
Toxicometabolomics: Small Molecules to Answer Big Toxicological Questions

 ,
,  ,
,  and
and 
Abstract
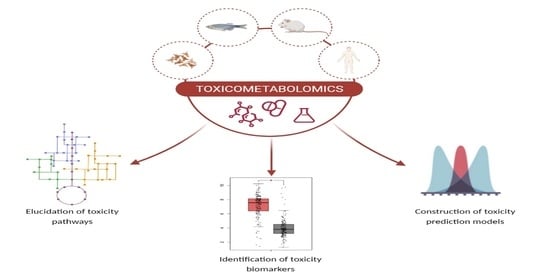
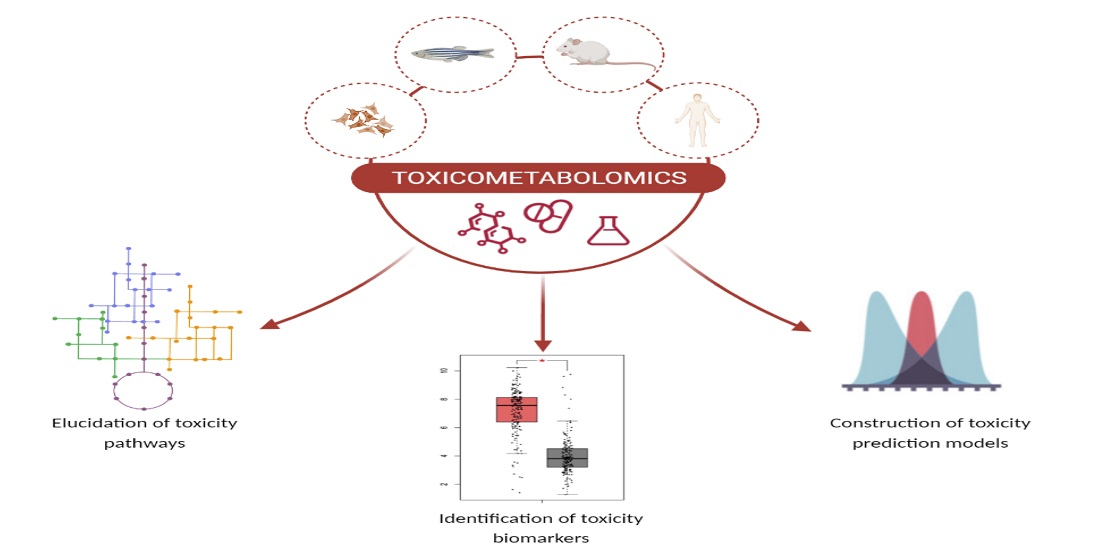
:
1. Introduction
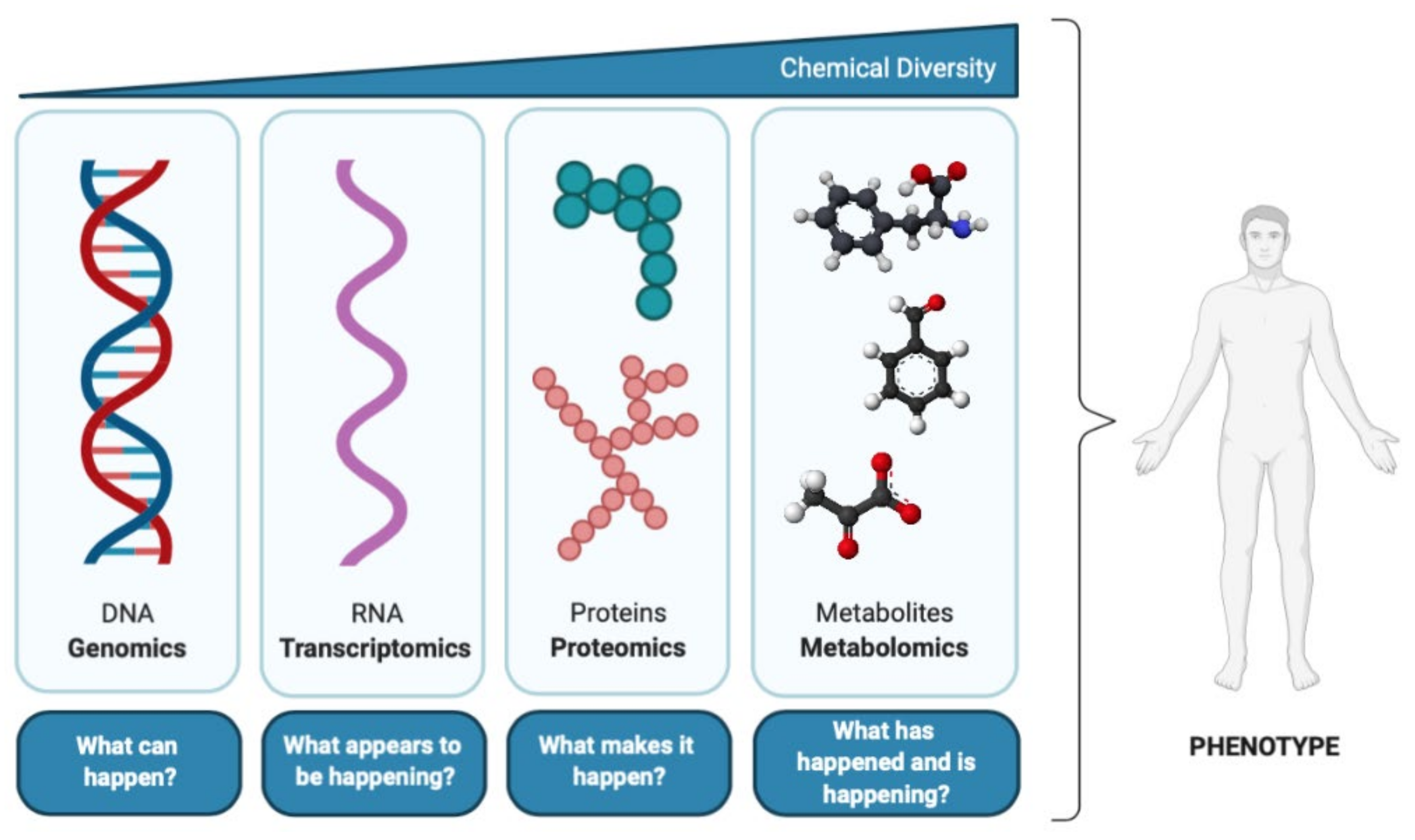
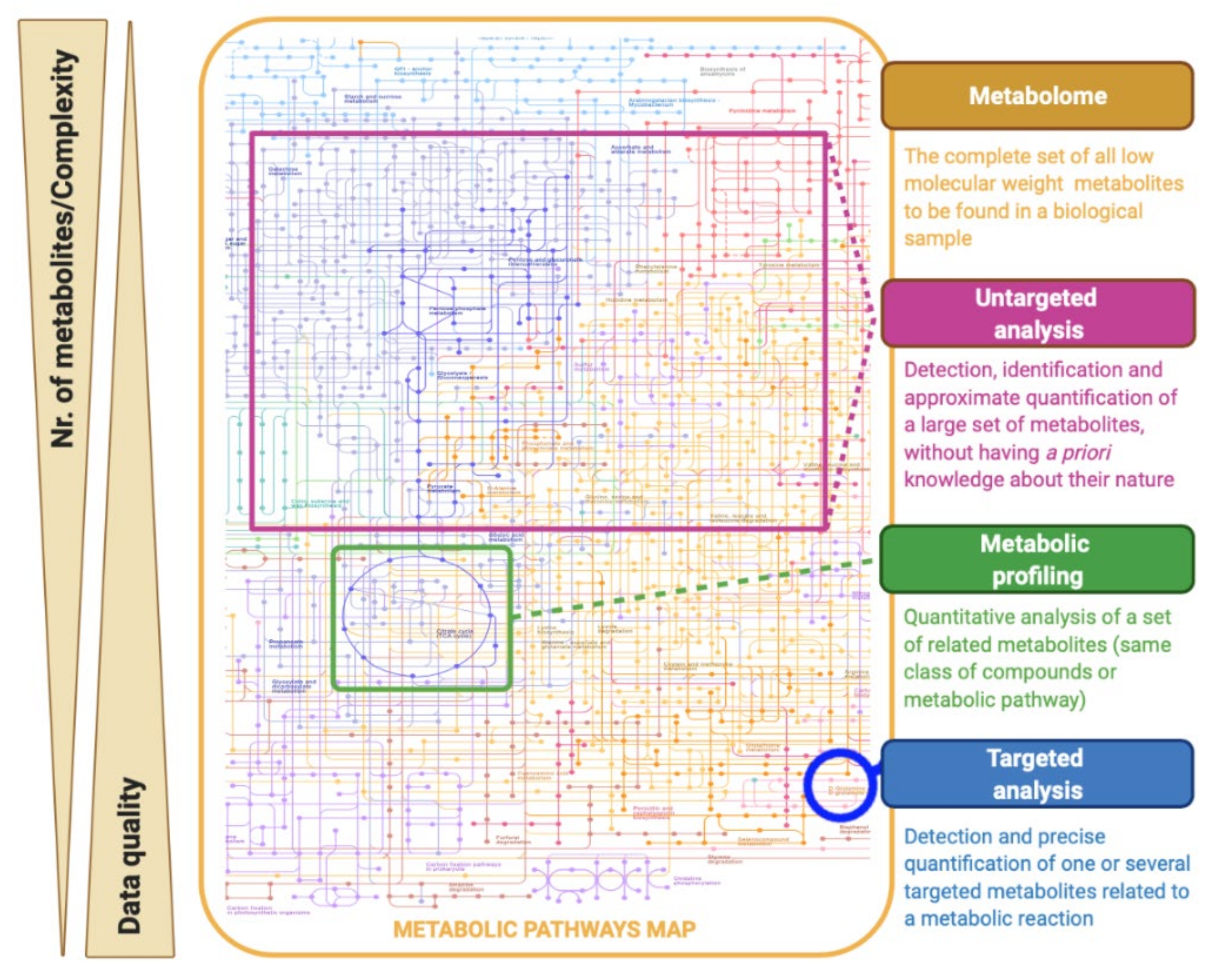
2. What Is the Metabolome?
3. Metabolomics: Concept and Strategies
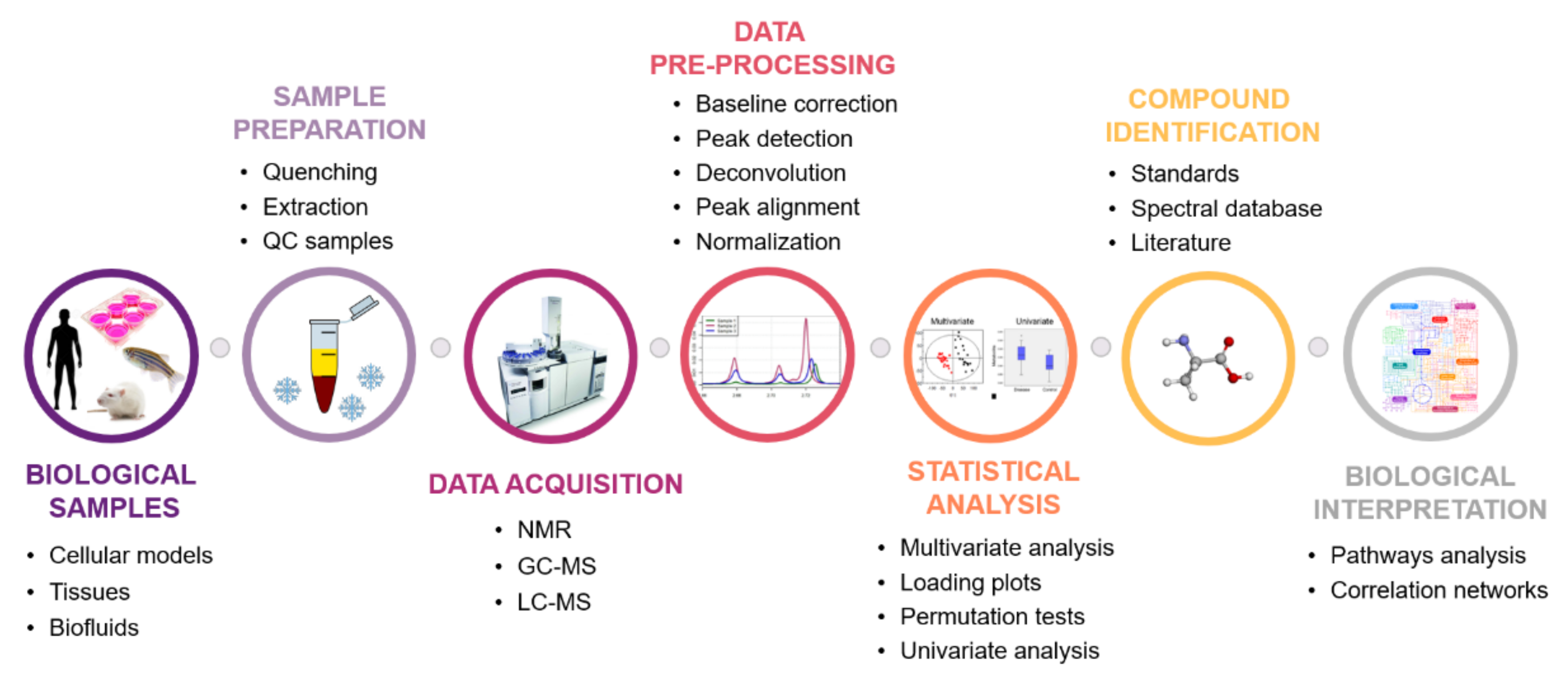
4. Metabolomics Workflow
4.1. Biological Question Formulation
4.2. Which Sample to Choose?
4.2.1. Cellular Models
4.2.2. Tissues
4.2.3. Urine and Blood
4.3. Sample Collection and Preparation
4.4. Analytical Platforms
4.5. Bioinformatics and Statistical Tools in Metabolomics
4.5.1. Data Preprocessing
4.5.2. Multivariate Analysis (MVA)
4.5.3. Univariate Statistical Analyses
4.5.4. The Multiple Testing Problem
4.6. Metabolites Identification
4.7. Biological Interpretation
5. Metabolomics: A New Route in Toxicological Research
6. Current Challenges and Future Perspectives
Author Contributions
Funding
Conflicts of Interest
References
- Miggiels, P.; Wouters, B.; van Westen, G.J.P.; Dubbelman, A.C.; Hankemeier, T. Novel technologies for metabolomics: More for less. Trends Analyt. Chem. 2019, 120, 115323. [Google Scholar] [CrossRef]
- Oliver, S.G.; Winson, M.K.; Kell, D.B.; Baganz, F. Systematic functional analysis of the yeast genome. Trends Biotechnol. 1998, 16, 373–378. [Google Scholar] [CrossRef]
- Gomase, V.S.; Changbhale, S.S.; Patil, S.A.; Kale, K.V. Metabolomics. Curr. Drug Metab. 2008, 9, 89–98. [Google Scholar] [CrossRef]
- German, J.B.; Hammock, B.D.; Watkins, S.M. Metabolomics: Building on a century of biochemistry to guide human health. Metabolomics 2005, 1, 3–9. [Google Scholar] [CrossRef]
- Wishart, D.S.; Feunang, Y.D.; Marcu, A.; Guo, A.C.; Liang, K.; Vazquez-Fresno, R.; Sajed, T.; Johnson, D.; Li, C.; Karu, N.; et al. HMDB 4.0: The human metabolome database for 2018. Nucleic Acids Res. 2018, 46, D608–D617. [Google Scholar] [CrossRef]
- Ruiz-Aracama, A.; Peijnenburg, A.; Kleinjans, J.; Jennen, D.; van Delft, J.; Hellfrisch, C.; Lommen, A. An untargeted multi-technique metabolomics approach to studying intracellular metabolites of HepG2 cells exposed to 2,3,7,8-tetrachlorodibenzo-p-dioxin. BMC Genom. 2011, 12, 251. [Google Scholar] [CrossRef] [Green Version]
- Hayton, S.; Maker, G.L.; Mullaney, I.; Trengove, R.D. Untargeted metabolomics of neuronal cell culture: A model system for the toxicity testing of insecticide chemical exposure. J. Appl. Toxicol. 2017, 37, 1481–1492. [Google Scholar] [CrossRef] [PubMed]
- Fiehn, O. Metabolomics—The link between genotypes and phenotypes. Plant Mol. Biol. 2002, 48, 155–171. [Google Scholar] [CrossRef] [PubMed]
- Weiss, R.H.; Kim, K. Metabolomics in the study of kidney diseases. Nat. Rev. Nephrol. 2011, 8, 22–33. [Google Scholar] [CrossRef] [PubMed]
- Manach, C.; Hubert, J.; Llorach, R.; Scalbert, A. The complex links between dietary phytochemicals and human health deciphered by metabolomics. Mol. Nutr. Food Res. 2009, 53, 1303–1315. [Google Scholar] [CrossRef]
- Nielsen, J.; Oliver, S. The next wave in metabolome analysis. Trends Biotechnol. 2005, 23, 544–546. [Google Scholar] [CrossRef] [PubMed]
- Paglia, G.; Hrafnsdottir, S.; Magnusdottir, M.; Fleming, R.M.; Thorlacius, S.; Palsson, B.O.; Thiele, I. Monitoring metabolites consumption and secretion in cultured cells using ultra-performance liquid chromatography quadrupole-time of flight mass spectrometry (UPLC-Q-ToF-MS). Anal. Bioanal. Chem. 2012, 402, 1183–1198. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Tang, J. Microbial metabolomics. Curr. Genom. 2011, 12, 391–403. [Google Scholar] [CrossRef]
- Johnson, C.H.; Patterson, A.D.; Idle, J.R.; Gonzalez, F.J. Xenobiotic metabolomics: Major impact on the metabolome. Annu. Rev. Pharmacol. Toxicol. 2012, 52, 37–56. [Google Scholar] [CrossRef] [PubMed]
- Nicholson, J.K.; Lindon, J.C.; Holmes, E. ’Metabonomics’: Understanding the metabolic responses of living systems to pathophysiological stimuli via multivariate statistical analysis of biological NMR spectroscopic data. Xenobiotica 1999, 29, 1181–1189. [Google Scholar] [CrossRef]
- Alonso, A.; Marsal, S.; Julia, A. Analytical methods in untargeted metabolomics: State of the art in 2015. Front Bioeng. Biotechnol. 2015, 3, 23. [Google Scholar] [CrossRef] [Green Version]
- Dettmer, K.; Aronov, P.A.; Hammock, B.D. Mass spectrometry-based metabolomics. Mass Spectrom. Rev. 2007, 26, 51–78. [Google Scholar] [CrossRef]
- Ryan, D.; Robards, K. Metabolomics: The greatest omics of them all? Anal. Chem. 2006, 78, 7954–7958. [Google Scholar] [CrossRef]
- Goodacre, R.; Vaidyanathan, S.; Dunn, W.B.; Harrigan, G.G.; Kell, D.B. Metabolomics by numbers: Acquiring and understanding global metabolite data. Trends Biotechnol. 2004, 22, 245–252. [Google Scholar] [CrossRef]
- Kell, D.B.; Oliver, S.G. The metabolome 18 years on: A concept comes of age. Metabolomics 2016, 12, 148. [Google Scholar] [CrossRef]
- Bouhifd, M.; Hartung, T.; Hogberg, H.T.; Kleensang, A.; Zhao, L. Review: Toxicometabolomics. J. Appl. Toxicol. 2013, 33, 1365–1383. [Google Scholar] [CrossRef]
- Krastanov, A. Metablomics—The state of art. Biotechnol. Biotechnol. Equip. 2010, 24, 1537–1543. [Google Scholar] [CrossRef] [Green Version]
- Zamboni, N.; Saghatelian, A.; Patti, G.J. Defining the metabolome: Size, flux, and regulation. Mol. Cell 2015, 58, 699–706. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Villas-Boas, S.G.; Mas, S.; Akesson, M.; Smedsgaard, J.; Nielsen, J. Mass spectrometry in metabolome analysis. Mass Spectrom. Rev. 2005, 24, 613–646. [Google Scholar] [CrossRef] [PubMed]
- Oldiges, M.; Lutz, S.; Pflug, S.; Schroer, K.; Stein, N.; Wiendahl, C. Metabolomics: Current state and evolving methodologies and tools. Appl. Microbiol. Biotechnol. 2007, 76, 495–511. [Google Scholar] [CrossRef] [PubMed]
- Roessner, U.; Bowne, J. What is metabolomics all about? Biotechniques 2009, 46, 363–365. [Google Scholar] [CrossRef] [PubMed]
- Allen, J.; Davey, H.M.; Broadhurst, D.; Heald, J.K.; Rowland, J.J.; Oliver, S.G.; Kell, D.B. High-throughput classification of yeast mutants for functional genomics using metabolic footprinting. Nat. Biotechnol. 2003, 21, 692–696. [Google Scholar] [CrossRef]
- Chetwynd, A.J.; Dunn, W.B.; Rodriguez-Blanco, G. Collection and Preparation of Clinical Samples for Metabolomics. Adv. Exp. Med. Biol. 2017, 965, 19–44. [Google Scholar] [CrossRef]
- Leon, Z.; Garcia-Canaveras, J.C.; Donato, M.T.; Lahoz, A. Mammalian cell metabolomics: Experimental design and sample preparation. Electrophoresis 2013, 34, 2762–2775. [Google Scholar] [CrossRef]
- Lindon, J.C.; Holmes, E.; Nicholson, J.K. So what’s the deal with metabonomics? Anal. Chem. 2003, 75, 384A–391A. [Google Scholar] [CrossRef]
- Cuperlovic-Culf, M.; Barnett, D.A.; Culf, A.S.; Chute, I. Cell culture metabolomics: Applications and future directions. Drug Discov. Today 2010, 15, 610–621. [Google Scholar] [CrossRef]
- Hartung, T.; Daston, G. Are in vitro tests suitable for regulatory use? Toxicol. Sci. 2009, 111, 233–237. [Google Scholar] [CrossRef] [Green Version]
- Daskalaki, E.; Pillon, N.J.; Krook, A.; Wheelock, C.E.; Checa, A. The influence of culture media upon observed cell secretome metabolite profiles: The balance between cell viability and data interpretability. Anal. Chim. Acta 2018, 1037, 338–350. [Google Scholar] [CrossRef]
- Tokarz, J.; Prehn, C.; Artati, A. Standard Operating Procedures (SOP) for Cell Culture Metabolomics at the GAC. Available online: https://www.helmholtz-muenchen.de/fileadmin/GAC/SOPs/2017_SOP_CellCulture_Metabolomics_V2.9.pdf (accessed on 12 September 2021).
- Goodacre, R.; Ellis, D.; Hollywood, K.; Trivedi, D.; Muhamadali, H. Laboratory Guide for Metabolomics Experiments. Available online: http://www.biospec.net/wordpress/wp-content/uploads/Metabolomics-laboratory-handbook.pdf (accessed on 12 September 2021).
- Halama, A. Metabolomics in cell culture—A strategy to study crucial metabolic pathways in cancer development and the response to treatment. Arch. Biochem. Biophys. 2014, 564, 100–109. [Google Scholar] [CrossRef]
- Garcia-Canaveras, J.C.; Castell, J.V.; Donato, M.T.; Lahoz, A. A metabolomics cell-based approach for anticipating and investigating drug-induced liver injury. Sci. Rep. 2016, 6, 27239. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Maker, G.L.; Green, T.; Mullaney, I.; Trengove, R.D. Untargeted Metabolomic Analysis of Rat Neuroblastoma Cells as a Model System to Study the Biochemical Effects of the Acute Administration of Methamphetamine. Metabolites 2018, 8, 38. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Mueller, D.; Muller-Vieira, U.; Biemel, K.M.; Tascher, G.; Nussler, A.K.; Noor, F. Biotransformation of diclofenac and effects on the metabolome of primary human hepatocytes upon repeated dose exposure. Eur. J. Pharm. Sci. 2012, 45, 716–724. [Google Scholar] [CrossRef] [PubMed]
- Kumar, A.; Kumar, Y.; Sevak, J.K.; Kumar, S.; Kumar, N.; Gopinath, S.D. Metabolomic analysis of primary human skeletal muscle cells during myogenic progression. Sci. Rep. 2020, 10, 11824. [Google Scholar] [CrossRef] [PubMed]
- Vernardis, S.I.; Terzoudis, K.; Panoskaltsis, N.; Mantalaris, A. Human embryonic and induced pluripotent stem cells maintain phenotype but alter their metabolism after exposure to ROCK inhibitor. Sci. Rep. 2017, 7, 42138. [Google Scholar] [CrossRef] [Green Version]
- Kim, J.; Kang, S.C.; Yoon, N.E.; Kim, Y.; Choi, J.; Park, N.; Jung, H.; Jung, B.H.; Ju, J.H. Metabolomic profiles of induced pluripotent stem cells derived from patients with rheumatoid arthritis and osteoarthritis. Stem. Cell Res. Ther. 2019, 10, 319. [Google Scholar] [CrossRef]
- Kaur, G.; Dufour, J.M. Cell lines: Valuable tools or useless artifacts. Spermatogenesis 2012, 2, 1–5. [Google Scholar] [CrossRef] [Green Version]
- Shyh-Chang, N.; Ng, H.H. The metabolic programming of stem cells. Genes Dev. 2017, 31, 336–346. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Sun, Q.; Zhang, Z.; Sun, Z. The potential and challenges of using stem cells for cardiovascular repair and regeneration. Genes Dis. 2014, 1, 113–119. [Google Scholar] [CrossRef] [Green Version]
- Pamies, D.; Hartung, T. 21st Century Cell Culture for 21st Century Toxicology. Chem. Res. Toxicol. 2017, 30, 43–52. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Kosmides, A.K.; Kamisoglu, K.; Calvano, S.E.; Corbett, S.A.; Androulakis, I.P. Metabolomic fingerprinting: Challenges and opportunities. Crit. Rev. Biomed. Eng. 2013, 41, 205–221. [Google Scholar] [CrossRef] [PubMed]
- Abaffy, T.; Moller, M.; Riemer, D.D.; Milikowski, C.; Defazio, R.A. A case report—Volatile metabolomic signature of malignant melanoma using matching skin as a control. J. Cancer Sci. Ther. 2011, 3, 140–144. [Google Scholar] [CrossRef]
- Rombouts, C.; De Spiegeleer, M.; Van Meulebroek, L.; De Vos, W.H.; Vanhaecke, L. Validated comprehensive metabolomics and lipidomics analysis of colon tissue and cell lines. Anal. Chim. Acta 2019, 1066, 79–92. [Google Scholar] [CrossRef]
- Want, E.J.; Masson, P.; Michopoulos, F.; Wilson, I.D.; Theodoridis, G.; Plumb, R.S.; Shockcor, J.; Loftus, N.; Holmes, E.; Nicholson, J.K. Global metabolic profiling of animal and human tissues via UPLC-MS. Nat. Protoc. 2013, 8, 17–32. [Google Scholar] [CrossRef]
- Overmyer, K.A.; Thonusin, C.; Qi, N.R.; Burant, C.F.; Evans, C.R. Impact of anesthesia and euthanasia on metabolomics of mammalian tissues: Studies in a C57BL/6J mouse model. PLoS ONE 2015, 10, e0117232. [Google Scholar] [CrossRef]
- Khamis, M.M.; Adamko, D.J.; El-Aneed, A. Mass spectrometric based approaches in urine metabolomics and biomarker discovery. Mass Spectrom. Rev. 2017, 36, 115–134. [Google Scholar] [CrossRef]
- Chan, E.C.; Pasikanti, K.K.; Nicholson, J.K. Global urinary metabolic profiling procedures using gas chromatography-mass spectrometry. Nat. Protoc. 2011, 6, 1483–1499. [Google Scholar] [CrossRef] [PubMed]
- Assfalg, M.; Bertini, I.; Colangiuli, D.; Luchinat, C.; Schafer, H.; Schutz, B.; Spraul, M. Evidence of different metabolic phenotypes in humans. Proc. Natl. Acad. Sci. USA 2008, 105, 1420–1424. [Google Scholar] [CrossRef] [Green Version]
- Walsh, M.C.; Brennan, L.; Malthouse, J.P.; Roche, H.M.; Gibney, M.J. Effect of acute dietary standardization on the urinary, plasma, and salivary metabolomic profiles of healthy humans. Am. J. Clin. Nutr. 2006, 84, 531–539. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Serkova, N.J.; Standiford, T.J.; Stringer, K.A. The emerging field of quantitative blood metabolomics for biomarker discovery in critical illnesses. Am. J. Respir. Crit. Care Med. 2011, 184, 647–655. [Google Scholar] [CrossRef] [PubMed]
- Zhang, A.; Sun, H.; Wang, P.; Han, Y.; Wang, X. Recent and potential developments of biofluid analyses in metabolomics. J. Proteom. 2012, 75, 1079–1088. [Google Scholar] [CrossRef]
- Lauridsen, M.; Hansen, S.H.; Jaroszewski, J.W.; Cornett, C. Human urine as test material in 1H NMR-based metabonomics: Recommendations for sample preparation and storage. Anal. Chem. 2007, 79, 1181–1186. [Google Scholar] [CrossRef]
- Stringer, K.A.; Younger, J.G.; McHugh, C.; Yeomans, L.; Finkel, M.A.; Puskarich, M.A.; Jones, A.E.; Trexel, J.; Karnovsky, A. Whole Blood Reveals More Metabolic Detail of the Human Metabolome than Serum as Measured by 1H-NMR Spectroscopy: Implications for Sepsis Metabolomics. Shock 2015, 44, 200–208. [Google Scholar] [CrossRef] [Green Version]
- Smith, L.; Villaret-Cazadamont, J.; Claus, S.P.; Canlet, C.; Guillou, H.; Cabaton, N.J.; Ellero-Simatos, S. Important Considerations for Sample Collection in Metabolomics Studies with a Special Focus on Applications to Liver Functions. Metabolites 2020, 10, 104. [Google Scholar] [CrossRef] [Green Version]
- Gonzalez-Dominguez, R.; Gonzalez-Dominguez, A.; Sayago, A.; Fernandez-Recamales, A. Recommendations and Best Practices for Standardizing the Pre-Analytical Processing of Blood and Urine Samples in Metabolomics. Metabolites 2020, 10, 229. [Google Scholar] [CrossRef]
- Yu, Z.; Kastenmuller, G.; He, Y.; Belcredi, P.; Moller, G.; Prehn, C.; Mendes, J.; Wahl, S.; Roemisch-Margl, W.; Ceglarek, U.; et al. Differences between human plasma and serum metabolite profiles. PLoS ONE 2011, 6, e21230. [Google Scholar] [CrossRef]
- Segers, K.; Declerck, S.; Mangelings, D.; Heyden, Y.V.; Eeckhaut, A.V. Analytical techniques for metabolomic studies: A review. Bioanalysis 2019, 11, 2297–2318. [Google Scholar] [CrossRef]
- Dyar, K.A.; Eckel-Mahan, K.L. Circadian Metabolomics in Time and Space. Front. Neurosci. 2017, 11, 369. [Google Scholar] [CrossRef] [Green Version]
- Slupsky, C.M.; Rankin, K.N.; Wagner, J.; Fu, H.; Chang, D.; Weljie, A.M.; Saude, E.J.; Lix, B.; Adamko, D.J.; Shah, S.; et al. Investigations of the effects of gender, diurnal variation, and age in human urinary metabolomic profiles. Anal. Chem. 2007, 79, 6995–7004. [Google Scholar] [CrossRef]
- Deprez, S.; Sweatman, B.C.; Connor, S.C.; Haselden, J.N.; Waterfield, C.J. Optimisation of collection, storage and preparation of rat plasma for 1H NMR spectroscopic analysis in toxicology studies to determine inherent variation in biochemical profiles. J. Pharm. Biomed. Anal. 2002, 30, 1297–1310. [Google Scholar] [CrossRef]
- Zhou, Z.; Chen, Y.; He, J.; Xu, J.; Zhang, R.; Mao, Y.; Abliz, Z. Systematic evaluation of serum and plasma collection on the endogenous metabolome. Bioanalysis 2017, 9, 239–250. [Google Scholar] [CrossRef]
- Gonzalez-Riano, C.; Garcia, A.; Barbas, C. Metabolomics studies in brain tissue: A review. J. Pharm. Biomed. Anal. 2016, 130, 141–168. [Google Scholar] [CrossRef]
- Ly-Verdu, S.; Schaefer, A.; Kahle, M.; Groeger, T.; Neschen, S.; Arteaga-Salas, J.M.; Ueffing, M.; de Angelis, M.H.; Zimmermann, R. The impact of blood on liver metabolite profiling—A combined metabolomic and proteomic approach. Biomed. Chromatogr. 2014, 28, 231–240. [Google Scholar] [CrossRef]
- Kapoore, R.V.; Coyle, R.; Staton, C.A.; Brown, N.J.; Vaidyanathan, S. Influence of washing and quenching in profiling the metabolome of adherent mammalian cells: A case study with the metastatic breast cancer cell line MDA-MB-231. Analyst 2017, 142, 2038–2049. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Zukunft, S.; Prehn, C.; Rohring, C.; Moller, G.; Hrabe de Angelis, M.; Adamski, J.; Tokarz, J. High-throughput extraction and quantification method for targeted metabolomics in murine tissues. Metabolomics 2018, 14, 18. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Ivanisevic, J.; Epstein, A.A.; Kurczy, M.E.; Benton, P.H.; Uritboonthai, W.; Fox, H.S.; Boska, M.D.; Gendelman, H.E.; Siuzdak, G. Brain region mapping using global metabolomics. Chem. Biol. 2014, 21, 1575–1584. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Teng, Q.; Huang, W.; Collette, T.W.; Ekman, D.R.; Tan, C. A direct cell quenching method for cell-culture based metabolomics. Metabolomics 2009, 5, 199. [Google Scholar] [CrossRef]
- Pinu, F.R.; Villas-Boas, S.G.; Aggio, R. Analysis of Intracellular Metabolites from Microorganisms: Quenching and Extraction Protocols. Metabolites 2017, 7, 53. [Google Scholar] [CrossRef] [Green Version]
- Sellick, C.A.; Hansen, R.; Stephens, G.M.; Goodacre, R.; Dickson, A.J. Metabolite extraction from suspension-cultured mammalian cells for global metabolite profiling. Nat. Protoc. 2011, 6, 1241–1249. [Google Scholar] [CrossRef]
- Liu, R.; Chou, J.; Hou, S.; Liu, X.; Yu, J.; Zhao, X.; Li, Y.; Liu, L.; Sun, C. Evaluation of two-step liquid-liquid extraction protocol for untargeted metabolic profiling of serum samples to achieve broader metabolome coverage by UPLC-Q-TOF-MS. Anal. Chim. Acta 2018, 1035, 96–107. [Google Scholar] [CrossRef]
- Martin, A.C.; Pawlus, A.D.; Jewett, E.M.; Wyse, D.L.; Angerhofer, C.K.; Hegeman, A.D. Evaluating solvent extraction systems using metabolomics approaches. RSC Adv. 2014, 4, 26325–26334. [Google Scholar] [CrossRef]
- Danielsson, A.P.; Moritz, T.; Mulder, H.; Spegel, P. Development and optimization of a metabolomic method for analysis of adherent cell cultures. Anal. Biochem. 2010, 404, 30–39. [Google Scholar] [CrossRef] [PubMed]
- Huie, C.W. A review of modern sample-preparation techniques for the extraction and analysis of medicinal plants. Anal. Bioanal. Chem. 2002, 373, 23–30. [Google Scholar] [CrossRef] [PubMed]
- Parab, G.S.; Rao, R.; Lakshminarayanan, S.; Bing, Y.V.; Moochhala, S.M.; Swarup, S. Data-driven optimization of metabolomics methods using rat liver samples. Anal. Chem. 2009, 81, 1315–1323. [Google Scholar] [CrossRef]
- Jaroch, K.; Boyaci, E.; Pawliszyn, J.; Bojko, B. The use of solid phase microextraction for metabolomic analysis of non-small cell lung carcinoma cell line (A549) after administration of combretastatin A4. Sci. Rep. 2019, 9, 402. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Bolten, C.J.; Kiefer, P.; Letisse, F.; Portais, J.C.; Wittmann, C. Sampling for metabolome analysis of microorganisms. Anal. Chem. 2007, 79, 3843–3849. [Google Scholar] [CrossRef] [PubMed]
- Prasannan, C.B.; Jaiswal, D.; Davis, R.; Wangikar, P.P. An improved method for extraction of polar and charged metabolites from cyanobacteria. PLoS ONE 2018, 13, e0204273. [Google Scholar] [CrossRef]
- Issaq, H.J.; Van, Q.N.; Waybright, T.J.; Muschik, G.M.; Veenstra, T.D. Analytical and statistical approaches to metabolomics research. J. Sep. Sci. 2009, 32, 2183–2199. [Google Scholar] [CrossRef]
- Ludwig, C.; Viant, M.R. Two-dimensional J-resolved NMR spectroscopy: Review of a key methodology in the metabolomics toolbox. Phytochem. Anal. 2010, 21, 22–32. [Google Scholar] [CrossRef]
- Gowda, G.A.; Raftery, D. Can NMR solve some significant challenges in metabolomics? J. Magn. Reson. 2015, 260, 144–160. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Ren, J.L.; Zhang, A.H.; Kong, L.; Wang, X.J. Advances in mass spectrometry-based metabolomics for investigation of metabolites. RSC Adv. 2018, 8, 22335–22350. [Google Scholar] [CrossRef] [Green Version]
- Chen, C.; Kim, S. LC-MS-based Metabolomics of Xenobiotic-induced Toxicities. Comput. Struct. Biotechnol. J. 2013, 4, e201301008. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Ramautar, R. Capillary Electrophoresis–Mass Spectrometry for Metabolomics–From Metabolite Analysis to Metabolic Profiling. In Capillary Electrophoresis–Mass Spectrometry for Metabolomics; Ramautar, R., Ed.; Royal Society of Chemistry: London, UK, 2018; pp. 1–20. [Google Scholar]
- Gagnebin, Y.; Julien, B.; Belen, P.; Serge, R. Metabolomics in chronic kidney disease: Strategies for extended metabolome coverage. J. Pharm. Biomed. Anal. 2018, 161, 313–325. [Google Scholar] [CrossRef]
- Zhang, X.; Li, Q.; Xu, Z.; Dou, J. Mass spectrometry-based metabolomics in health and medical science: A systematic review. RSC Adv. 2020, 10, 3092–3104. [Google Scholar] [CrossRef] [Green Version]
- Cascante, M.; Marin, S. Metabolomics and fluxomics approaches. Essays Biochem. 2008, 45, 67–81. [Google Scholar] [CrossRef]
- Balashova, E.E.; Maslov, D.L.; Lokhov, P.G. A Metabolomics Approach to Pharmacotherapy Personalization. J. Pers. Med. 2018, 8, 28. [Google Scholar] [CrossRef] [Green Version]
- Weljie, A.M.; Newton, J.; Mercier, P.; Carlson, E.; Slupsky, C.M. Targeted profiling: Quantitative analysis of 1H NMR metabolomics data. Anal. Chem. 2006, 78, 4430–4442. [Google Scholar] [CrossRef] [PubMed]
- Burton, L.; Ivosev, G.; Tate, S.; Impey, G.; Wingate, J.; Bonner, R. Instrumental and experimental effects in LC-MS-based metabolomics. J. Chromatogr. B Analyt. Technol. Biomed. Life Sci. 2008, 871, 227–235. [Google Scholar] [CrossRef]
- Liland, K.H. Multivariate methods in metabolomics—From pre-processing to dimension reduction and statistical analysis. Trends Anal. Chem. 2011, 30, 827–841. [Google Scholar] [CrossRef]
- Katajamaa, M.; Oresic, M. Data processing for mass spectrometry-based metabolomics. J. Chromatogr. A 2007, 1158, 318–328. [Google Scholar] [CrossRef]
- Lommen, A.; Kools, H.J. MetAlign 3.0: Performance enhancement by efficient use of advances in computer hardware. Metabolomics 2012, 8, 719–726. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Pluskal, T.; Castillo, S.; Villar-Briones, A.; Oresic, M. MZmine 2: Modular framework for processing, visualizing, and analyzing mass spectrometry-based molecular profile data. BMC Bioinform. 2010, 11, 395. [Google Scholar] [CrossRef] [Green Version]
- Tautenhahn, R.; Patti, G.J.; Rinehart, D.; Siuzdak, G. XCMS Online: A web-based platform to process untargeted metabolomic data. Anal. Chem. 2012, 84, 5035–5039. [Google Scholar] [CrossRef] [Green Version]
- Vu, T.N.; Laukens, K. Getting your peaks in line: A review of alignment methods for NMR spectral data. Metabolites 2013, 3, 259–276. [Google Scholar] [CrossRef]
- Du, X.; Zeisel, S.H. Spectral deconvolution for gas chromatography mass spectrometry-based metabolomics: Current status and future perspectives. Comput. Struct. Biotechnol. J. 2013, 4, e201301013. [Google Scholar] [CrossRef] [Green Version]
- Wu, Y.; Li, L. Sample normalization methods in quantitative metabolomics. J. Chromatogr. A 2016, 1430, 80–95. [Google Scholar] [CrossRef]
- Silva, L.P.; Lorenzi, P.L.; Purwaha, P.; Yong, V.; Hawke, D.H.; Weinstein, J.N. Measurement of DNA concentration as a normalization strategy for metabolomic data from adherent cell lines. Anal. Chem. 2013, 85, 9536–9542. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Dieterle, F.; Ross, A.; Schlotterbeck, G.; Senn, H. Probabilistic quotient normalization as robust method to account for dilution of complex biological mixtures. Application in 1H NMR metabonomics. Anal. Chem. 2006, 78, 4281–4290. [Google Scholar] [CrossRef] [PubMed]
- Cuevas-Delgado, P.; Dudzik, D.; Miguel, V.; Lamas, S.; Barbas, C. Data-dependent normalization strategies for untargeted metabolomics—A case study. Anal. Bioanal. Chem. 2020, 412, 6391–6405. [Google Scholar] [CrossRef] [PubMed]
- Wulff, J.E.; Mitchell, M.W. A comparison of various normalization methods for LC/MS metabolomics data. Adv. Biosci. Biotechnol. 2018, 9, 339–351. [Google Scholar] [CrossRef] [Green Version]
- Van den Berg, R.A.; Hoefsloot, H.C.; Westerhuis, J.A.; Smilde, A.K.; van der Werf, M.J. Centering, scaling, and transformations: Improving the biological information content of metabolomics data. BMC Genom. 2006, 7, 142. [Google Scholar] [CrossRef] [Green Version]
- Lindon, J.C.; Holmes, E.; Nicholson, J.K. Pattern recognition methods and applications in biomedical magnetic resonance. Prog. Nucl. Magn. Reson. Spectrosc. 2001, 39, 1–40. [Google Scholar] [CrossRef]
- Wheelock, A.M.; Wheelock, C.E. Trials and tribulations of ’omics data analysis: Assessing quality of SIMCA-based multivariate models using examples from pulmonary medicine. Mol. Biosyst. 2013, 9, 2589–2596. [Google Scholar] [CrossRef] [Green Version]
- Trygg, J.; Holmes, E.; Lundstedt, T. Chemometrics in metabonomics. J. Proteome Res. 2007, 6, 469–479. [Google Scholar] [CrossRef]
- Blekherman, G.; Laubenbacher, R.; Cortes, D.F.; Mendes, P.; Torti, F.M.; Akman, S.; Torti, S.V.; Shulaev, V. Bioinformatics tools for cancer metabolomics. Metabolomics 2011, 7, 329–343. [Google Scholar] [CrossRef] [Green Version]
- Broadhurst, D.; Kell, D.B. Statistical strategies for avoiding false discoveries in metabolomics and related experiments. Metabolomics 2006, 2, 171–176. [Google Scholar] [CrossRef] [Green Version]
- Smolinska, A.; Blanchet, L.; Buydens, L.M.; Wijmenga, S.S. NMR and pattern recognition methods in metabolomics: From data acquisition to biomarker discovery: A review. Anal. Chim. Acta 2012, 750, 82–97. [Google Scholar] [CrossRef] [PubMed]
- Westerhuis, J.A.; Hoefsloot, H.C.; Smit, S.; Vis, D.J.; Smilde, A.K.; van Velzen, E.J.J.; van Duijnhoven, J.P.M.; van Dorsten, F.A. Assessment of PLSDA cross validation. Metabolomics 2008, 4, 81–89. [Google Scholar] [CrossRef] [Green Version]
- Vinaixa, M.; Samino, S.; Saez, I.; Duran, J.; Guinovart, J.J.; Yanes, O. A Guideline to Univariate Statistical Analysis for LC/MS-Based Untargeted Metabolomics-Derived Data. Metabolites 2012, 2, 775–795. [Google Scholar] [CrossRef]
- Benjamini, Y.; Hochberg, Y. Controlling the false discovery rate: A practical and powerful approach to multiple testing. J. R. Statist. Soc. B 1995, 57, 289–300. [Google Scholar] [CrossRef]
- Hendriks, M.M.W.B.; van Eeuwijk, F.A.; Jellema, R.H.; Westerhuis, J.A.; Reijmers, T.H.; Hoefsloot, H.C.J.; Smilde, A.K. Data-processing strategies for metabolomics studies. Trends Analyt. Chem. 2011, 30, 1685–1698. [Google Scholar] [CrossRef]
- Kanehisa, M.; Furumichi, M.; Tanabe, M.; Sato, Y.; Morishima, K. KEGG: New perspectives on genomes, pathways, diseases and drugs. Nucleic Acids Res. 2017, 45, D353–D361. [Google Scholar] [CrossRef] [Green Version]
- Chong, J.; Soufan, O.; Li, C.; Caraus, I.; Li, S.; Bourque, G.; Wishart, D.S.; Xia, J. MetaboAnalyst 4.0: Towards more transparent and integrative metabolomics analysis. Nucleic Acids Res. 2018, 46, W486–W494. [Google Scholar] [CrossRef] [Green Version]
- Caspi, R.; Billington, R.; Fulcher, C.A.; Keseler, I.M.; Kothari, A.; Krummenacker, M.; Latendresse, M.; Midford, P.E.; Ong, Q.; Ong, W.K.; et al. The MetaCyc database of metabolic pathways and enzymes. Nucleic Acids Res. 2018, 46, D633–D639. [Google Scholar] [CrossRef] [Green Version]
- Frolkis, A.; Knox, C.; Lim, E.; Jewison, T.; Law, V.; Hau, D.D.; Liu, P.; Gautam, B.; Ly, S.; Guo, A.C.; et al. SMPDB: The Small Molecule Pathway Database. Nucleic Acids Res. 2010, 38, D480–D487. [Google Scholar] [CrossRef] [Green Version]
- Kale, N.S.; Haug, K.; Conesa, P.; Jayseelan, K.; Moreno, P.; Rocca-Serra, P.; Nainala, V.C.; Spicer, R.A.; Williams, M.; Li, X.; et al. MetaboLights: An Open-Access Database Repository for Metabolomics Data. Curr. Protoc. Bioinform. 2016, 53, 14.13.1–14.13.18. [Google Scholar] [CrossRef] [PubMed]
- Kumar, B.; Prakash, A.; Ruhela, R.K.; Medhi, B. Potential of metabolomics in preclinical and clinical drug development. Pharmacol. Rep. 2014, 66, 956–963. [Google Scholar] [CrossRef]
- Beger, R.D.; Sun, J.; Schnackenberg, L.K. Metabolomics approaches for discovering biomarkers of drug-induced hepatotoxicity and nephrotoxicity. Toxicol. Appl. Pharmacol. 2010, 243, 154–166. [Google Scholar] [CrossRef] [PubMed]
- Araujo, A.M.; Carvalho, M.; Costa, V.M.; Duarte, J.A.; Dinis-Oliveira, R.J.; Bastos, M.L.; Guedes de Pinho, P.; Carvalho, F. In vivo toxicometabolomics reveals multi-organ and urine metabolic changes in mice upon acute exposure to human-relevant doses of 3,4-methylenedioxypyrovalerone (MDPV). Arch. Toxicol. 2021, 95, 509–527. [Google Scholar] [CrossRef] [PubMed]
- Roux, A.; Lison, D.; Junot, C.; Heilier, J.F. Applications of liquid chromatography coupled to mass spectrometry-based metabolomics in clinical chemistry and toxicology: A review. Clin. Biochem. 2011, 44, 119–135. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Shi, C.; Wu, C.Q.; Cao, A.M.; Sheng, H.Z.; Yan, X.Z.; Liao, M.Y. NMR-spectroscopy-based metabonomic approach to the analysis of Bay41-4109, a novel anti-HBV compound, induced hepatotoxicity in rats. Toxicol. Lett. 2007, 173, 161–167. [Google Scholar] [CrossRef]
- Huo, T.; Chen, X.; Lu, X.; Qu, L.; Liu, Y.; Cai, S. An effective assessment of valproate sodium-induced hepatotoxicity with UPLC-MS and (1)HNMR-based metabonomics approach. J. Chromatogr. B Analyt. Technol. Biomed. Life Sci. 2014, 969, 109–116. [Google Scholar] [CrossRef] [PubMed]
- Hanna, M.H.; Segar, J.L.; Teesch, L.M.; Kasper, D.C.; Schaefer, F.S.; Brophy, P.D. Urinary metabolomic markers of aminoglycoside nephrotoxicity in newborn rats. Pediatric Res. 2013, 73, 585–591. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Boudonck, K.J.; Mitchell, M.W.; Nemet, L.; Keresztes, L.; Nyska, A.; Shinar, D.; Rosenstock, M. Discovery of metabolomics biomarkers for early detection of nephrotoxicity. Toxicol. Pathol. 2009, 37, 280–292. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Andreadou, I.; Papaefthimiou, M.; Zira, A.; Constantinou, M.; Sigala, F.; Skaltsounis, A.L.; Tsantili-Kakoulidou, A.; Iliodromitis, E.K.; Kremastinos, D.T.; Mikros, E. Metabonomic identification of novel biomarkers in doxorubicin cardiotoxicity and protective effect of the natural antioxidant oleuropein. NMR Biomed. 2009, 22, 585–592. [Google Scholar] [CrossRef]
- Li, Y.; Ju, L.; Hou, Z.; Deng, H.; Zhang, Z.; Wang, L.; Yang, Z.; Yin, J.; Zhang, Y. Screening, verification, and optimization of biomarkers for early prediction of cardiotoxicity based on metabolomics. J. Proteome Res. 2015, 14, 2437–2445. [Google Scholar] [CrossRef] [PubMed]
- Van Vliet, E.; Morath, S.; Eskes, C.; Linge, J.; Rappsilber, J.; Honegger, P.; Hartung, T.; Coecke, S. A novel in vitro metabolomics approach for neurotoxicity testing, proof of principle for methyl mercury chloride and caffeine. Neurotoxicology 2008, 29, 1–12. [Google Scholar] [CrossRef]
- Liu, B.; Gu, Y.; Xiao, H.; Lei, X.; Liang, W.; Zhang, J. Altered metabolomic profiles may be associated with sevoflurane-induced neurotoxicity in neonatal rats. Neurochem. Res. 2015, 40, 788–799. [Google Scholar] [CrossRef]
- Huang, J.W.; Kuo, C.H.; Kuo, H.C.; Shih, J.Y.; Tsai, T.W.; Chang, L.C. Cell metabolomics analyses revealed a role of altered fatty acid oxidation in neurotoxicity pattern difference between nab-paclitaxel and solvent-based paclitaxel. PLoS ONE 2021, 16, e0248942. [Google Scholar] [CrossRef]
- Cappello, T.; Giannetto, A.; Parrino, V.; De Marco, G.; Mauceri, A.; Maisano, M. Food safety using NMR-based metabolomics: Assessment of the Atlantic bluefin tuna, Thunnus thynnus, from the Mediterranean Sea. Food Chem. Toxicol. 2018, 115, 391–397. [Google Scholar] [CrossRef] [PubMed]
- Stella, R.; Bovo, D.; Mastrorilli, E.; Manuali, E.; Pezzolato, M.; Bozzetta, E.; Lega, F.; Angeletti, R.; Biancotto, G. A novel tool to screen for treatments with clenbuterol in bovine: Identification of two hepatic markers by metabolomics investigation. Food Chem. 2021, 353, 129366. [Google Scholar] [CrossRef] [PubMed]
- Deng, P.; Li, X.; Petriello, M.C.; Wang, C.; Morris, A.J.; Hennig, B. Application of metabolomics to characterize environmental pollutant toxicity and disease risks. Rev. Environ. Health 2019, 34, 251–259. [Google Scholar] [CrossRef]
- Garcia-Sevillano, M.A.; Garcia-Barrera, T.; Gomez-Ariza, J.L. Environmental metabolomics: Biological markers for metal toxicity. Electrophoresis 2015, 36, 2348–2365. [Google Scholar] [CrossRef] [PubMed]
- Szeremeta, M.; Pietrowska, K.; Niemcunowicz-Janica, A.; Kretowski, A.; Ciborowski, M. Applications of Metabolomics in Forensic Toxicology and Forensic Medicine. Int. J. Mol. Sci. 2021, 22, 3010. [Google Scholar] [CrossRef] [PubMed]
- Steuer, A.E.; Brockbals, L.; Kraemer, T. Metabolomic Strategies in Biomarker Research-New Approach for Indirect Identification of Drug Consumption and Sample Manipulation in Clinical and Forensic Toxicology? Front. Chem. 2019, 7, 319. [Google Scholar] [CrossRef]
- Olesti, E.; Gonzalez-Ruiz, V.; Wilks, M.F.; Boccard, J.; Rudaz, S. Approaches in metabolomics for regulatory toxicology applications. Analyst 2021, 146, 1820–1834. [Google Scholar] [CrossRef]
- Sun, L.; Fang, L.; Lian, B.; Xia, J.J.; Zhou, C.J.; Wang, L.; Mao, Q.; Wang, X.F.; Gong, X.; Liang, Z.H.; et al. Biochemical effects of venlafaxine on astrocytes as revealed by (1)H NMR-based metabolic profiling. Mol. Biosyst. 2017, 13, 338–349. [Google Scholar] [CrossRef]
- Yuan, Y.; Fan, S.; Shu, L.; Huang, W.; Xie, L.; Bi, C.; Yu, H.; Wang, Y.; Li, Y. Exploration the Mechanism of Doxorubicin-Induced Heart Failure in Rats by Integration of Proteomics and Metabolomics Data. Front. Pharmacol. 2020, 11, 600561. [Google Scholar] [CrossRef]
- Xia, W.; Liu, G.; Shao, Z.; Xu, E.; Yuan, H.; Liu, J.; Gao, L. Toxicology of tramadol following chronic exposure based on metabolomics of the cerebrum in mice. Sci. Rep. 2020, 10, 11130. [Google Scholar] [CrossRef] [PubMed]
- Araújo, A.M.; Enea, M.; Fernandes, E.; Carvalho, F.; Bastos, M.L.; Carvalho, M.; Guedes de Pinho, P. MDMA hepatotoxicity under heat stress condition: Novel insights from in vitro metabolomic studies. J. Proteome Res. 2020, 19, 1222–1234. [Google Scholar] [CrossRef] [PubMed]
- Zeng, T.; Liang, Y.; Chen, J.; Cao, G.; Yang, Z.; Zhao, X.; Tian, J.; Xin, X.; Lei, B.; Cai, Z. Urinary metabolic characterization with nephrotoxicity for residents under cadmium exposure. Environ. Int. 2021, 154, 106646. [Google Scholar] [CrossRef] [PubMed]
- Sarma, S.N.; Saleem, A.; Lee, J.Y.; Tokumoto, M.; Hwang, G.W.; Man Chan, H.; Satoh, M. Effects of long-term cadmium exposure on urinary metabolite profiles in mice. J. Toxicol. Sci. 2018, 43, 89–100. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- An, Z.; Li, C.; Lv, Y.; Li, P.; Wu, C.; Liu, L. Metabolomics of Hydrazine-Induced Hepatotoxicity in Rats for Discovering Potential Biomarkers. Dis. Markers 2018, 2018, 8473161. [Google Scholar] [CrossRef] [Green Version]
- Jiang, J.; Li, S.; Wang, Y.; Xiao, X.; Jin, Y.; Wang, Y.; Yang, Z.; Yan, S.; Li, Y. Potential neurotoxicity of prenatal exposure to sevoflurane on offspring: Metabolomics investigation on neurodevelopment and underlying mechanism. Int. J. Dev. Neurosci. 2017, 62, 46–53. [Google Scholar] [CrossRef] [PubMed]
- Yin, J.; Xie, J.; Guo, X.; Ju, L.; Li, Y.; Zhang, Y. Plasma metabolic profiling analysis of cyclophosphamide-induced cardiotoxicity using metabolomics coupled with UPLC/Q-TOF-MS and ROC curve. J. Chromatogr. B Analyt. Technol. Biomed. Life Sci. 2016, 1033–1034, 428–435. [Google Scholar] [CrossRef]
- Ezaki, T.; Nishiumi, S.; Azuma, T.; Yoshida, M. Metabolomics for the early detection of cisplatin-induced nephrotoxicity. Toxicol. Res. 2017, 6, 843–853. [Google Scholar] [CrossRef] [Green Version]
- Ryu, S.H.; Kim, J.W.; Yoon, D.; Kim, S.; Kim, K.B. Serum and urine toxicometabolomics following gentamicin-induced nephrotoxicity in male Sprague-Dawley rats. J. Toxicol. Environ. Health A 2018, 81, 408–420. [Google Scholar] [CrossRef]
- Kostopoulou, S.; Ntatsi, G.; Arapis, G.; Aliferis, K.A. Assessment of the effects of metribuzin, glyphosate, and their mixtures on the metabolism of the model plant Lemna minor L. applying metabolomics. Chemosphere 2020, 239, 124582. [Google Scholar] [CrossRef] [PubMed]
- Wu, S.Y.; Phan, N.N.; Ho, S.H.; Lai, Y.H.; Tsai, C.H.; Yang, C.H.; Yu, H.G.; Wang, J.C.; Huang, P.L.; Lin, Y.C. Metabolomic assessment of arsenite toxicity and novel biomarker discovery in early development of zebrafish embryos. Toxicol. Lett. 2018, 290, 116–122. [Google Scholar] [CrossRef] [PubMed]
- Palmer, J.A.; Smith, A.M.; Gryshkova, V.; Donley, E.L.R.; Valentin, J.P.; Burrier, R.E. A Targeted Metabolomics-Based Assay Using Human Induced Pluripotent Stem Cell-Derived Cardiomyocytes Identifies Structural and Functional Cardiotoxicity Potential. Toxicol. Sci. 2020, 174, 218–240. [Google Scholar] [CrossRef]
- Sun, J.; Slavov, S.; Schnackenberg, L.K.; Ando, Y.; Greenhaw, J.; Yang, X.; Salminen, W.; Mendrick, D.L.; Beger, R.D. Identification of a Metabolic Biomarker Panel in Rats for Prediction of Acute and Idiosyncratic Hepatotoxicity. Comput. Struct. Biotechnol. J. 2014, 10, 78–89. [Google Scholar] [CrossRef] [PubMed] [Green Version]




{kind=link}
{kind=link}
{kind=link}
{kind=link}
| Omics Technology | Strengths | Limitations |
|---|---|---|
| Genomics |
|
|
| Transcriptomics |
|
|
| Proteomics |
|
|
| Metabolomics |
|
|
| Analytical Technique | Strengths | Weaknesses | |
|---|---|---|---|
| Nuclear magnetic resonance (NMR) |
|
| |
| Mass spectrometry (MS) | Direct infusion-mass spectrometry (DI-MS) |
|
|
| Capillary electrophoresis-mass spectrometry (CE-MS) |
|
| |
| Liquid chromatography-mass spectrometry (LC-MS) |
|
| |
| Gas chromatography-mass spectrometry (GC-MS) |
|
| |
| High-resolution mass spectrometry (HRMS) |
|
| |
| Toxicant Classification/ Name | Study Model/ Matrix | Study Design | Analytical Platform | Study Outcomes | Ref. |
|---|---|---|---|---|---|
| A. ELUCIDATION OF TOXICITY PATHWAYS | |||||
| Antidepressant drug: Venlafaxine (VEN) | In vitro model: Primary rat astrocytes | Astrocytes were treated with 10 µM VEN (n = 30), or with DMSO as a vehicle control (n = 30) for 72 h. The intracellular metabolites were profiled. | 1H NMR | Metabolic pathways significantly disturbed:
| [144] |
| Antineoplastic drug: Doxorubicin (DOX) | Animal model: Plasma of male Wistar rats | Rats were randomly divided into a treatment group injected i.p. with 3 mg DOX/kg once a week, for 6 weeks (n = 9), and a control group injected i.p. with saline (n = 8) | UPLC-Q-TOF-MS | Metabolic pathways significantly disturbed:
| [145] |
| Opioid analgesic: Tramadol | Animal model: Cerebrum of Kunming mice | Mice were treated with 0, 20, or 50 mg tramadol/kg/day, via oral gavage for 5 weeks (n = 6/group) | GC-TOF-MS | Metabolic pathways significantly disturbed:
| [146] |
| Drug of abuse: MDMA | In vitro model: Primary mouse hepatocytes | Hepatocytes were exposed to subtoxic (LC01: 203 µM and LC10: 472 µM) and toxic concentrations (LC30: 757 µM) for 24 h (n = 10/group). The intracellular metabolites were profiled. | GC-MS | Metabolic pathways significantly disturbed:
| [147] |
| Drug of abuse: MDPV | Animal model: Organs (liver, kidney, heart, and brain) and urine of male CD-1 mice | Mice were exposed to human-relevant doses (3 × 2.5 mg/kg and 3 × 5 mg/kg, i.p.) and sacrificed 24 h after the first administration (n = 10/group) | GC-MS | Metabolic pathways significantly disturbed in:
| [126] |
| Heavy metal (environmental pollutant): Cadmium (Cd) | Human model: Urine | 144 volunteers (n = 99 females and n = 45 males) living in three nearby villages with different levels of Cd: control area (<0.05 mg/kg), low-polluted area (0.2–0.4 mg/kg), and high-polluted area (>0.4 mg/kg) (according to the Cd content found in rice and vegetables growing in the area) | UHPLC-Q-Exactive Orbitrap MS | Metabolic pathways significantly disturbed:
| [148] |
| Heavy metal (environmental pollutant): Cd | Animal model: Urine of wild-type 129/Sv female mice | Mice were fed with 300 ppm Cd-containing chow (n = 5) for 67 weeks and compared to the control group (n = 4) | UPLC-QTOF-MS | Metabolic pathways significantly disturbed:
| [149] |
| B. IDENTIFICATION OF TOXICITY BIOMARKERS | |||||
| Hepatotoxic agent: Hydrazine | Animal model: Serum and urine of male Wistar rats | Rats were randomly divided into four groups: two control groups (n = 12/group) and two hydrazine-treated groups (n = 18/group). One control group and one hydrazine-treated group were allocated for sampling at 24 h postdosing, while the remaining two groups were for sampling at 48 h postdosing. The hydrazine-treated groups were orally administrated with a single dose of hydrazine (150 mg/kg), at which hydrazine could induce an obvious histopathological effect and hepatocellular lipid accumulation. | RRLC-MS |
| [150] |
| Neurotoxic agent: Sevoflurane (SEVO) | Animal model: Serum samples of offspring rats born from maternal Sprague Dawley rats | Rats within 18–19 days of gestation were randomly divided into control (CTR) or sevoflurane (SEVO) groups (n = 4/group). In the SEVO group, animals were treated with 2% sevoflurane carried by 100% oxygen for 6 h. For the control group, animals were placed in an identical condition without sevoflurane. Then, 26 postnatal-7-day rats were randomly selected from offspring generation groups (n = 13/group) and decapitated, and samples were collected for metabolomic analysis. | UPLC-TOF-MS |
| [151] |
| Cardiotoxic agent: Cyclophosphamide (CY) | Animal model: Plasma of male Wistar rats | Rats were randomly divided into four groups: control (n = 15), CY-1d, CY-3d, and CY-5d groups (n = 10/group). CY was administered i.p. to the rats on the first day, and the dosage was set at 200 mg/kg. The control group was administered i.p. with 1 mL saline on the first day. Rat plasma samples were collected one, three, and five days after CY administration. | UPLC-QTOF-MS |
| [152] |
| Nephrotoxic agent: Cisplatin | Animal model: Plasma and kidney tissue of male Crl:CD (SD) rats | Rats were randomly divided into three groups: the high-dose group (10 mg cisplatin/kg i.p., n = 13), low-dose group (5 mg/kg of cisplatin i.p., n = 10), and untreated group (n = 10). Blood samples were collected from the tail vein at 24, 48, and 96 h after the injection of cisplatin. Rats were sacrificed at 96 h, and kidney samples were also harvested for the metabolome analysis | GC-MS and LC-MS |
| [153] |
| Nephrotoxic agent: Gentamicin (GM) | Animal model: Serum and urine of male Sprague Dawley rats | Rats were given 0, 30, or 300 mg GM/kg/day i.p. for 3 consecutive days (n = 4–5/group) and were sacrificed 2 days (D2) or 8 days (D8) after last administration. | 1H NMR |
| [154] |
| Herbicides: Metribuzin, glyphosate and their mixtures | Aquatic plant model: Lemna minor L. | Plants were exposed for 72 h to concentrations of metribuzin or glyphosate equal to their corresponding EC50 values, or their mixtures (25–75, 50–50, or 75–25% of their corresponding EC50 values) (n = 6/group). | GC/EI/MS |
| [155] |
| Heavy metal: Arsenite | Animal model: Zebrafish (ZF) embryos | ZF embryos were exposed to sodium arsenite under different concentrations (0.5, 1.0, 2.0, and 5.0 mg/L) 24, 48, and 72 h postfertilization. ZF embryonic homogenate was used for metabolomic analysis. | UPLC-QTOF-MS |
| [156] |
| C. CONSTRUCTION OF TOXICITY PREDICTION MODELS | |||||
| Drugs from several different therapeutic classes with cardiotoxic potential | In vitro model: Pluripotent stem cell-derived cardiomyocytes (hiPSC-CM) | Phase 1: 66 drugs tested at a single, noncytotoxic concentration were used to identify predictive metabolites that could discriminate cardiotoxicants from noncardiotoxicants independent of changes. Phase 2: the discriminatory metabolites identified in phase 1 were used to create an exposure-based, targeted assay for identifying a drug’s cardiotoxicity potential. The predictivity was evaluated with 81 drugs (52 cardiotoxic and 29 noncardiotoxic). | UPLC-HRMS |
| [157] |
| Cardiotoxic drugs: DOX, isoproterenol (ISO) and 5-fluorouracil (5-FU) | Animal model: Plasma of male Wistar rats | Phase 1: 100 rats were randomly divided into 10 groups (n = 10/group) to screen the potential biomarkers for the early prediction of cardiotoxicity. For each drug, different doses and sampling times were tested. Phase 2: 70 rats were randomly divided into seven groups, which included control, two cardiotoxicity groups (ISO and 5-FU), two hepatotoxicity groups (Radix Bupleuri and carbon tetrachloride), and two nephrotoxicity groups (gentamicin and etimicin), to examine the specificity of the selected biomarkers. Phase 3: the discriminatory metabolites selected in phase 2 were used to create a predictive model of drug-induced cardiotoxicity in its early stages. | UPLC-Q-TOF-MS |
| [133] |
| Two overt hepatotoxicants (acetaminophen (APAP) and carbon tetrachloride (CCl4)), two idiosyncratic hepatotoxicants (felbamate (FEL) and dantrolene(DAN)), and three nonhepatotoxicants (meloxicam (MEL), penicillin (PEN) and metformin (MET)) | Animal model: Blood of male Sprague Dawley rats | Rats were orally gavaged with a single dose of vehicle (n = 4 for APAP study and n = 5 for other compound studies), low dose or high dose of the compounds (100 or 1250 mg APAP/kg, 50 or 2000 mg CCl4/kg, 300 or 1920 mg FEL/kg, 100 or 1000 mg DAN/kg, 100 or 1500 mg MET/kg, 0.4 or 12 mg MEL/kg, and 100 or 2400 mg PEN/kg (n = 7/APAP groups and n = 5/for all other groups)). At 6 and 24 h postdosing, blood was collected for metabolomics analysis. | LC-QTOF-MS |
| [158] |
Publisher’s Note: MDPI stays neutral with regard to jurisdictional claims in published maps and institutional affiliations. |
© 2021 by the authors. Licensee MDPI, Basel, Switzerland. This article is an open access article distributed under the terms and conditions of the Creative Commons Attribution (CC BY) license (https://creativecommons.org/licenses/by/4.0/).
Share and Cite
Araújo, A.M.; Carvalho, F.; Guedes de Pinho, P.; Carvalho, M. Toxicometabolomics: Small Molecules to Answer Big Toxicological Questions. Metabolites 2021, 11, 692. https://doi.org/10.3390/metabo11100692
Araújo AM, Carvalho F, Guedes de Pinho P, Carvalho M. Toxicometabolomics: Small Molecules to Answer Big Toxicological Questions. Metabolites. 2021; 11(10):692. https://doi.org/10.3390/metabo11100692
Chicago/Turabian StyleAraújo, Ana Margarida, Félix Carvalho, Paula Guedes de Pinho, and Márcia Carvalho. 2021. "Toxicometabolomics: Small Molecules to Answer Big Toxicological Questions" Metabolites 11, no. 10: 692. https://doi.org/10.3390/metabo11100692
APA StyleAraújo, A. M., Carvalho, F., Guedes de Pinho, P., & Carvalho, M. (2021). Toxicometabolomics: Small Molecules to Answer Big Toxicological Questions. Metabolites, 11(10), 692. https://doi.org/10.3390/metabo11100692