acid | |||
| Glycerolipids (Endocannabinoid) | Monoacylglycerols | 2-arachidonoylglycerol (2-AG) 2- oleoylglycerol (2-OG) 2- palmitoyl-glycerol (2-PG) | Akkermansia muciniphila | [58,59,60,61,62] |
Publisher’s Note: MDPI stays neutral with regard to jurisdictional claims in published maps and institutional affiliations. |
© 2021 by the authors. Licensee MDPI, Basel, Switzerland. This article is an open access article distributed under the terms and conditions of the Creative Commons Attribution (CC BY) license (http://creativecommons.org/licenses/by/4.0/).
Share and Cite
Lamichhane, S.; Sen, P.; Alves, M.A.; Ribeiro, H.C.; Raunioniemi, P.; Hyötyläinen, T.; Orešič, M. Linking Gut Microbiome and Lipid Metabolism: Moving beyond Associations. Metabolites 2021, 11, 55. https://doi.org/10.3390/metabo11010055
Lamichhane S, Sen P, Alves MA, Ribeiro HC, Raunioniemi P, Hyötyläinen T, Orešič M. Linking Gut Microbiome and Lipid Metabolism: Moving beyond Associations. Metabolites. 2021; 11(1):55. https://doi.org/10.3390/metabo11010055
Chicago/Turabian StyleLamichhane, Santosh, Partho Sen, Marina Amaral Alves, Henrique C. Ribeiro, Peppi Raunioniemi, Tuulia Hyötyläinen, and Matej Orešič. 2021. "Linking Gut Microbiome and Lipid Metabolism: Moving beyond Associations" Metabolites 11, no. 1: 55. https://doi.org/10.3390/metabo11010055
APA StyleLamichhane, S., Sen, P., Alves, M. A., Ribeiro, H. C., Raunioniemi, P., Hyötyläinen, T., & Orešič, M. (2021). Linking Gut Microbiome and Lipid Metabolism: Moving beyond Associations. Metabolites, 11(1), 55. https://doi.org/10.3390/metabo11010055