
The “Common Soil Hypothesis” Revisited—Risk Factors for Type 2 Diabetes and Cardiovascular Disease


Abstract
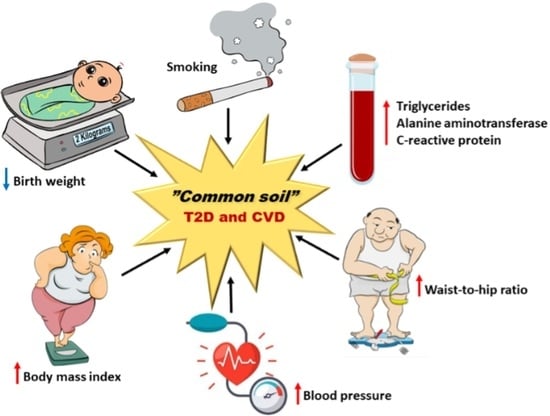
:
1. Introduction
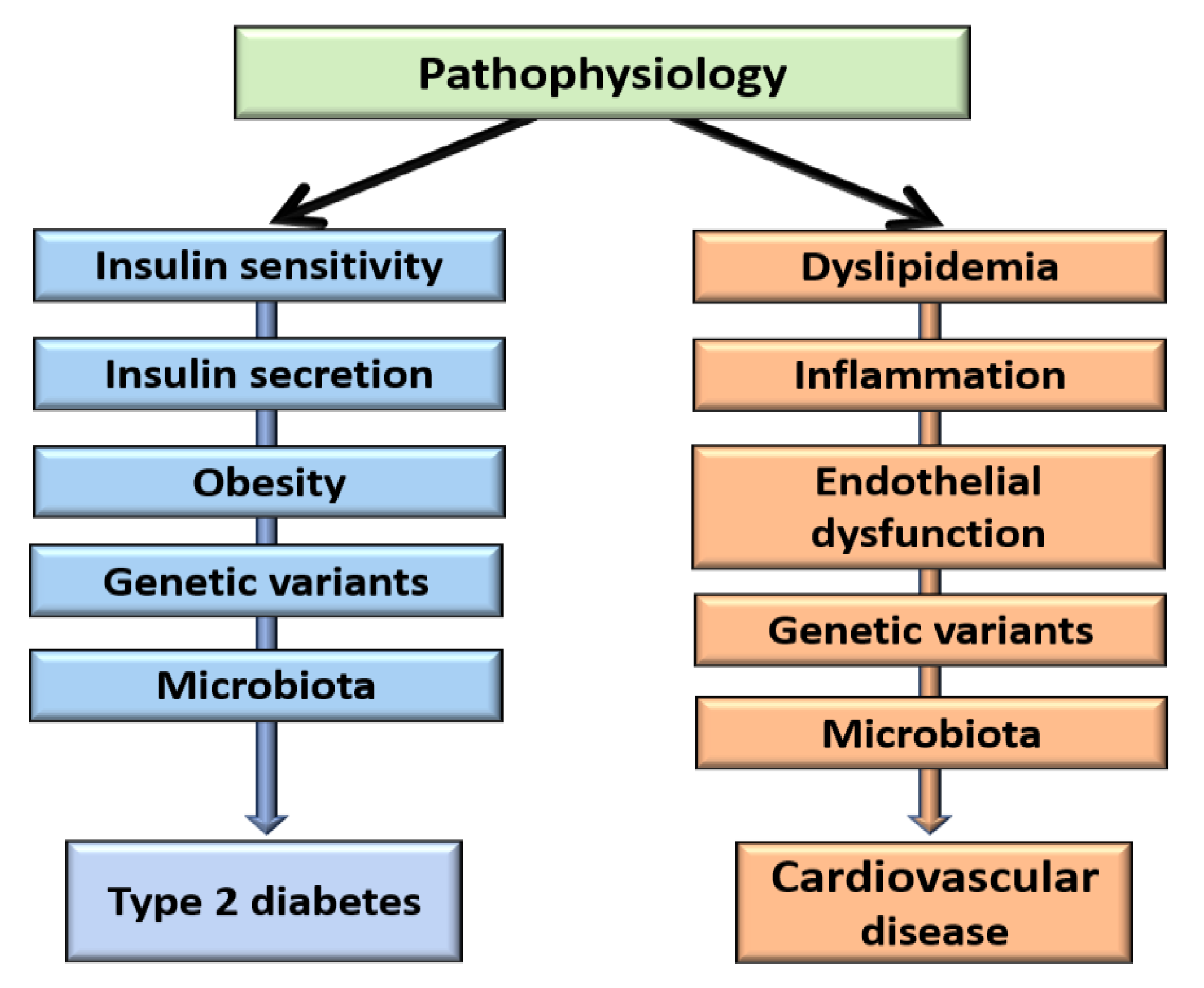
2. Pathophysiology of Type 2 Diabetes
3. Pathophysiology of Cardiovascular Diseases
4. Microbiota in Type 2 Diabetes and Cardiovascular Disease
4.1. Type 2 Diabetes
4.2. Cardiovascular Disease
5. Approaches to Identify Risk Factors for Type 2 Diabetes and Cardiovascular Disease
5.1. Population-Based Studies
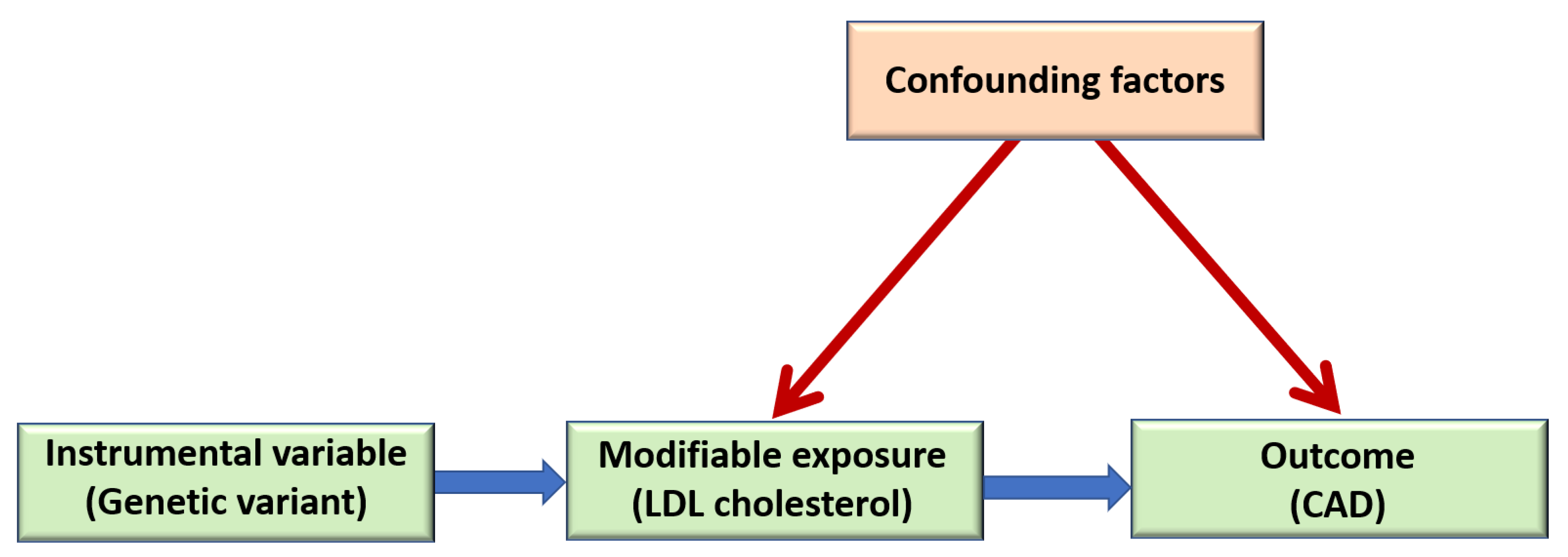
5.2. Mendelian Randomization Studies
6. Is Type 2 Diabetes a Causal Risk Factor for Cardiovascular Disease?
7. Risk Factors for Type 2 Diabetes and Cardiovascular Disease: Evidence from Mendelian Randomization Studies
7.1. Type 2 Diabetes
7.2. Cardiovascular Disease
7.3. Mendelian Randomization Studies in Type 2 Diabetes and Cardiovascular Disease: Are There Differences?
8. Mendelian Randomization Studies on Metabolites in Type 2 Diabetes and Cardiovascular Disease
9. Towards “Precision Medicine”
10. “Common Soil” Hypothesis—Does It Apply to T2D and CVD?
11. Future Perspectives
Author Contributions
Funding
Conflicts of Interest
References
- International Diabetes Federation. Available online: https://www.idf.org/ (accessed on 20 July 2021).
- Laakso, M.; Kuusisto, J. Insulin resistance and hyperglycaemia in cardiovascular disease development. Nat. Rev. Endocrinol. 2014, 10, 293–302. [Google Scholar] [CrossRef] [PubMed]
- World Health Organization. Available online: https://www.who.int/health-topics/cardiovascular-diseases (accessed on 20 July 2021).
- Tzoulaki, I.; Elliott, P.; Kontis, V.; Ezzati, M. Worldwide exposures to cardiovascular risk factors and associated health effects: Current knowledge and data gaps. Circulation 2016, 133, 2314–2333. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Stern, M.P. Diabetes and cardiovascular disease. The “common soil” hypothesis. Diabetes 1995, 44, 369–374. [Google Scholar] [CrossRef]
- Peters, T.M.; Holmes, M.V.; Richards, J.B.; Palmer, T.; Vincenzo, F.; Lindgren, C.M.; Folkert, W.; Asselbergs, F.W.; Nelson, C.P.; Samani, N.J.; et al. Sex differences in the risk of coronary heart disease associated with type 2 diabetes: A Mendelian randomization analysis. Diabetes Care 2020, 44, 556–562. [Google Scholar] [CrossRef]
- Herder, C.; Karakas, M.; Koenig, W. Biomarkers for the prediction of type 2 diabetes and cardiovascular disease. Clin. Pharmacol. Ther. 2011, 90, 52–66. [Google Scholar] [CrossRef]
- Mahajan, A.; Taliun, D.; Thurner, M.; Robertson, N.R.; Torres, J.M.; Rayner, N.W.; Payne, A.J.; Steinthorsdottir, V.; Scott, R.A.; Grarup, N.; et al. Fine-mapping of an expanded set of type 2 diabetes loci to single-variant resolution using high-density imputation and islet-specific epigenome maps. Nat. Genet. 2018, 50, 1505–1513. [Google Scholar] [CrossRef] [Green Version]
- Strawbridge, R.J.; van Zuydam, N.R. Shared genetic contribution of type 2 diabetes and cardiovascular disease: Implications for prognosis and treatment. Curr. Diabetes Rep. 2018, 18, 59. [Google Scholar] [CrossRef] [Green Version]
- Saxena, R.; Voight, B.F.; Lyssenko, V.; Burtt, N.P.; de Bakker, P.I.; Chen, H.; Roix, J.J.; Kathiresan, S.; Hirschhorn, J.N.; Daly, M.J.; et al. Genome-wide association analysis identifies loci for type 2 diabetes and triglyceride levels. Science 2007, 316, 1331–1336. [Google Scholar]
- Helgadottir, A.; Thorleifsson, G.; Manolescu, A.; Gretarsdottir, S.; Blondal, T.; Jonasdottir, A.; Jonasdottir, A.; Sigurdsson, A.; Baker, A.; Palsson, A. A common variant on chromosome 9p21 affects the risk of myocardial infarction. Science 2007, 316, 1491–1493. [Google Scholar] [CrossRef] [PubMed]
- Klarin, D.; Zhu, Q.M.; Emdin, C.A.; Chaffin, M.; Horner, S.; McMillan, B.J.; Leed, A.; Weale, M.E.; Spencer, C.C.A.; Aguet, F.; et al. Genetic analysis in UK Biobank links insulin resistance and transendothelial migration pathways to coronary artery disease. Nat. Genet. 2017, 49, 1392–1397. [Google Scholar] [CrossRef]
- Goodarzi, M.O.; Rotter, J.I. Genetics insights in the relationship between type 2 diabetes and coronary heart disease. Circ. Res. 2020, 126, 1526–1548. [Google Scholar] [CrossRef] [PubMed]
- Laakso, M. Is insulin resistance a feature of or a primary risk factor for cardiovascular disease? Curr. Diabetes Rep. 2015, 15, 105. [Google Scholar] [CrossRef]
- Shu, L.; Chan, K.H.K.; Zhang, G.; Huan, T.; Kurt, Z.; Zhao, Y.; Codoni, V.; Trégouët, D.A.; Consortium, C.; Yang, J.; et al. Shared genetic regulatory networks for cardiovascular disease and type 2 diabetes in multiple populations of diverse ethnicities in the United States. PLoS Genet. 2017, 13, e1007040. [Google Scholar] [CrossRef] [Green Version]
- Chen, Y.; Qin, Z.; Wang, Y.; Li, X.; Zheng, Y.; Liu, Y. Role of Inflammation in Vascular Disease-Related Perivascular Adipose Tissue Dysfunction. Front. Endocrinol. 2021, 12, 710842. [Google Scholar] [CrossRef] [PubMed]
- Salunkhe, V.A.; Veluthakal, R.; Kahn, S.E.; Thurmond, D.C. Novel approaches to restore beta cell function in prediabetes and type 2 diabetes. Diabetologia 2018, 61, 1895–1901. [Google Scholar] [CrossRef] [Green Version]
- Tura, A.; Bagger, J.I.; Ferrannini, E.; Holst, J.J.; Knop, F.K.; Vilsbøll, T.; Mary, A. Impaired beta cell sensitivity to incretins in type 2 diabetes is insufficiently compensated by higher incretin response. Nutr. Metab. Cardiovasc. Dis. 2017, 27, 1123–1129. [Google Scholar] [CrossRef]
- DeFronzo, R.A. From the triumvirate to the ominous octet: A new paradigm for the treatment of type 2 diabetes mellitus. Diabetes 2009, 58, 773–795. [Google Scholar] [CrossRef] [Green Version]
- Deeb, S.S.; Fajas, L.; Nemoto, M.; Pihlajamäki, J.; Mykkänen, L.; Kuusisto, J.; Laakso, M.; Fujimoto, W.; Auwerx, J. A Pro12Ala substitution in PPARgamma2 associated with decreased receptor activity, lower body mass index and improved insulin sensitivity. Nat. Genet. 1998, 20, 284–287. [Google Scholar] [CrossRef] [PubMed]
- James, D.E.; Stöckli, J.; Birnbaum, M.J. The aetiology and molecular landscape of insulin resistance. Nat. Rev. Mol. Cell Biol. 2021. [Google Scholar] [CrossRef]
- Stančáková, A.; Kuulasmaa, T.; Kuusisto, J.; Mohlke, K.L.; Collins, F.S.; Boehnke, M.; Laakso, M. Genetic risk scores in the prediction of plasma glucose, impaired insulin secretion, insulin resistance and incident type 2 diabetes in the METSIM study. Diabetologia 2017, 60, 1722–1730. [Google Scholar] [CrossRef]
- Nayor, M.; Brown, K.J.; Vasan, R.S. The molecular basis of predicting atherosclerotic cardiovascular disease risk. Circ. Res. 2021, 128, 287–303. [Google Scholar] [CrossRef]
- Doran, S.; Arif, M.; Lam, S.; Bayraktar, A.; Turkez, H.; Uhlen, M.; Boren, J.; Mardinoglu, A. Multi-omics approaches for revealing the complexity of cardiovascular disease. Brief. Bioinform. 2021, 17, bbab061. [Google Scholar] [CrossRef] [PubMed]
- Semaev, S.; Shakhtshneider, E. Genetic risk score for coronary heart disease: Review. J. Pers. Med. 2020, 10, 239. [Google Scholar] [CrossRef]
- Inouye, M.; Abraham, G.; Nelson, C.P.; Wood, A.M.; Sweeting, M.J.; Dudbridge, F.; Lai, F.Y.; Kaptoge, S.; Brozynska, M.; Wang, T.; et al. Genomic risk prediction of coronary artery disease in 480,000 adults: Implications for primary prevention. J. Am. Coll. Cardiol. 2018, 72, 1883–1893. [Google Scholar] [CrossRef] [PubMed]
- Lindstrom, J.; Tuomilehto, J. The diabetes risk score: A practical tool to predict type 2 diabetes risk. Diabetes Care 2003, 26, 725–731. [Google Scholar] [CrossRef] [Green Version]
- Saaristo, T.; Peltonen, M.; Lindström, J.; Saarikoski, L.; Sundvall, J.; Eriksson, J.G.; Tuomilehto, J. Cross-sectional evaluation of the Finnish Diabetes Risk Score: A tool to identify undetected type 2 diabetes, abnormal glucose tolerance and metabolic syndrome. Diabetes Vasc. Dis. Res. 2005, 2, 67–72. [Google Scholar] [CrossRef] [PubMed]
- Fizelova, M.; Jauhiainen, R.; Stančáková, A.; Kuusisto, J.; Laakso, M. Finnish Diabetes Risk Score Is Associated with Impaired Insulin Secretion and Insulin Sensitivity, Drug-Treated Hypertension and Cardiovascular Disease: A Follow-Up Study of the METSIM Cohort. PLoS ONE 2016, 11, e0166584. [Google Scholar] [CrossRef]
- Sonnenburg, J.L.; Bäckhed, F. Diet-microbiota interactions as moderators of human metabolism. Nature 2016, 535, 56–64. [Google Scholar] [CrossRef]
- Tang, W.H.; Kitai, T.; Hazen, S.L. Gut microbiota in cardiovascular health and disease. Circ. Res. 2017, 120, 1183–1196. [Google Scholar] [CrossRef] [Green Version]
- Lee, C.J.; Sears, C.L.; Maruthur, N. Gut microbiome and its role in obesity and insulin resistance. Ann. N. Y. Acad. Sci. 2020, 1461, 37–52. [Google Scholar] [CrossRef]
- Sircana, A.; Framarin, L.; Leone, N.; Berrutti, M.; Castellino, F.; Parente, R.; De Michieli, F.; Paschetta, E.; Musso, G. Altered gut microbiota in type 2 diabetes: Just a coincidence? Curr. Diabetes Rep. 2018, 18, 98. [Google Scholar] [CrossRef]
- Witkowski, M.; Weeks, T.L.; Hazen, S.L. Gut microbiota and cardiovascular disease. Circ. Res. 2020, 127, 553–570. [Google Scholar] [CrossRef] [PubMed]
- Vangipurapu, J.; Fernandes Silva, L.; Kuulasmaa, T.; Smith, U.; Laakso, M. Microbiota-related metabolites and the risk of type 2 diabetes. Diabetes Care 2020, 43, 1319–1325. [Google Scholar] [CrossRef] [PubMed]
- Zhuang, R.; Ge, X.; Han, L.; Yu, P.; Gong, X.; Meng, Q.; Zhang, Y.; Fan, H.; Zheng, L.; Liu, Z.; et al. Gut microbe-generated metabolite trimethylamine N-oxide and the risk of diabetes: A systematic review and dose-response meta-analysis. Obes. Rev. 2019, 20, 883–894. [Google Scholar] [CrossRef]
- Zhu, T.; Goodarzi, M.O. Metabolites linking the gut microbiome with risk for type 2 diabetes. Curr. Nutr. Rep. 2020, 9, 83–93. [Google Scholar] [CrossRef] [PubMed]
- Sanna, S.; van Zuydam, N.R.; Mahajan, A.; Kurilshikov, A.; Vich Vila, A.; Võsa, U.; Mujagic, Z.; Masclee, A.A.M.; Jonkers, D.M.A.E.; Oosting, M.; et al. Causal relationships among the gut microbiome, short-chain fatty acids and metabolic diseases. Nat. Genet. 2019, 51, 600–605. [Google Scholar] [CrossRef]
- Le Chatelier, E.; Nielsen, T.; Qin, J.; Prifti, E.; Hildebrand, F.; Falony, G.; Almeida, M.; Arumugam, M.; Batto, J.M.; Kennedy, S.; et al. Richness of human gut microbiome correlates with metabolic markers. Nature 2013, 500, 541–546. [Google Scholar] [CrossRef] [PubMed]
- Craciun, S.; Balskus, E.P. Microbial conversion of choline to trimethylamine requires a glycyl radical enzyme. Proc. Natl. Acad. Sci. USA 2012, 109, 21307–21312. [Google Scholar] [CrossRef] [Green Version]
- Tang, W.H.; Wang, Z.; Levison, B.S.; Koeth, R.A.; Britt, E.B.; Fu, X.; Wu, Y.; Hazen, S.L. Intestinal microbial metabolism of phosphatidylcholine and cardiovascular risk. N. Engl. J. Med. 2013, 368, 1575–1584. [Google Scholar] [CrossRef] [Green Version]
- Schiattarella, G.G.; Sannino, A.; Toscano, E.; Giugliano, G.; Gargiulo, G.; Franzone, A.; Trimarco, B.; Esposito, G.; Perrino, C. Gut microbe-generated metabolite trimethylamine-N-oxide as cardiovascular risk biomarker: A systematic review and dose-response meta-analysis. Eur. Heart J. 2017, 38, 2948–2956. [Google Scholar] [CrossRef] [Green Version]
- Haghikia, A.; Li, X.S.; Liman, T.G.; Bledau, N.; Schmidt, D.; Zimmermann, F.; Kränkel, N.; Widera, C.; Sonnenschein, K.; Haghikia, A.; et al. Gut microbiota-dependent trimethylamine n-oxide predicts risk of cardiovascular events in patients with stroke and is related to proinflammatory monocytes. Arterioscler. Thromb. Vasc. Biol. 2018, 38, 2225–2235. [Google Scholar] [CrossRef] [Green Version]
- Jia, J.; Dou, P.; Gao, M.; Kong, X.; Li, C.; Liu, Z.; Huang, T. Assessment of causal direction between gut microbiota-dependent metabolites and cardiometabolic health: A bidirectional Mendelian randomization analysis. Diabetes 2019, 68, 1747–1755. [Google Scholar] [CrossRef] [PubMed]
- Mamic, P.; Chaikijurajai, T.; Wilson Tang, W.H. Gut microbiome—A potential mediator of pathogenesis in heart failure and its comorbidities: State-of-the-art review. J. Mol. Cell. Cardiol. 2021, 152, 105–117. [Google Scholar] [CrossRef] [PubMed]
- Laakso, M.; Kuusisto, J.; Stančáková, A.; Kuulasmaa, T.; Pajukanta, P.; Lusis, A.J.; Collins, F.S.; Mohlke, K.L.; Boehnke, M. The Metabolic Syndrome in Men study: A resource for studies of metabolic and cardiovascular diseases. J. Lipid Res. 2017, 58, 481–493. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- American Diabetes Association. Diagnosis and classification of diabetes mellitus. Diabetes Care 2013, 36, S67–S74. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Laakso, M. Biomarkers for type 2 diabetes. Mol. Metab. 2019, 27, S139–S146. [Google Scholar] [CrossRef] [PubMed]
- Vangipurapu, J.; Stancáková, A.; Smith, U.; Kuusisto, J.; Laakso, M. Nine amino acids are associated with decreased insulin secretion and elevated glucose levels in a 7.4-year follow-up study of 5181 Finnish men. Diabetes 2019, 68, 1353–1358. [Google Scholar] [CrossRef]
- Bellou, V.; Belbasis, L.; Tzoulaki, I.; Evangelou, E. Risk factors for type 2 diabetes mellitus: An exposure-wide umbrella review of meta-analyses. PLoS ONE 2018, 13, e0194127. [Google Scholar] [CrossRef]
- Mardinoglu, A.; Stančáková, A.; Lotta, L.A.; Kuusisto, J.; Boren, J.; Blüher, M.; Wareham, N.J.; Ferrannini, E.; Groop, P.H.; Laakso, M.; et al. Plasma mannose levels are associated with incident type 2 diabetes and cardiovascular disease. Cell Metab. 2017, 26, 281–283. [Google Scholar] [CrossRef] [Green Version]
- Lee, S.; Zhang, C.; Kilicarslan, M.; Piening, B.D.; Bjornson, E.; Hallström, B.M.; Groen, A.K.; Ferrannini, E.; Laakso, M.; Snyder, M.; et al. Integrated network analysis reveals an association between plasma mannose levels and insulin resistance. Cell Metab. 2016, 24, 172–184. [Google Scholar] [CrossRef] [Green Version]
- Porcu, E.; Gilardi, F.; Darrous, L.; Yengo, L.; Bararpour, N.; Gasser, M.; Marques-Vidal, P.; Froguel, P.; Waeber, G.; Thomas, A.; et al. Triangulating evidence from longitudinal and Mendelian randomization studies of metabolomic biomarkers for type 2 diabetes. Sci. Rep. 2021, 11, 6197. [Google Scholar] [CrossRef]
- Holmes, M.V.; Ala-Korpela, M.; Smith, G.D. Mendelian randomization in cardiometabolic disease: Challenges in evaluating causality. Nat. Rev. Cardiol. 2017, 14, 577–590. [Google Scholar] [CrossRef] [Green Version]
- Swerdlow, D.I.; Hingorani, A.D.; Humphries, S.E. Genetic risk factors and Mendelian randomization in cardiovascular disease. Curr. Cardiol. Rep. 2015, 17, 33. [Google Scholar] [CrossRef]
- Burgess, S.; Foley, C.N.; Allara, E.; Staley, J.R.; Howson, J.M.M. A robust and efficient method for Mendelian randomization with hundreds of genetic variants. Nat. Commun. 2020, 11, 376. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Sheehan, N.A.; Didelez, V.; Burton, P.R.; Tobin, M.D. Mendelian randomisation and causal inference in observational epidemiology. PLoS Med. 2008, 5, e177. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Deloukas, P.; Kanoni, S.; Willenborg, C.; Farrall, M.; Assimes, T.L.; Thompson, J.R.; Ingelsson, E.; Saleheen, D.; Erdmann, J.; Goldstein, B.A.; et al. Large-scale association analysis identifies new risk loci for coronary artery disease. Nat. Genet. 2013, 45, 25–33. [Google Scholar] [CrossRef]
- Ahmad, O.S.; Morris, J.A.; Mujammami, M.; Forgetta, V.; Leong, A.; Li, R.; Turgeon, M.; Greenwood, C.M.; Thanassoulis, G.; Meigs, J.B.; et al. A Mendelian randomization study of the effect of type-2 diabetes on coronary heart disease. Nat. Commun. 2015, 6, 7060. [Google Scholar] [CrossRef]
- Gan, W.; Bragg, F.; Walters, R.G.; Millwood, I.Y.; Lin, K.; Chen, Y.; Guo, Y.; Vaucher, J.; Bian, Z.; Bennett, D.; et al. Genetic predisposition to type 2 diabetes and risk of subclinical atherosclerosis and cardiovascular diseases among 160,000 Chinese adults. Diabetes 2019, 68, 2155–2164. [Google Scholar] [CrossRef]
- Larsson, S.C.; Scott, R.A.; Traylor, M.; Langenberg, C.C.; Hindy, G.; Melander, O.; Orho-Melander, M.; Seshadri, S.; Wareham, N.J.; Markus, H.S.; et al. Type 2 diabetes, glucose, insulin, BMI, and ischemic stroke subtypes: Mendelian randomization study. Neurology 2017, 89, 454–460. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Tikkanen, E.; Pirinen, M.; Sarin, A.P.; Havulinna, A.S.; Männistö, S.; Saltevo, J.; Lokki, M.L.; Sinisalo, J.; Lundqvist, A.; Jula, A.; et al. Genetic support for the causal role of insulin in coronary heart disease. Diabetologia 2016, 59, 2369–2377. [Google Scholar] [CrossRef] [Green Version]
- Zhan, Y.; Karlsson, I.K.; Karlsson, R.; Tillander, A.; Reynolds, C.A.; Pedersen, N.L.; Hägg, S. Exploring the causal pathway from telomere length to coronary heart disease: A network Mendelian randomization study. Circ. Res. 2017, 121, 214–219. [Google Scholar] [CrossRef] [Green Version]
- Zhao, J.V.; Luo, S.; Schooling, C.M. Sex-specific Mendelian randomization study of genetically predicted insulin and cardiovascular events in the UK Biobank. Commun. Biol. 2019, 2, 332. [Google Scholar] [CrossRef] [Green Version]
- Ross, S.; Gerstein, H.C.; Eikelboom, J.; Anand, S.S.; Yusuf, S.; Paré, G. Mendelian randomization analysis supports the causal role of dysglycaemia and diabetes in the risk of coronary artery disease. Eur. Heart J. 2015, 36, 1454–1462. [Google Scholar] [CrossRef] [Green Version]
- Laakso, M. How good a marker is insulin level for insulin resistance? Am. J. Epidemiol. 1993, 137, 959–965. [Google Scholar] [CrossRef]
- Chatterjee, S.; Khunti, K.; Davies, M.J. Type 2 diabetes. Lancet 2017, 389, 2239–2251. [Google Scholar] [CrossRef]
- Wang, T.; Huang, T.; Li, Y.; Zheng, Y.; Manson, J.E.; Hu, F.B.; Qi, L. Low birthweight and risk of type 2 diabetes: A Mendelian randomisation study. Diabetologia 2016, 59, 1920–1927. [Google Scholar] [CrossRef] [PubMed]
- Zanetti, D.; Tikkanen, E.; Gustafsson, S.; Priest, J.R.; Burgess, S.; Ingelsson, E. Birthweight, type 2 diabetes mellitus, and cardiovascular disease: Addressing the barker hypothesis with Mendelian randomization. Circ. Genom. Precis. Med. 2018, 11, e002054. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Corbin, L.J.; Richmond, R.C.; Wade, K.H.; Burgess, S.; Bowden, J.; Smith, G.D.; Timpson, N.J. BMI as a modifiable risk factor for type 2 diabetes: Refining and understanding causal estimates using Mendelian randomization. Diabetes 2016, 65, 3002–3007. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Gill, D.; Zuber, V.; Dawson, J.; Pearson-Stuttard, J.; Carter, A.R.; Sanderson, E.; Karhunen, V.; Levin, M.G.; Wootton, R.E.; Klarin, D.; et al. Risk factors mediating the effect of body mass index and waist-to-hip ratio on cardiovascular outcomes: Mendelian randomization analysis. Int. J. Obes. 2021, 45, 1428–1438. [Google Scholar] [CrossRef] [PubMed]
- Dale, C.E.; Fatemifar, G.; Palmer, T.M.; White, J.; Prieto-Merino, D.; Zabaneh, D.; Engmann, J.E.L.; Shah, T.; Wong, A.; Warren, H.R.; et al. Causal associations of adiposity and body fat distribution with coronary heart disease, stroke subtypes, and type 2 diabetes mellitus: A Mendelian randomization analysis. Circulation 2017, 135, 2373–2388. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Aikens, R.C.; Zhao, W.; Saleheen, D.; Reilly, M.P.; Epstein, S.E.; Tikkanen, E.; Salomaa, V.; Voight, B.F. Systolic blood pressure and risk of type 2 diabetes: A Mendelian randomization study. Diabetes 2017, 66, 543–550. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Malik, R.; Georgakis, M.K.; Vujkovic, M.; Damrauer, S.M.; Elliott, P.; Karhunen, V.; Giontella, A.; Fava, C.; Hellwege, J.N.; Shuey, M.M.; et al. Relationship between blood pressure and incident cardiovascular disease: Linear and nonlinear Mendelian randomization analyses. Hypertension 2021, 77, 2004–2013. [Google Scholar] [CrossRef]
- Yuan, S.; Larsson, S.C. A causal relationship between cigarette smoking and type 2 diabetes mellitus: A Mendelian randomization study. Sci. Rep. 2019, 9, 19342. [Google Scholar] [CrossRef]
- Levin, M.G.; Klarin, D.; Assimes, T.L.; Freiberg, M.S.; Ingelsson, E.; Lynch, J.; Natarajan, P.; O’Donnell, C.; Rader, D.J.; Tsao, P.S.; et al. Genetics of smoking and risk of atherosclerotic cardiovascular diseases: A Mendelian randomization study. JAMA Netw. Open 2021, 4, e2034461. [Google Scholar] [CrossRef]
- Zanetti, D.; Gustafsson, S.; Assimes, T.L.; Ingelsson, E. Comprehensive investigation of circulating biomarkers and their causal role in atherosclerosis-related risk factors and clinical events. Circ. Genom. Precis. Med. 2020, 13, e002996. [Google Scholar] [CrossRef]
- Niu, W.; Liu, Y.; Qi, Y.; Wu, Z.; Zhu, D.; Jin, W. Association of interleukin-6 circulating levels with coronary artery disease: A meta-analysis implementing mendelian randomization approach. Int. J. Cardiol. 2012, 157, 243–252. [Google Scholar] [CrossRef]
- Richardson, T.G.; Sanderson, E.; Palmer, T.M.; Ala-Korpela, M.; Ference, B.A.; Davey Smith, G.; Holmes, M.V. Evaluating the relationship between circulating lipoprotein lipids and apolipoproteins with risk of coronary heart disease: A multivariable Mendelian randomisation analysis. PLoS Med. 2020, 17, e1003062. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Glezeva, N.; Chisale, M.; McDonald, K.; Ledwidge, M.; Gallagher, J.; Watson, C.J. Diabetes and complications of the heart in Sub-Saharan Africa: An urgent need for improved awareness, diagnostics and management. Diabetes Res. Clin. Pract. 2018, 137, 10–19. [Google Scholar] [CrossRef]
- Ling, C.; Rönn, T. Epigenetics in human obesity and type 2 Diabetes. Cell Metab. 2019, 29, 1028–1044. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Wang, X.; Pan, Y.; Zhu, H.; Hao, G.; Huang, Y.; Barnes, V.; Shi, H.; Snieder, H.; Pankow, J.; North, K.; et al. An epigenome-wide study of obesity in African American youth and young adults: Novel findings, replication in neutrophils, and relationship with gene expression. Clin. Epigenet. 2018, 10, 3. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Subramaniam, N.; Nair, R.; Marsden, P.A. Epigenetic Regulation of the Vascular Endothelium by Angiogenic LncRNAs. Front. Genet. 2021, 12, 668313. [Google Scholar] [CrossRef] [PubMed]
- Cederberg, H.; Stančáková, A.; Yaluri, N.; Modi, S.; Kuusisto, J.; Laakso, M. Increased risk of diabetes with statin treatment is associated with impaired insulin sensitivity and insulin secretion: A 6-year follow-up study of the METSIM cohort. Diabetologia 2015, 58, 1109–1117. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Laakso, M.; Kuusisto, J. Diabetes secondary to treatment with statins. Curr. Diabetes Rep. 2017, 17, 10. [Google Scholar] [CrossRef]
- Swerdlow, D.I.; Preiss, D.; Kuchenbaecker, K.B.; Holmes, M.V.; Engmann, J.E.; Shah, T.; Sofat, R.; Stender, S.; Johnson, P.C.; Scott, R.A.; et al. HMG-coenzyme A reductase inhibition, type 2 diabetes, and bodyweight: Evidence from genetic analysis and randomised trials. Lancet 2015, 385, 351–361. [Google Scholar] [CrossRef] [Green Version]
- Besseling, J.; Kastelein, J.J.; Defesche, J.C.; Hutten, B.A.; Hovingh, G.K. Association between familial hypercholesterolemia and prevalence of type 2 diabetes mellitus. JAMA 2015, 313, 1029–1036. [Google Scholar] [CrossRef] [Green Version]
- Arnold, N.; Lechner, K.; Waldeyer, C.; Shapiro, M.D.; Koenig, W. Inflammation and cardiovascular disease: The future. Eur. Cardiol. 2021, 16, e20. [Google Scholar] [CrossRef]
- Libby, P. The changing landscape of atherosclerosis. Nature 2021, 592, 524–533. [Google Scholar] [CrossRef]
- Kasikara, C.; Doran, A.C.; Cai, B.; Tabas, I. The role of non-resolving inflammation in atherosclerosis. J. Clin. Investig. 2018, 128, 2713–2723. [Google Scholar] [CrossRef] [Green Version]
- Ridker, P.M.; Everett, B.M.; Thuren, T.; MacFadyen, J.G.; Chang, W.H.; Ballantyne, C.; Fonseca, F.; Nicolau, J.; Koenig, W.; Anker, S.D.; et al. Antiinflammatory therapy with canakinumab for atherosclerotic disease. N. Engl. J. Med. 2017, 377, 1119–1131. [Google Scholar] [CrossRef]
- Bouabdallaoui, N.; Tardif, J.C.; Waters, D.D.; Pinto, F.J.; Maggioni, A.P.; Diaz, R.; Berry, C.; Koenig, W.; Lopez-Sendon, J.; Gamra, H.; et al. Time-to-treatment initiation of colchicine and cardiovascular outcomes after myocardial infarction in the Colchicine Cardiovascular Outcomes Trial (COLCOT). Eur. Heart J. 2020, 41, 4092–4099. [Google Scholar] [CrossRef]
- Aderemi, A.V.; Ayeleso, A.O.; Oyedapo, O.O.; Mukwevho, E. Metabolomics: A scoping review of its role as a tool for disease biomarker discovery in selected non-communicable diseases. Metabolites 2021, 11, 418. [Google Scholar] [CrossRef]
- Emwas, A.H. The strengths and weaknesses of NMR spectroscopy and mass spectrometry with particular focus on metabolomics research. Methods Mol. Biol. 2015, 1277, 161–193. [Google Scholar]
- Klein, M.S.; Shearer, J. Metabolomics and type 2 diabetes: Translating basic research into clinical application. J. Diabetes Res. 2016, 2016, 3898502. [Google Scholar] [CrossRef] [Green Version]
- Newgard, C.B.; An, J.; Bain, J.R.; Muehlbauer, M.J.; Stevens, R.D.; Lien, L.F.; Haqq, A.M.; Shah, S.H.; Arlotto, M.; Slentz, C.A.; et al. A branched-chain amino acid-related metabolic signature that differentiates obese and lean humans and contributes to insulin resistance. Cell Metab. 2009, 9, 311–326. [Google Scholar] [CrossRef] [Green Version]
- Lotta, L.A.; Scott, R.A.; Sharp, S.J.; Burgess, S.; Luan, J.; Tillin, T.; Schmidt, A.F.; Imamura, F.; Stewart, I.D.; Perry, J.R.; et al. Genetic predisposition to an impaired metabolism of the branched-chain amino acids and risk of type 2 diabetes: A Mendelian randomisation analysis. PLoS Med. 2016, 13, e1002179. [Google Scholar] [CrossRef]
- Wang, Q.; Holmes, M.V.; Davey Smith, G.; Ala-Korpela, M. Genetic support for a causal role of insulin resistance on circulating branched-chain amino acids and inflammation. Diabetes Care 2017, 40, 1779–1786. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Merino, J.; Leong, A.; Liu, C.T.; Porneala, B.; Walford, G.A.; von Grotthuss, M.; Wang, T.J.; Flannick, J.; Dupuis, J.; Levy, D.; et al. Metabolomics insights into early type 2 diabetes pathogenesis and detection in individuals with normal fasting glucose. Diabetologia 2018, 61, 1315–1324. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Feofanova, E.V.; Chen, H.; Dai, Y.; Jia, P.; Grove, M.L.; Morrison, A.C.; Qi, Q.; Daviglus, M.; Cai, J.; North, K.E.; et al. A genome-wide association study discovers 46 loci of the human metabolome in the Hispanic Community Health Study/Study of Latinos. Am. J. Hum. Genet. 2020, 107, 849–863. [Google Scholar] [CrossRef] [PubMed]
- Yun, H.; Sun, L.; Wu, Q.; Zong, G.; Qi, Q.; Li, H.; Zheng, H.; Zeng, R.; Liang, L.; Lin, X. Associations among circulating sphingolipids, β-cell function, and risk of developing type 2 diabetes: A population-based cohort study in China. PLoS Med. 2020, 17, e1003451. [Google Scholar] [CrossRef] [PubMed]
- Dimas, A.S.; Lagou, V.; Barker, A.; Knowles, J.W.; Mägi, R.; Hivert, M.F.; Benazzo, A.; Rybin, D.; Jackson, A.U.; Stringham, H.M.; et al. Impact of type 2 diabetes susceptibility variants on quantitative glycemic traits reveals mechanistic heterogeneity. Diabetes 2014, 63, 2158–2171. [Google Scholar] [CrossRef] [Green Version]
- Udler, M.S.; Kim, J.; von Grotthuss, M.; Bonàs-Guarch, S.; Cole, J.B.; Chiou, J.; Anderson, C.D.; Boehnke, M.; Laakso, M.; Atzmon, G.; et al. Type 2 diabetes genetic loci informed by multi-trait associations point to disease mechanisms and subtypes: A soft clustering analysis. PLoS Med. 2018, 15, e1002654. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Ahlqvist, E.; Storm, P.; Käräjämäki, A.; Martinell, M.; Dorkhan, M.; Carlsson, A.; Vikman, P.; Prasad, R.B.; Aly, D.M.; Almgren, P.; et al. Novel subgroups of adult-onset diabetes and their association with outcomes: A data-driven cluster analysis of six variables. Lancet Diabetes Endocrinol. 2018, 6, 361–369. [Google Scholar] [CrossRef] [Green Version]
- Ahlqvist, E.; Prasad, R.B.; Groop, L. Subtypes of type 2 diabetes determined from clinical parameters. Diabetes 2020, 69, 2086–2093. [Google Scholar] [CrossRef] [PubMed]
- Wagner, R.; Heni, M.; Tabák, A.G.; Machann, J.; Schick, F.; Randrianarisoa, E.; Hrabě de Angelis, M.; Birkenfeld, A.L.; Stefan, N.; Peter, A.; et al. Pathophysiology-based subphenotyping of individuals at elevated risk for type 2 diabetes. Nat. Med. 2021, 27, 49–57. [Google Scholar] [CrossRef]
- Udler, M.S.; McCarthy, M.I.; Florez, J.C.; Mahajan, A. Genetic risk scores for diabetes diagnosis and precision medicine. Endocr. Rev. 2019, 40, 1500–1520. [Google Scholar] [CrossRef] [Green Version]
- Zeng, L.; Talukdar, H.A.; Koplev, S.; Giannarelli, C.; Ivert, T.; Gan, L.M.; Ruusalepp, A.; Schadt, E.E.; Kovacic, J.C.; Lusis, A.J.; et al. Contribution of gene regulatory networks to heritability of coronary artery disease. J. Am. Coll. Cardiol. 2019, 73, 2946–2957. [Google Scholar] [CrossRef] [PubMed]




{kind=link}
{kind=link}
{kind=link}
{kind=link}
| Type 2 Diabetes | Cardiovascular Disease | ||||||
|---|---|---|---|---|---|---|---|
| Exposure | Genetic Instrument | Cases/Controls | Causal Effect Size or (95% CI) | Reference | Cases/Controls | Causal Effect Size or (95% CI) | Reference |
| Birth weight | PRS | 3627/12,974 | 2.94 (1.70–5.16) | [68] | 5542 /237,631 | 0.69 (0.60–0.80) | [69] |
| BMI | PRS | 12,171/56,862 | 1.26 (1.17–1.34) | [70] | 60,801/123,504 | 1.49 (1.39–1.60) | [71] |
| Waist:hip ratio | PRS | 34,840/149,821 | 1.82 (1.38–2.42) | [72] | 60,801/123,504 | 1.54 (1.38–1.71) | [71] |
| Blood pressure | PRS | 37,293/125,686 | 1.02 (1.01–1.03) | [73] | 255,714/10 mmHg increase | 1.49 (1.38–1.63) | [74] |
| Smoking | PRS | 74,124/824,006 | 1.28 (1.20–1.37) | [75] | 60,801/123,504 | 1.48 (1.25–1.75) | [76] |
| LDLC | PRS | 33,323/418,610 | 0.82 (0.63–1.00) | [77] | 9647/451,933 | 1.44 (1.42–1.47) | [77] |
| Total TG | PRS | 33,323/418,610 | 1.35 (1.19–1.51) | [77] | 9647/451,933 | 1.31 (1.24–1.38) | [77] |
| ALT | PRS | 33,323/418,610 | 1.73 (1.52–1.94) | [77] | 9647/451,933 | 1.45 (1.10–1.92) | [77] |
| CRP | PRS | 33,323/418,610 | 0.92 (0.76–1.09) | [77] | 9647/451,933 | 1.12 (1.10–1.15) | [77] |
| IL-6 | PRS | Not available | − | − | 9417/15982 | 1.60 (1.60–1.72) | [78] |
| Apo-B | PRS | Not available | − | − | 60,801/123,504 | 1.73 (1.56–1.91) | [79] |
Publisher’s Note: MDPI stays neutral with regard to jurisdictional claims in published maps and institutional affiliations. |
© 2021 by the authors. Licensee MDPI, Basel, Switzerland. This article is an open access article distributed under the terms and conditions of the Creative Commons Attribution (CC BY) license (https://creativecommons.org/licenses/by/4.0/).
Share and Cite
Fernandes Silva, L.; Vangipurapu, J.; Laakso, M. The “Common Soil Hypothesis” Revisited—Risk Factors for Type 2 Diabetes and Cardiovascular Disease. Metabolites 2021, 11, 691. https://doi.org/10.3390/metabo11100691
Fernandes Silva L, Vangipurapu J, Laakso M. The “Common Soil Hypothesis” Revisited—Risk Factors for Type 2 Diabetes and Cardiovascular Disease. Metabolites. 2021; 11(10):691. https://doi.org/10.3390/metabo11100691
Chicago/Turabian StyleFernandes Silva, Lilian, Jagadish Vangipurapu, and Markku Laakso. 2021. "The “Common Soil Hypothesis” Revisited—Risk Factors for Type 2 Diabetes and Cardiovascular Disease" Metabolites 11, no. 10: 691. https://doi.org/10.3390/metabo11100691
APA StyleFernandes Silva, L., Vangipurapu, J., & Laakso, M. (2021). The “Common Soil Hypothesis” Revisited—Risk Factors for Type 2 Diabetes and Cardiovascular Disease. Metabolites, 11(10), 691. https://doi.org/10.3390/metabo11100691