
Comparison of Kit-Based Metabolomics with Other Methodologies in a Large Cohort, towards Establishing Reference Values

 , , , ,
, , , ,
Abstract
:1. Introduction
2. Results
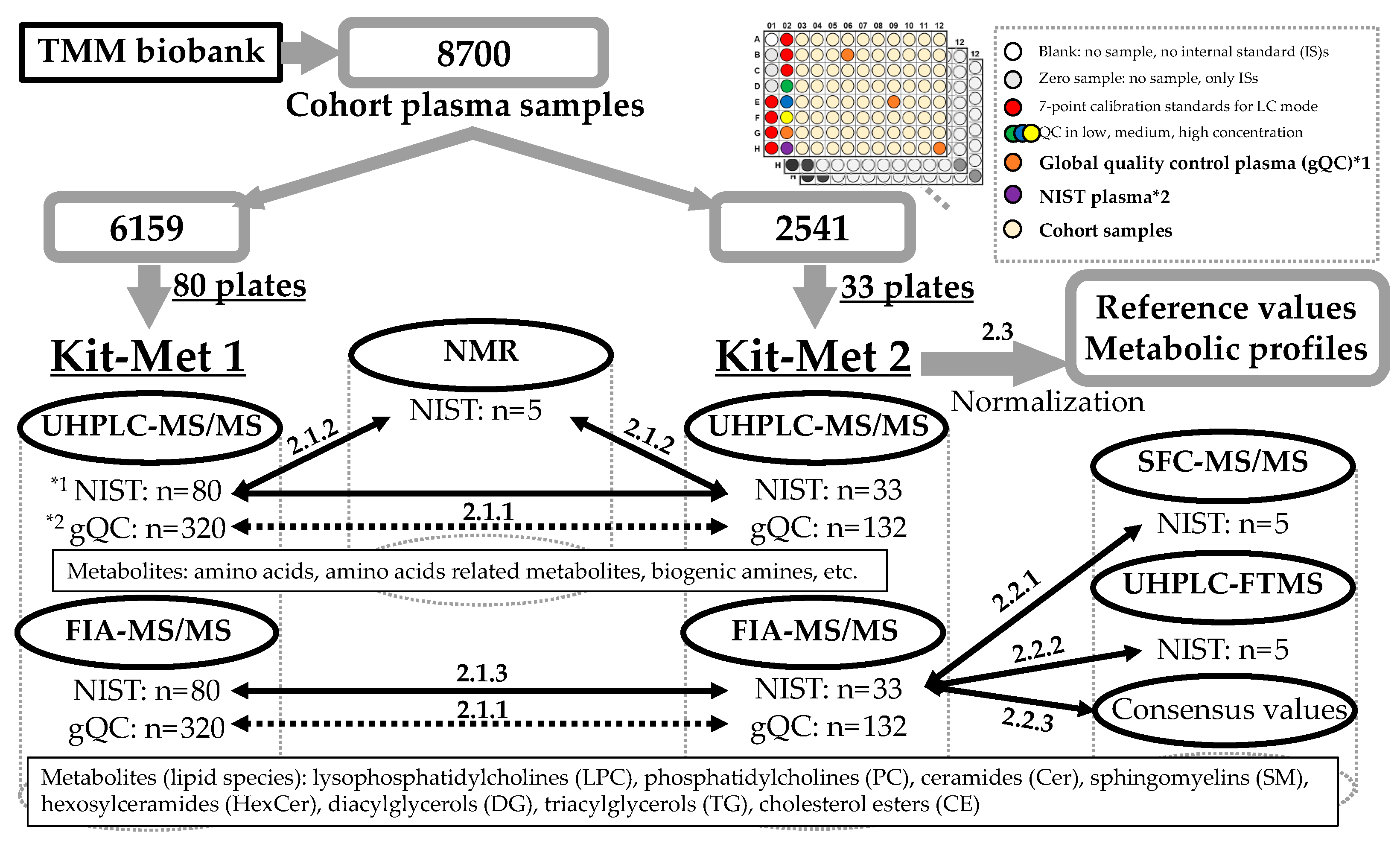
2.1. Evaluation of System Difference in the Large-Scale Analysis
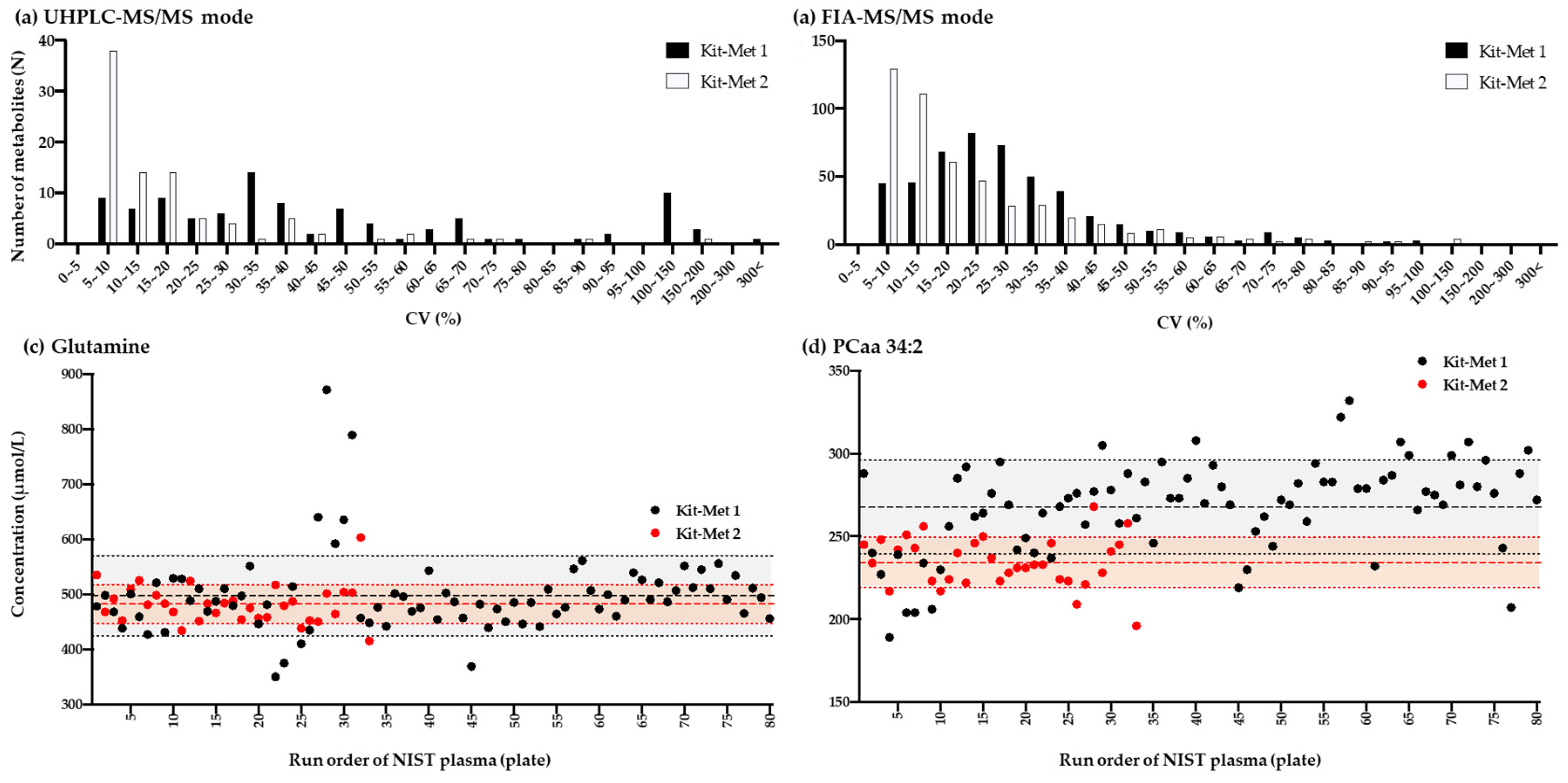
2.1.1. Comparison of the Variation Performed Using Kit-Met 1 and Kit-Met 2
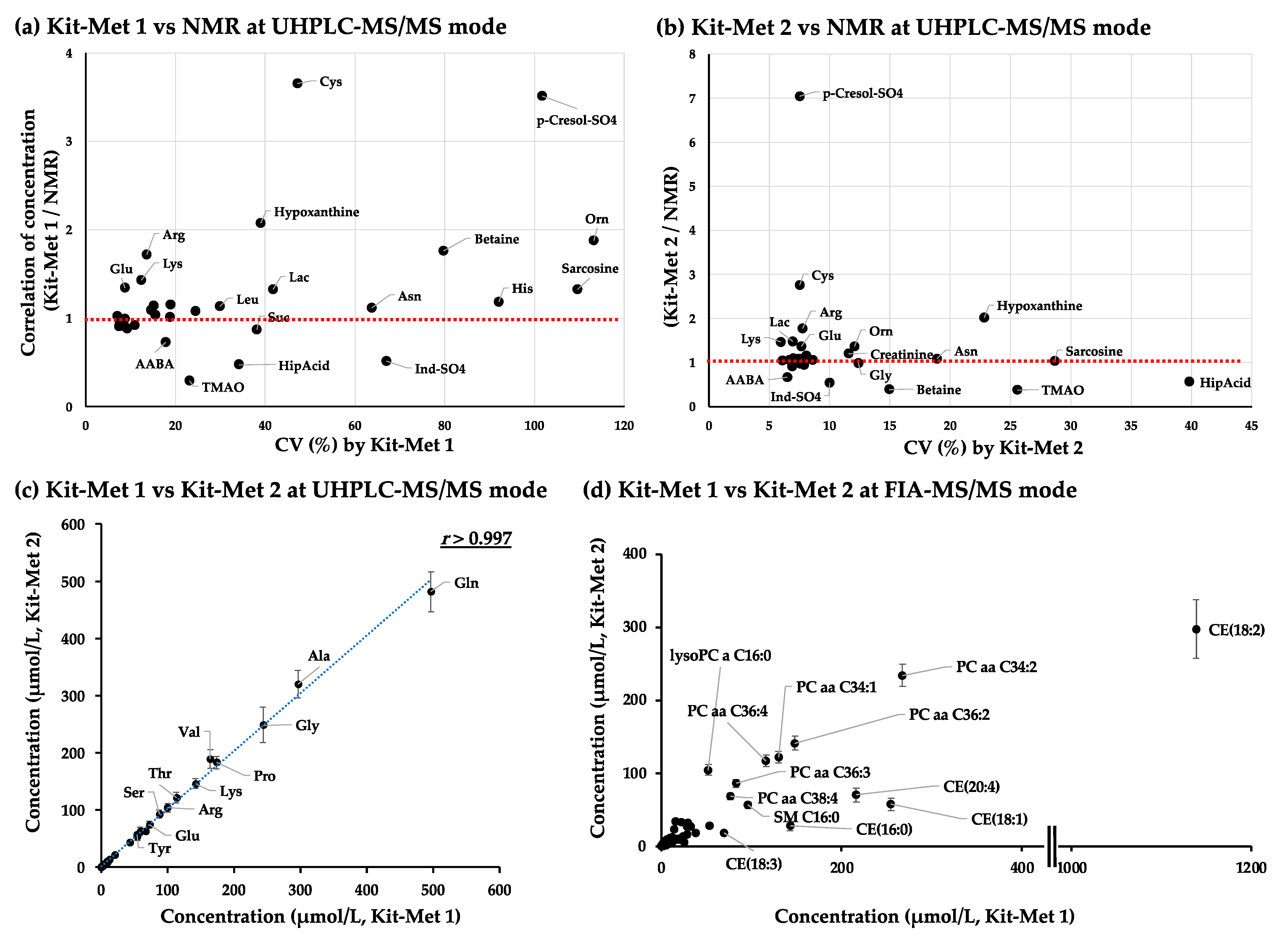
2.1.2. Correlation Analysis of the Quantified Values of Metabolites in the NIST Plasma Samples Detected by NMR and Kit-Met 1 and Kit-Met 2 in UHPLC-MS/MS Mode
2.1.3. Correlation Analysis of the Quantified Values of Metabolites in the NIST Plasma Samples Detected by Kit-Met 1 and Kit-Met 2 in FIA-MS/MS Mode
2.2. Evaluation of the Quantified Values of the Lipid Species in the NIST Plasma Samples
2.2.1. Correlation Analysis of the Quantified Values of the Lipid Species in the NIST Plasma Samples by SFC-MS/MS and FIA-MS/MS
2.2.2. Correlation Analysis of the Quantified Values of the Lipid Species in the NIST Plasma Samples by UHPLC-FTMS and FIA-MS/MS
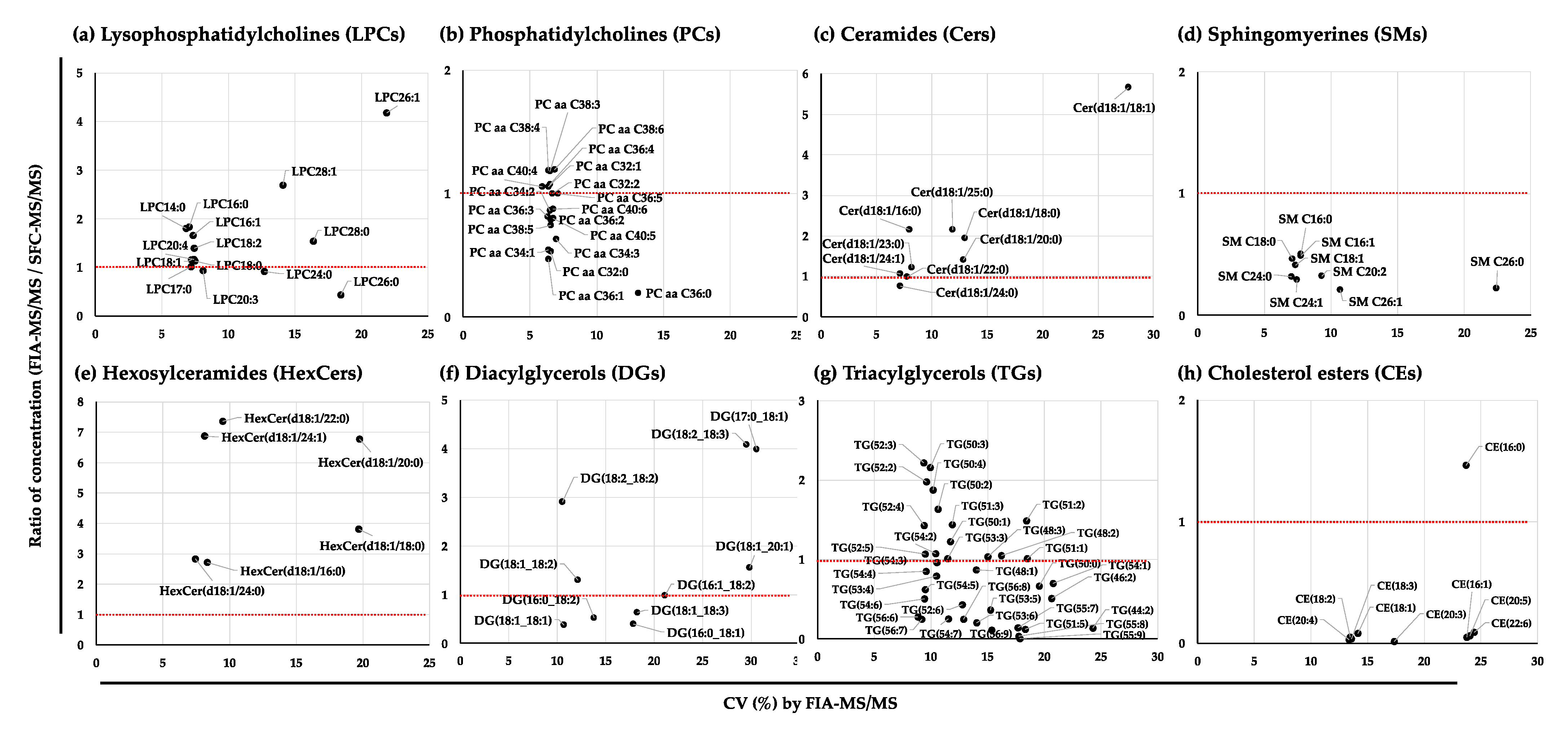
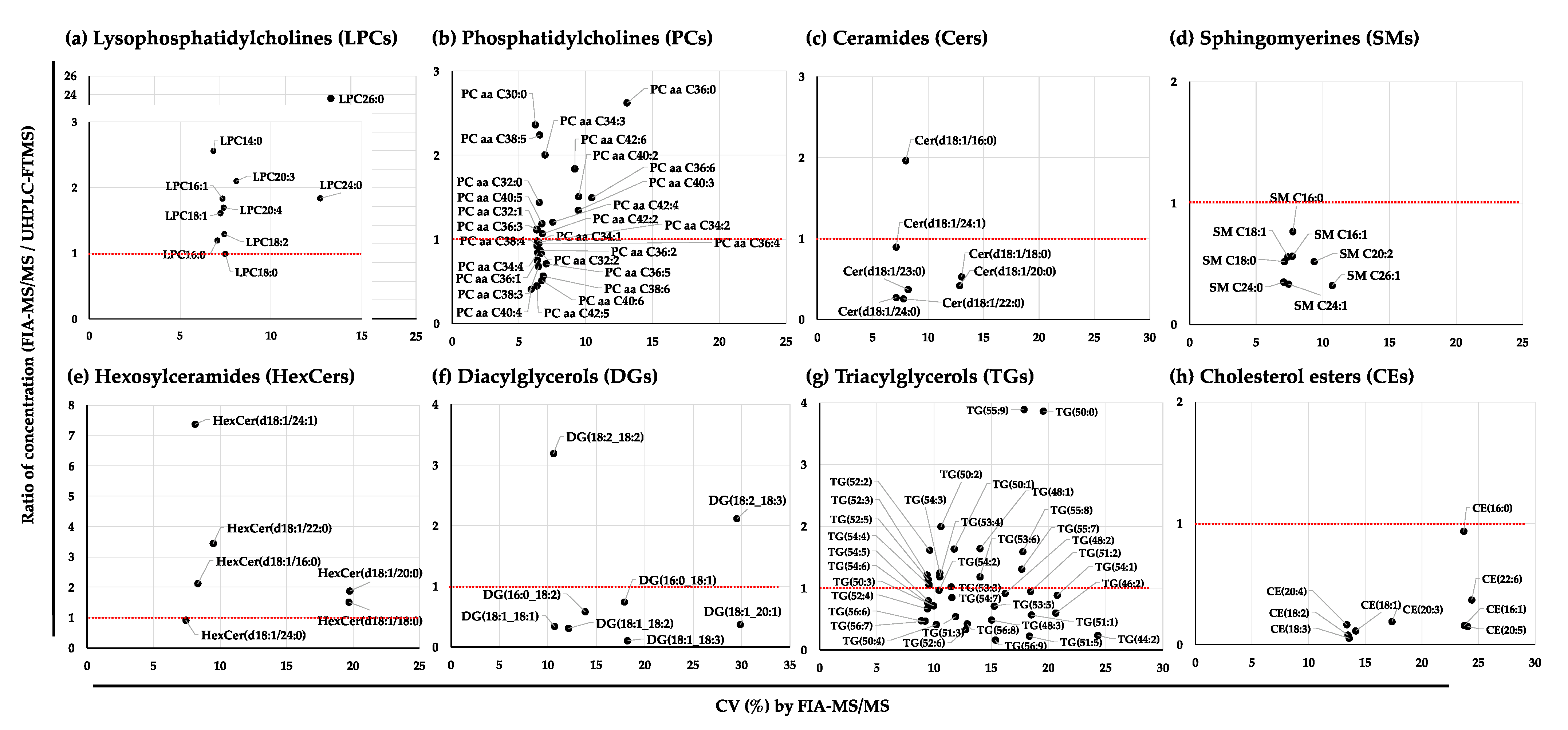
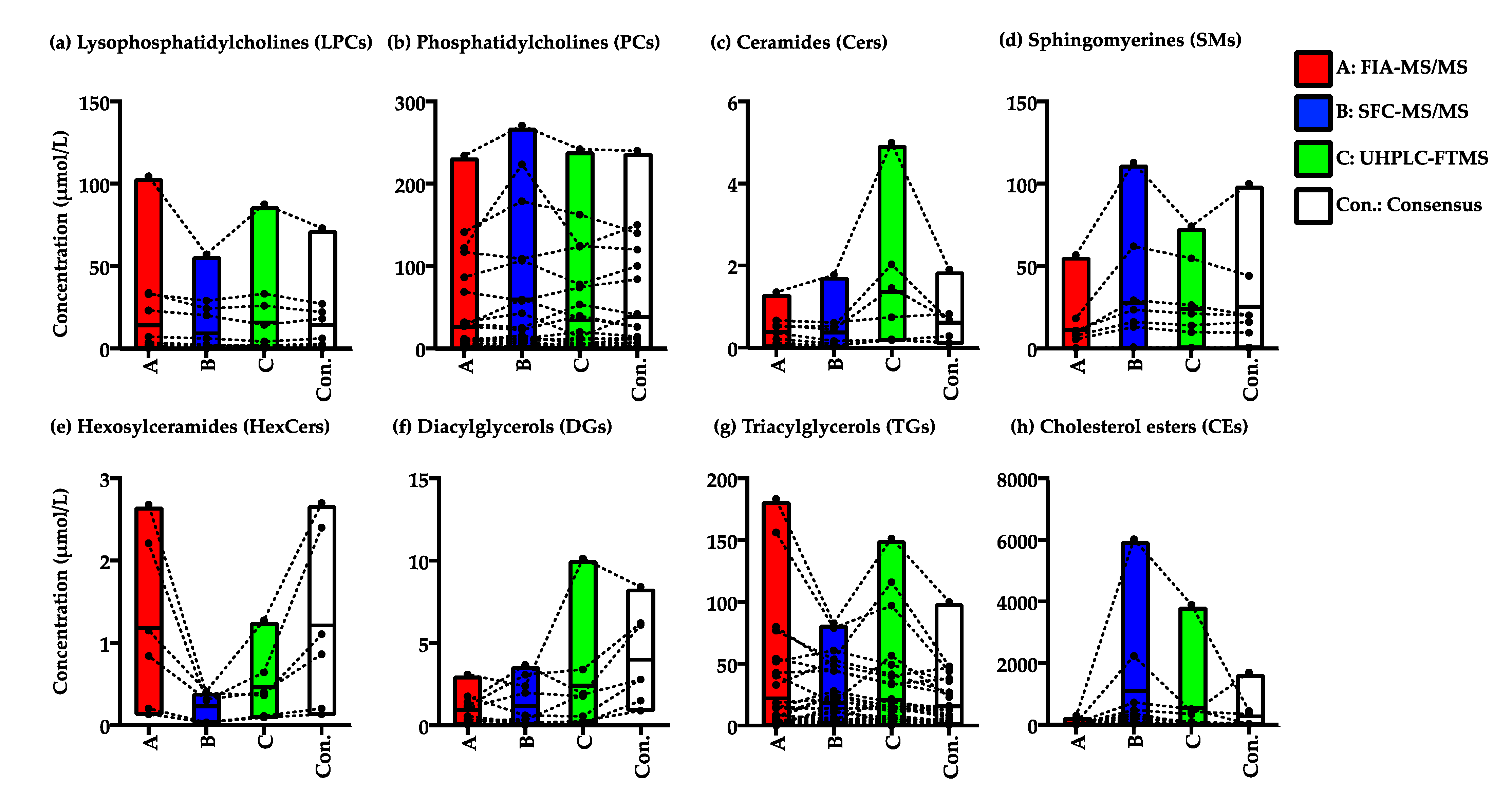
2.2.3. Variation in the Quantified Values of the Lipid Species in the NIST Plasma Samples by FIA-MS/MS, SFC-MS/MS, and UHPLC-FTMS
2.3. Effect of Normalization with the gQC Samples for Metabolic Profiling in a Large-Scale Cohort
3. Discussion
4. Materials and Methods
4.1. Reagents
4.2. Study Population and Plasma Collection of Metabolic Profiling
4.3. UHPLC-MS/MS Analysis
4.3.1. Sample Preparation
4.3.2. Data Acquisition and Data Processing
4.4. NMR Analysis
4.4.1. Sample Preparation, Data Acquisition, and Data Processing
4.4.2. Manual Quantification of the Metabolites in Plasma
4.5. SFC-MS/MS Analysis
4.5.1. Sample Preparation
4.5.2. Data Acquisition and Data Processing
4.6. UHPLC-FTMS Analysis
4.6.1. Sample Preparation
4.6.2. Data Acquisition and Data Processing
5. Conclusions
Supplementary Materials
Author Contributions
Funding
Institutional Review Board Statement
Informed Consent Statement
Data Availability Statement
Acknowledgments
Conflicts of Interest
References
- Yugi, K.; Kubota, H.; Hatano, A.; Kuroda, S. Trans-OMICS: How to Reconstruct Biochemical Networks across Multiple ‘Omic’ Layers. Trends Biotechnol. 2016, 34, 276–290. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Holmes, E.; Wilson, I.D.; Nicholson, J.K. Metabolic phenotyping in health and disease. Cell 2008, 134, 714–717. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Gieger, C.; Geistlinger, L.; Altmaier, E.; Hrabé de Angelis, M.; Kronenberg, F.; Meitinger, T.; Mewes, H.W.; Wichmann, H.E.; Weinberger, K.M.; Adamski, J.; et al. Genetics meets metabolomics: A genome-wide association study of metabolite profiles in human serum. PLoS Genet. 2008, 4, e1000282. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Nicholson, J.K.; Holmes, E.; Kinross, J.M.; Darzi, A.W.; Takats, Z.; Lindon, J.C. Metabolic phenotyping in clinical and surgical environments. Nature 2012, 491, 384–392. [Google Scholar] [CrossRef]
- Rinschen, M.M.; Ivanisevic, J.; Giera, M.; Siuzdak, G. Identification of bioactive metabolites using activity metabolomics. Nat. Rev. Mol. Cell Biol. 2019, 20, 353–367. [Google Scholar] [CrossRef]
- Saigusa, D.; Matsukawa, N.; Hishinuma, E.; Koshiba, S. Identification of biomarkers to diagnose diseases and find adverse drug reactions by metabolomics. Drug Metab. Pharmacokinet. 2021, 37, 100373. [Google Scholar] [CrossRef]
- Holmes, E.; Loo, R.L.; Stamler, J.; Bictash, M.; Yap, I.K.; Chan, Q.; Ebbels, T.; De Iorio, M.; Brown, I.J.; Veselkov, K.A.; et al. Human metabolic phenotype diversity and its association with diet and blood pressure. Nature 2008, 453, 396–400. [Google Scholar] [CrossRef]
- Emwas, A.H.; Luchinat, C.; Turano, P.; Tenori, L.; Roy, R.; Salek, R.M.; Ryan, D.; Merzaban, J.S.; Kaddurah-Daouk, R.; Zeri, A.C.; et al. Standardizing the experimental conditions for using urine in NMR-based metabolomic studies with a particular focus on diagnostic studies: A review. Metabolomics 2015, 11, 872–894. [Google Scholar] [CrossRef] [Green Version]
- Fiehn, O.; Kind, T. Metabolite profiling in blood plasma. Methods Mol. Biol. 2007, 358, 3–17. [Google Scholar] [CrossRef]
- Ismail, I.T.; Showalter, M.R.; Fiehn, O. Inborn Errors of Metabolism in the Era of Untargeted Metabolomics and Lipidomics. Metabolites 2019, 9, 242. [Google Scholar] [CrossRef] [Green Version]
- Want, E.J.; Wilson, I.D.; Gika, H.; Theodoridis, G.; Plumb, R.S.; Shockcor, J.; Holmes, E.; Nicholson, J.K. Global metabolic profiling procedures for urine using UPLC-MS. Nat. Protoc. 2010, 5, 1005–1018. [Google Scholar] [CrossRef]
- Saigusa, D.; Okamura, Y.; Motoike, I.N.; Katoh, Y.; Kurosawa, Y.; Saijyo, R.; Koshiba, S.; Yasuda, J.; Motohashi, H.; Sugawara, J.; et al. Establishment of Protocols for Global Metabolomics by LC-MS for Biomarker Discovery. PLoS ONE 2016, 11, e0160555. [Google Scholar] [CrossRef]
- Siskos, A.P.; Jain, P.; Römisch-Margl, W.; Bennett, M.; Achaintre, D.; Asad, Y.; Marney, L.; Richardson, L.; Koulman, A.; Griffin, J.L.; et al. Interlaboratory Reproducibility of a Targeted Metabolomics Platform for Analysis of Human Serum and Plasma. Anal. Chem. 2017, 89, 656–665. [Google Scholar] [CrossRef]
- Thompson, J.W.; Adams, K.J.; Adamski, J.; Asad, Y.; Borts, D.; Bowden, J.A.; Byram, G.; Dang, V.; Dunn, W.B.; Fernandez, F.; et al. International Ring Trial of a High Resolution Targeted Metabolomics and Lipidomics Platform for Serum and Plasma Analysis. Anal. Chem. 2019, 91, 14407–14416. [Google Scholar] [CrossRef]
- Weng, W.C.; Huang, W.Y.; Tang, H.Y.; Cheng, M.L.; Chen, K.H. The Differences of Serum Metabolites between Patients with Early-Stage Alzheimer’s Disease and Mild Cognitive Impairment. Front. Neurol. 2019, 10, 1223. [Google Scholar] [CrossRef]
- Tynkkynen, J.; Chouraki, V.; van der Lee, S.J.; Hernesniemi, J.; Yang, Q.; Li, S.; Beiser, A.; Larson, M.G.; Sääksjärvi, K.; Shipley, M.J.; et al. Association of branched-chain amino acids and other circulating metabolites with risk of incident dementia and Alzheimer’s disease: A prospective study in eight cohorts. Alzheimers. Dement. 2018, 14, 723–733. [Google Scholar] [CrossRef]
- Chang, K.H.; Cheng, M.L.; Tang, H.Y.; Huang, C.Y.; Wu, Y.R.; Chen, C.M. Alternations of Metabolic Profile and Kynurenine Metabolism in the Plasma of Parkinson’s Disease. Mol. Neurobiol. 2018, 55, 6319–6328. [Google Scholar] [CrossRef]
- Baranyi, A.; Meinitzer, A.; Rothenhäusler, H.B.; Amouzadeh-Ghadikolai, O.; Lewinski, D.V.; Breitenecker, R.J.; Herrmann, M. Metabolomics approach in the investigation of depression biomarkers in pharmacologically induced immune-related depression. PLoS ONE 2018, 13, e0208238. [Google Scholar] [CrossRef]
- Graham, S.F.; Turkoglu, O.; Yilmaz, A.; Ustun, I.; Ugur, Z.; Bjorndhal, T.; Han, B.; Mandal, R.; Wishart, D.; Bahado-Singh, R.O. Targeted metabolomics highlights perturbed metabolism in the brain of autism spectrum disorder sufferers. Metabolomics 2020, 16, 59. [Google Scholar] [CrossRef]
- Kilk, K.; Aug, A.; Ottas, A.; Soomets, U.; Altraja, S.; Altraja, A. Phenotyping of Chronic Obstructive Pulmonary Disease Based on the Integration of Metabolomes and Clinical Characteristics. Int. J. Mol. Sci. 2018, 19, 666. [Google Scholar] [CrossRef] [Green Version]
- Ryan, P.M.; London, L.E.; Bjorndahl, T.C.; Mandal, R.; Murphy, K.; Fitzgerald, G.F.; Shanahan, F.; Ross, R.P.; Wishart, D.S.; Caplice, N.M.; et al. Microbiome and metabolome modifying effects of several cardiovascular disease interventions in apo-E-/- mice. Microbiome 2017, 5, 30. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Lai, M.; Liu, Y.; Ronnett, G.V.; Wu, A.; Cox, B.J.; Dai, F.F.; Röst, H.L.; Gunderson, E.P.; Wheeler, M.B. Amino acid and lipid metabolism in post-gestational diabetes and progression to type 2 diabetes: A metabolic profiling study. PLoS Med. 2020, 17, e1003112. [Google Scholar] [CrossRef] [PubMed]
- Lee, H.; Jang, H.B.; Yoo, M.G.; Park, S.I.; Lee, H.J. Amino Acid Metabolites Associated with Chronic Kidney Disease: An Eight-Year Follow-Up Korean Epidemiology Study. Biomedicines 2020, 8, 222. [Google Scholar] [CrossRef] [PubMed]
- Leruez, S.; Marill, A.; Bresson, T.; de Saint Martin, G.; Buisset, A.; Muller, J.; Tessier, L.; Gadras, C.; Verny, C.; Gohier, P.; et al. A Metabolomics Profiling of Glaucoma Points to Mitochondrial Dysfunction, Senescence, and Polyamines Deficiency. Invest. Ophthalmol. Vis. Sci. 2018, 59, 4355–4361. [Google Scholar] [CrossRef]
- Klupczynska, A.; Plewa, S.; Kasprzyk, M.; Dyszkiewicz, W.; Kokot, Z.J.; Matysiak, J. Serum lipidome screening in patients with stage I non-small cell lung cancer. Clin. Exp. Med. 2019, 19, 505–513. [Google Scholar] [CrossRef] [Green Version]
- Assi, N.; Gunter, M.J.; Thomas, D.C.; Leitzmann, M.; Stepien, M.; Chajès, V.; Philip, T.; Vineis, P.; Bamia, C.; Boutron-Ruault, M.C.; et al. Metabolic signature of healthy lifestyle and its relation with risk of hepatocellular carcinoma in a large European cohort. Am. J. Clin. Nutr. 2018, 108, 117–126. [Google Scholar] [CrossRef]
- Lario, S.; Ramírez-Lázaro, M.J.; Sanjuan-Herráez, D.; Brunet-Vega, A.; Pericay, C.; Gombau, L.; Junquera, F.; Quintás, G.; Calvet, X. Plasma sample based analysis of gastric cancer progression using targeted metabolomics. Sci. Rep. 2017, 7, 17774. [Google Scholar] [CrossRef] [Green Version]
- Jelonek, K.; Krzywon, A.; Jablonska, P.; Slominska, E.M.; Smolenski, R.T.; Polanska, J.; Rutkowski, T.; Mrochem-Kwarciak, J.; Skladowski, K.; Widlak, P. Systemic Effects of Radiotherapy and Concurrent Chemo-Radiotherapy in Head and Neck Cancer Patients-Comparison of Serum Metabolome Profiles. Metabolites 2020, 10, 60. [Google Scholar] [CrossRef] [Green Version]
- Yuan, B.; Schafferer, S.; Tang, Q.; Scheffler, M.; Nees, J.; Heil, J.; Schott, S.; Golatta, M.; Wallwiener, M.; Sohn, C.; et al. A plasma metabolite panel as biomarkers for early primary breast cancer detection. Int. J. Cancer 2019, 144, 2833–2842. [Google Scholar] [CrossRef]
- Carayol, M.; Leitzmann, M.F.; Ferrari, P.; Zamora-Ros, R.; Achaintre, D.; Stepien, M.; Schmidt, J.A.; Travis, R.C.; Overvad, K.; Tjønneland, A.; et al. Blood Metabolic Signatures of Body Mass Index: A Targeted Metabolomics Study in the EPIC Cohort. J. Proteome Res. 2017, 16, 3137–3146. [Google Scholar] [CrossRef]
- Chak, C.M.; Lacruz, M.E.; Adam, J.; Brandmaier, S.; Covic, M.; Huang, J.; Meisinger, C.; Tiller, D.; Prehn, C.; Adamski, J.; et al. Ageing Investigation Using Two-Time-Point Metabolomics Data from KORA and CARLA Studies. Metabolites 2019, 9, 44. [Google Scholar] [CrossRef] [Green Version]
- Yu, B.; Zanetti, K.A.; Temprosa, M.; Albanes, D.; Appel, N.; Barrera, C.B.; Ben-Shlomo, Y.; Boerwinkle, E.; Casas, J.P.; Clish, C.; et al. The Consortium of Metabolomics Studies (COMETS): Metabolomics in 47 Prospective Cohort Studies. Am. J. Epidemiol. 2019, 188, 991–1012. [Google Scholar] [CrossRef] [Green Version]
- Bowden, J.A.; Heckert, A.; Ulmer, C.Z.; Jones, C.M.; Koelmel, J.P.; Abdullah, L.; Ahonen, L.; Alnouti, Y.; Armando, A.M.; Asara, J.M.; et al. Harmonizing lipidomics: NIST interlaboratory comparison exercise for lipidomics using SRM 1950-Metabolites in Frozen Human Plasma. J. Lipid Res. 2017, 58, 2275–2288. [Google Scholar] [CrossRef] [Green Version]
- Izumi, Y.; Matsuda, F.; Hirayama, A.; Ikeda, K.; Kita, Y.; Horie, K.; Saigusa, D.; Saito, K.; Sawada, Y.; Nakanishi, H.; et al. Inter-Laboratory Comparison of Metabolite Measurements for Metabolomics Data Integration. Metabolites 2019, 9, 257. [Google Scholar] [CrossRef] [Green Version]
- Kuriyama, S.; Yaegashi, N.; Nagami, F.; Arai, T.; Kawaguchi, Y.; Osumi, N.; Sakaida, M.; Suzuki, Y.; Nakayama, K.; Hashizume, H.; et al. The Tohoku Medical Megabank Project: Design and Mission. J. Epidemiol. 2016, 26, 493–511. [Google Scholar] [CrossRef] [Green Version]
- Fuse, N.; Sakurai-Yageta, M.; Katsuoka, F.; Danjoh, I.; Shimizu, R.; Tamiya, G.; Nagami, F.; Kawame, H.; Higuchi, S.; Kinoshita, K.; et al. Establishment of Integrated Biobank for Precision Medicine and Personalized Healthcare: The Tohoku Medical Megabank Project. JMA J. 2019, 2, 113–122. [Google Scholar] [CrossRef]
- Koshiba, S.; Motoike, I.; Saigusa, D.; Inoue, J.; Shirota, M.; Katoh, Y.; Katsuoka, F.; Danjoh, I.; Hozawa, A.; Kuriyama, S.; et al. Omics research project on prospective cohort studies from the Tohoku Medical Megabank Project. Genes Cells. 2018, 23, 406–417. [Google Scholar] [CrossRef] [Green Version]
- Tadaka, S.; Hishinuma, E.; Komaki, S.; Motoike, I.N.; Kawashima, J.; Saigusa, D.; Inoue, J.; Takayama, J.; Okamura, Y.; Aoki, Y.; et al. jMorp updates in 2020: Large enhancement of multi-omics data resources on the general Japanese population. Nucleic Acids Res. 2021, 49, D536–D544. [Google Scholar] [CrossRef]
- Koshiba, S.; Motoike, I.; Kojima, K.; Hasegawa, T.; Shirota, M.; Saito, T.; Saigusa, D.; Danjoh, I.; Katsuoka, F.; Ogishima, S.; et al. The structural origin of metabolic quantitative diversity. Sci. Rep. 2016, 6, 31463. [Google Scholar] [CrossRef] [Green Version]
- Takeda, H.; Izumi, Y.; Takahashi, M.; Paxton, T.; Tamura, S.; Koike, T.; Yu, Y.; Kato, N.; Nagase, K.; Shiomi, M.; et al. Widely-targeted quantitative lipidomics method by supercritical fluid chromatography triple quadrupole mass spectrometry. J. Lipid Res. 2018, 59, 1283–1293. [Google Scholar] [CrossRef] [Green Version]
- Burla, B.; Arita, M.; Arita, M.; Bendt, A.K.; Cazenave-Gassiot, A.; Dennis, E.A.; Ekroos, K.; Han, X.; Ikeda, K.; Liebisch, G.; et al. MS-based lipidomics of human blood plasma: A community-initiated position paper to develop accepted guidelines. J. Lipid Res. 2018, 59, 2001–2017. [Google Scholar] [CrossRef] [Green Version]
- Knittelfelder, O.L.; Weberhofer, B.P.; Eichmann, T.O.; Kohlwein, S.D.; Rechberger, G.N. A versatile ultra-high performance LC-MS method for lipid profiling. J. Chromatogr. B Analyt. Technol. Biomed. Life Sci. 2014, 951–952, 119–128. [Google Scholar] [CrossRef] [Green Version]
- Schmidt, J.A.; Fensom, G.K.; Rinaldi, S.; Scalbert, A.; Appleby, P.N.; Achaintre, D.; Gicquiau, A.; Gunter, M.J.; Ferrari, P.; Kaaks, R.; et al. Patterns in metabolite profile are associated with risk of more aggressive prostate cancer: A prospective study of 3057 matched case-control sets from EPIC. Int. J. Cancer 2020, 146, 720–730. [Google Scholar] [CrossRef] [Green Version]
- His, M.; Viallon, V.; Dossus, L.; Gicquiau, A.; Achaintre, D.; Scalbert, A.; Ferrari, P.; Romieu, I.; Onland-Moret, N.C.; Weiderpass, E.; et al. Prospective analysis of circulating metabolites and breast cancer in EPIC. BMC Med. 2019, 17, 178. [Google Scholar] [CrossRef] [PubMed]
- Saigusa, D.; Suzuki, N.; Takahashi, M.; Shiba, K.; Tanaka, S.; Abe, T.; Hishinuma, T.; Tomioka, Y. Simultaneous determination of guanidinosuccinic acid and guanidinoacetic acid in urine using high performance liquid chromatography/tandem mass spectrometry. Anal. Chim. Acta 2010, 677, 169–175. [Google Scholar] [CrossRef]
- Kanemitsu, Y.; Asaji, K.; Matsumoto, Y.; Tsukamoto, H.; Saigusa, D.; Mukawa, C.; Tachikawa, T.; Abe, T.; Tomioka, Y. Simultaneous quantitative analysis of uremic toxins by LC-MS/MS with a reversed-phase/cation-exchange/anion-exchange tri-modal mixed-mode column. J. Chromatogr. B Analyt. Technol. Biomed. Life Sci. 2017, 1068–1069, 1–8. [Google Scholar] [CrossRef] [PubMed]
- Simons, B.; Kauhanen, D.; Sylvänne, T.; Tarasov, K.; Duchoslav, E.; Ekroos, K. Shotgun Lipidomics by Sequential Precursor Ion Fragmentation on a Hybrid Quadrupole Time-of-Flight Mass Spectrometer. Metabolites 2012, 2, 195–213. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Wormwood Moser, K.L.; Van Aken, G.; DeBord, D.; Hatcher, N.G.; Maxon, L.; Sherman, M.; Yao, L.; Ekroos, K. High-defined quantitative snapshots of the ganglioside lipidome using high resolution ion mobility SLIM assisted shotgun lipidomics. Anal. Chim. Acta 2021, 1146, 77–87. [Google Scholar] [CrossRef]
- Oberacher, H.; Arnhard, K.; Linhart, C.; Diwo, A.; Marksteiner, J.; Humpel, C. Targeted Metabolomic Analysis of Soluble Lysates from Platelets of Patients with Mild Cognitive Impairment and Alzheimer’s Disease Compared to Healthy Controls: Is PC aeC40:4 a Promising Diagnostic Tool? J. Alzheimers. Dis. 2017, 57, 493–504. [Google Scholar] [CrossRef]
- Liebisch, G.; Ekroos, K.; Hermansson, M.; Ejsing, C.S. Reporting of lipidomics data should be standardized. Biochim. Biophys. Acta Mol. Cell Biol. Lipids. 2017, 1862, 747–751. [Google Scholar] [CrossRef]
- Lange, M.; Fedorova, M. Evaluation of lipid quantification accuracy using HILIC and RPLC MS on the example of NIST® SRM® 1950 metabolites in human plasma. Anal. Bioanal. Chem. 2020, 412, 3573–3584. [Google Scholar] [CrossRef] [Green Version]
- Okudaira, M.; Inoue, A.; Shuto, A.; Nakanaga, K.; Kano, K.; Makide, K.; Saigusa, D.; Tomioka, Y.; Aoki, J. Separation and quantification of 2-acyl-1-lysophospholipids and 1-acyl-2-lysophospholipids in biological samples by LC-MS/MS. J. Lipid Res. 2014, 55, 2178–2192. [Google Scholar] [CrossRef] [Green Version]
- Saigusa, D.; Okudaira, M.; Wang, J.; Kano, K.; Kurano, M.; Uranbileg, B.; Ikeda, H.; Yatomi, Y.; Motohashi, H.; Aoki, J. Simultaneous Quantification of Sphingolipids in Small Quantities of Liver by LC-MS/MS. Mass Spectrom. 2014, 3, S0046. [Google Scholar] [CrossRef] [Green Version]
- Schwalbe-Herrmann, M.; Willmann, J.; Leibfritz, D. Separation of phospholipid classes by hydrophilic interaction chromatography detected by electrospray ionization mass spectrometry. J. Chromatogr. A 2010, 1217, 5179–5183. [Google Scholar] [CrossRef]
- Cífková, E.; Holčapek, M.; Lísa, M.; Ovčačíková, M.; Lyčka, A.; Lynen, F.; Sandra, P. Nontargeted quantitation of lipid classes using hydrophilic interaction liquid chromatography-electrospray ionization mass spectrometry with single internal standard and response factor approach. Anal. Chem. 2012, 84, 10064–10070. [Google Scholar] [CrossRef]
- Lísa, M.; Holčapek, M. High-Throughput and Comprehensive Lipidomic Analysis Using Ultrahigh-Performance Supercritical Fluid Chromatography-Mass Spectrometry. Anal. Chem. 2015, 87, 7187–7195. [Google Scholar] [CrossRef]
- Takeda, H.; Takahashi, M.; Hara, T.; Izumi, Y.; Bamba, T. Improved quantitation of lipid classes using supercritical fluid chromatography with a charged aerosol detector. J. Lipid Res. 2019, 60, 1465–1474. [Google Scholar] [CrossRef]
- Liebisch, G.; Ahrends, R.; Arita, M.; Arita, M.; Bowden, J.A.; Ejsing, C.S.; Griffiths, W.J.; Holčapek, M.; Köfeler, H.; Mitchell, T.W.; et al. Lipidomics needs more standardization. Nat. Metab. 2019, 1, 745–747. [Google Scholar] [CrossRef] [Green Version]
- Hishinuma, E.; Shimada, M.; Matsukawa, N.; Saigusa, D.; Li, B.; Kudo, K.; Tsuji, K.; Shigeta, S.; Tokunaga, H.; Kumada, K.; et al. Wide-Targeted Metabolome Analysis Identifies Potential Biomarkers for Prognosis Prediction of Epithelial Ovarian Cancer. Toxins 2021, 13, 461. [Google Scholar] [CrossRef]
- Koshiba, S.; Motoike, I.N.; Saigusa, D.; Inoue, J.; Aoki, Y.; Tadaka, S.; Shirota, M.; Katsuoka, F.; Tamiya, G.; Minegishi, N.; et al. Identification of critical genetic variants associated with metabolic phenotypes of the Japanese population. Commun. Biol. 2020, 3, 662. [Google Scholar] [CrossRef]
- Simón-Manso, Y.; Lowenthal, M.S.; Kilpatrick, L.E.; Sampson, M.L.; Telu, K.H.; Rudnick, P.A.; Mallard, W.G.; Bearden, D.W.; Schock, T.B.; Tchekhovskoi, D.V.; et al. Metabolite profiling of a NIST Standard Reference Material for human plasma (SRM 1950): GC-MS, LC-MS, NMR, and clinical laboratory analyses, libraries, and web-based resources. Anal. Chem. 2013, 85, 11725–11731. [Google Scholar] [CrossRef]
- Bligh, E.G.; Dyer, W.J. A rapid method of total lipid extraction and purification. Can. J. Biochem. Physiol. 1959, 37, 911–917. [Google Scholar] [CrossRef] [Green Version]
- Folch, J.; Lees, M.; Sloane Stanley, G.H. A simple method for the isolation and purification of total lipides from animal tissues. J. Biol. Chem. 1957, 226, 497–509. [Google Scholar] [CrossRef]








{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
| Method | Age (N) | BMI | Cre | Glc | |
|---|---|---|---|---|---|
| Kit-Met 1 | All | 59.6 ± 12.9 (6159) | 22.7 ± 3.3 (6148) | 0.690 ± 0.170 (6158) | 88.4 ± 16.3 (6156) |
| M | 62.5 ± 12.4 (1926) | 23.7 ± 2.9 (1921) | 0.842 ± 0.189 (1925) | 93.1 ± 19.5 (1925) | |
| F | 58.2 ± 12.8 (4233) | 22.2 ± 3.3 (4227) | 0.621 ± 0.102 (4233) | 86.3 ± 14.2 (4231) | |
| Kit-Met 2 | All | 59.1 ± 13.8 (2541) | 22.9 ± 3.3 (2539) | 0.718 ± 0.289 (2541) | 90.2 ± 17.5 (2539) |
| M | 63.7 ± 11.7 (1085) | 23.7 ± 2.8 (1085) | 0.849 ± 0.350 (1085) | 94.7 ± 20.6 (1085) | |
| F | 55.7 ± 14.2 (1456) | 22.4 ± 3.6 (1454) | 0.620 ± 0.180 (1456) | 86.9 ± 14.0 (1454) |
| Method | Cohort Plasma Samples (77/Plate) | NIST Plasma Sample (1/Plate) | gQC Plasma Sample (4/Plate) |
|---|---|---|---|
| Kit-Met 1 | 6159 (80 plates *) | 80 | 320 |
| Kit-Met 2 | 2541 (33 plates) | 33 | 132 |
Publisher’s Note: MDPI stays neutral with regard to jurisdictional claims in published maps and institutional affiliations. |
© 2021 by the authors. Licensee MDPI, Basel, Switzerland. This article is an open access article distributed under the terms and conditions of the Creative Commons Attribution (CC BY) license (https://creativecommons.org/licenses/by/4.0/).
Share and Cite
Saigusa, D.; Hishinuma, E.; Matsukawa, N.; Takahashi, M.; Inoue, J.; Tadaka, S.; Motoike, I.N.; Hozawa, A.; Izumi, Y.; Bamba, T.; et al. Comparison of Kit-Based Metabolomics with Other Methodologies in a Large Cohort, towards Establishing Reference Values. Metabolites 2021, 11, 652. https://doi.org/10.3390/metabo11100652
Saigusa D, Hishinuma E, Matsukawa N, Takahashi M, Inoue J, Tadaka S, Motoike IN, Hozawa A, Izumi Y, Bamba T, et al. Comparison of Kit-Based Metabolomics with Other Methodologies in a Large Cohort, towards Establishing Reference Values. Metabolites. 2021; 11(10):652. https://doi.org/10.3390/metabo11100652
Chicago/Turabian StyleSaigusa, Daisuke, Eiji Hishinuma, Naomi Matsukawa, Masatomo Takahashi, Jin Inoue, Shu Tadaka, Ikuko N. Motoike, Atsushi Hozawa, Yoshihiro Izumi, Takeshi Bamba, and et al. 2021. "Comparison of Kit-Based Metabolomics with Other Methodologies in a Large Cohort, towards Establishing Reference Values" Metabolites 11, no. 10: 652. https://doi.org/10.3390/metabo11100652
APA StyleSaigusa, D., Hishinuma, E., Matsukawa, N., Takahashi, M., Inoue, J., Tadaka, S., Motoike, I. N., Hozawa, A., Izumi, Y., Bamba, T., Kinoshita, K., Ekroos, K., Koshiba, S., & Yamamoto, M. (2021). Comparison of Kit-Based Metabolomics with Other Methodologies in a Large Cohort, towards Establishing Reference Values. Metabolites, 11(10), 652. https://doi.org/10.3390/metabo11100652