
Molecular Mechanisms of the SLC13A5 Gene Transcription


Abstract
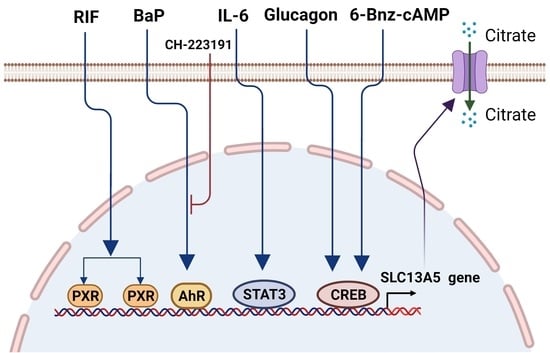
:
1. Introduction
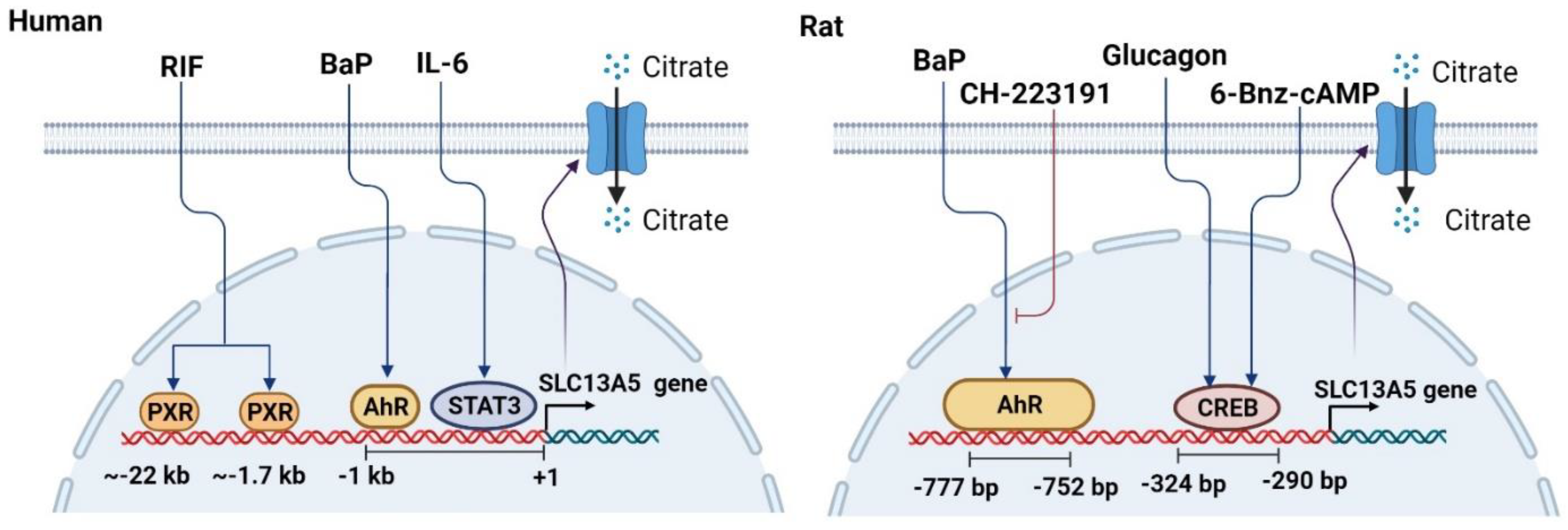
2. Upregulation of the SLC13A5 Expression
2.1. PXR in the SLC13A5 Transcription
2.2. AhR in the SLC13A5 Transcription
2.3. CREB in the SLC13A5 Transcription
2.4. STAT3 in the SLC13A5 Expression
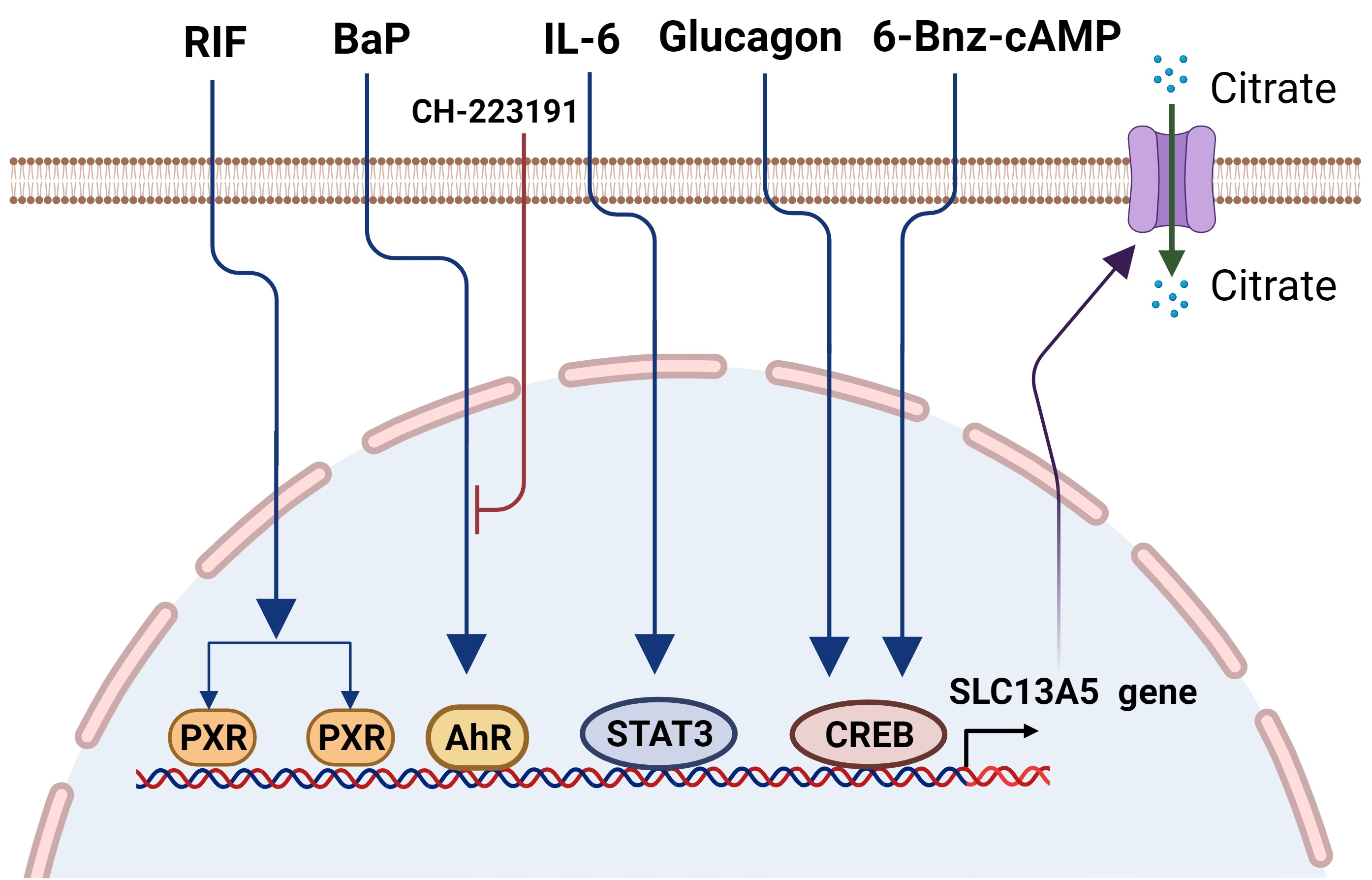
3. Downregulation of the SLC13A5 Expression
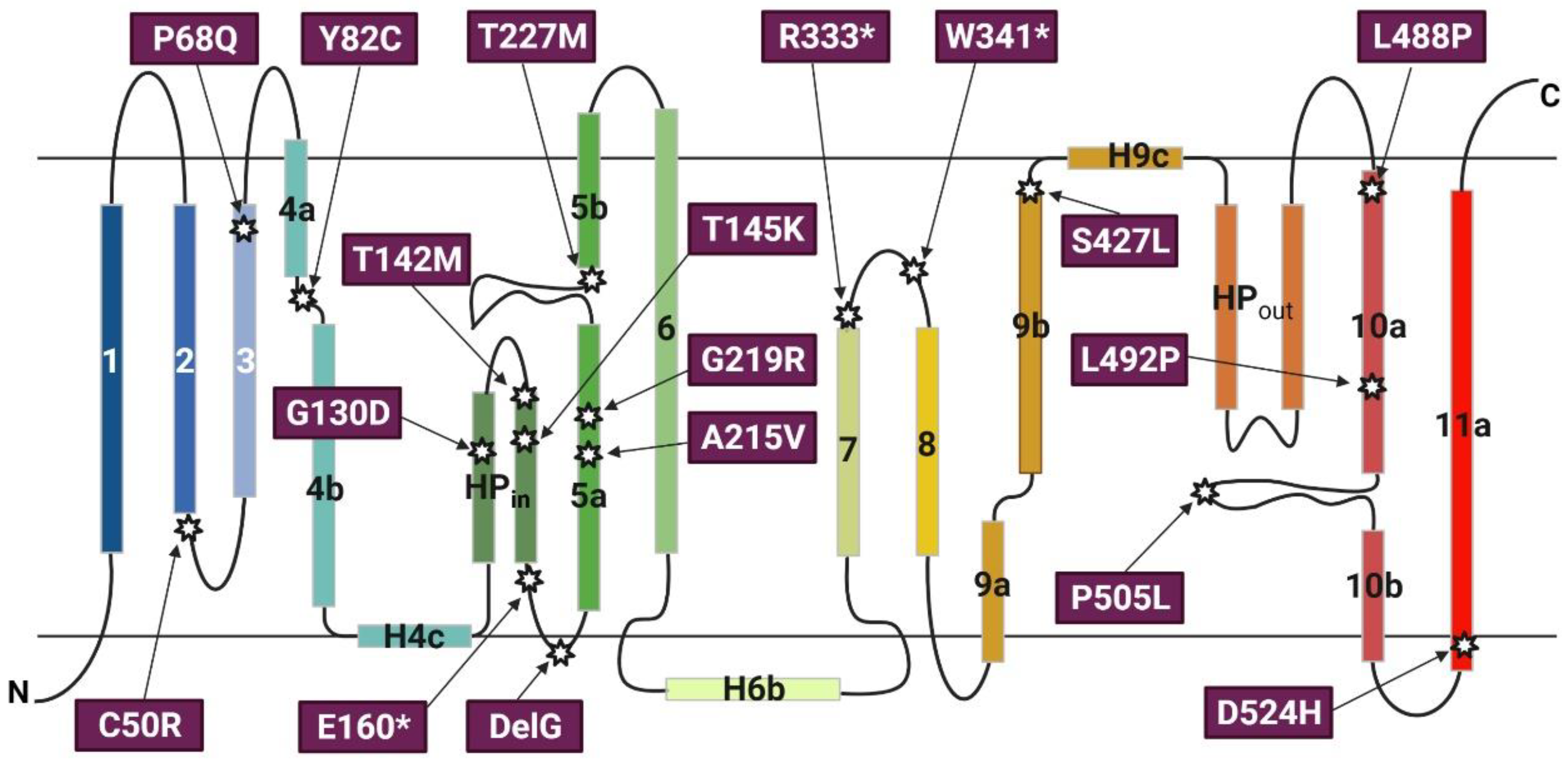
3.1. Naturally Occurring Mutations of the SLC13A5 Gene
3.2. Epigenetic Regulation of SLC13A5
4. Conclusions and Perspectives
Author Contributions
Funding
Acknowledgments
Conflicts of Interest
References
- Inoue, K.; Zhuang, L.; Ganapathy, V. Human Na+-coupled citrate transporter: Primary structure, genomic organization, and transport function. Biochem. Biophys. Res. Commun. 2002, 299, 465–471. [Google Scholar] [CrossRef]
- Inoue, K.; Zhuang, L.; Maddox, D.M.; Smith, S.B.; Ganapathy, V. Structure, function, and expression pattern of a novel sodium-coupled citrate transporter (NaCT) cloned from mammalian brain. J. Biol. Chem. 2002, 277, 39469–39476. [Google Scholar] [CrossRef] [Green Version]
- Iacobazzi, V.; Infantino, V. Citrate--new functions for an old metabolite. Biol. Chem. 2014, 395, 387–399. [Google Scholar] [CrossRef]
- Icard, P.; Poulain, L.; Lincet, H. Understanding the central role of citrate in the metabolism of cancer cells. Biochim. Biophys. Acta 2012, 1825, 111–116. [Google Scholar] [CrossRef]
- Gnoni, G.V.; Priore, P.; Geelen, M.J.; Siculella, L. The mitochondrial citrate carrier: Metabolic role and regulation of its activity and expression. IUBMB Life 2009, 61, 987–994. [Google Scholar] [CrossRef]
- Rogers, R.P.; Rogina, B. The role of INDY in metabolism, health and longevity. Front. Genet. 2015, 6, 204. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Chypre, M.; Zaidi, N.; Smans, K. ATP-citrate lyase: A mini-review. Biochem. Biophys. Res. Commun. 2012, 422, 1–4. [Google Scholar] [CrossRef] [PubMed]
- Zaidi, N.; Swinnen, J.V.; Smans, K. ATP-citrate lyase: A key player in cancer metabolism. Cancer Res. 2012, 72, 3709–3714. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Huang, L.; Wang, C.; Xu, H.; Peng, G. Targeting citrate as a novel therapeutic strategy in cancer treatment. Biochim. Biophys. Acta. Rev. Cancer 2020, 1873, 188332. [Google Scholar] [CrossRef]
- Costello, L.C.; Franklin, R.B. Plasma Citrate Homeostasis: How It Is Regulated; And Its Physiological and Clinical Implications. An Important, But Neglected, Relationship in Medicine. HSOA J. Hum. Endocrinol. 2016, 1. [Google Scholar] [CrossRef]
- Pajor, A.M. Sodium-coupled dicarboxylate and citrate transporters from the SLC13 family. Pflugers Arch. 2014, 466, 119–130. [Google Scholar] [CrossRef]
- Rogina, B.; Reenan, R.A.; Nilsen, S.P.; Helfand, S.L. Extended life-span conferred by cotransporter gene mutations in Drosophila. Science 2000, 290, 2137–2140. [Google Scholar] [CrossRef]
- Fei, Y.J.; Liu, J.C.; Inoue, K.; Zhuang, L.; Miyake, K.; Miyauchi, S.; Ganapathy, V. Relevance of NAC-2, an Na+-coupled citrate transporter, to life span, body size and fat content in Caenorhabditis elegans. Biochem. J. 2004, 379, 191–198. [Google Scholar] [CrossRef]
- Inoue, K.; Fei, Y.J.; Zhuang, L.; Gopal, E.; Miyauchi, S.; Ganapathy, V. Functional features and genomic organization of mouse NaCT, a sodium-coupled transporter for tricarboxylic acid cycle intermediates. Biochem. J. 2004, 378, 949–957. [Google Scholar] [CrossRef] [Green Version]
- Birkenfeld, A.L.; Lee, H.Y.; Guebre-Egziabher, F.; Alves, T.C.; Jurczak, M.J.; Jornayvaz, F.R.; Zhang, D.; Hsiao, J.J.; Martin-Montalvo, A.; Fischer-Rosinsky, A.; et al. Deletion of the mammalian INDY homolog mimics aspects of dietary restriction and protects against adiposity and insulin resistance in mice. Cell Metab. 2011, 14, 184–195. [Google Scholar] [CrossRef] [Green Version]
- Mantila Roosa, S.M.; Liu, Y.; Turner, C.H. Gene expression patterns in bone following mechanical loading. J. Bone Miner. Res. 2011, 26, 100–112. [Google Scholar] [CrossRef] [Green Version]
- Willmes, D.M.; Kurzbach, A.; Henke, C.; Schumann, T.; Zahn, G.; Heifetz, A.; Jordan, J.; Helfand, S.L.; Birkenfeld, A.L. The longevity gene INDY (I’m Not Dead Yet) in metabolic control: Potential as pharmacological target. Pharmacol. Ther. 2018, 185, 1–11. [Google Scholar] [CrossRef]
- Gopal, E.; Miyauchi, S.; Martin, P.M.; Ananth, S.; Srinivas, S.R.; Smith, S.B.; Prasad, P.D.; Ganapathy, V. Expression and functional features of NaCT, a sodium-coupled citrate transporter, in human and rat livers and cell lines. Am. J. Physiol. Gastrointest. Liver Physiol. 2007, 292, G402–G408. [Google Scholar] [CrossRef] [PubMed]
- Bhutia, Y.D.; Kopel, J.J.; Lawrence, J.J.; Neugebauer, V.; Ganapathy, V. Plasma Membrane Na(+)-Coupled Citrate Transporter (SLC13A5) and Neonatal Epileptic Encephalopathy. Molecules 2017, 22, 378. [Google Scholar] [CrossRef] [PubMed]
- Li, Z.; Li, D.; Choi, E.Y.; Lapidus, R.; Zhang, L.; Huang, S.M.; Shapiro, P.; Wang, H. Silencing of solute carrier family 13 member 5 disrupts energy homeostasis and inhibits proliferation of human hepatocarcinoma cells. J. Biol. Chem. 2017, 292, 13890–13901. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Huard, K.; Brown, J.; Jones, J.C.; Cabral, S.; Futatsugi, K.; Gorgoglione, M.; Lanba, A.; Vera, N.B.; Zhu, Y.; Yan, Q.; et al. Discovery and characterization of novel inhibitors of the sodium-coupled citrate transporter (NaCT or SLC13A5). Sci. Rep. 2015, 5, 17391. [Google Scholar] [CrossRef] [Green Version]
- Brachs, S.; Winkel, A.F.; Tang, H.; Birkenfeld, A.L.; Brunner, B.; Jahn-Hofmann, K.; Margerie, D.; Ruetten, H.; Schmoll, D.; Spranger, J. Inhibition of citrate cotransporter Slc13a5/mINDY by RNAi improves hepatic insulin sensitivity and prevents diet-induced non-alcoholic fatty liver disease in mice. Mol. Metab. 2016, 5, 1072–1082. [Google Scholar] [CrossRef]
- Pesta, D.H.; Perry, R.J.; Guebre-Egziabher, F.; Zhang, D.; Jurczak, M.; Fischer-Rosinsky, A.; Daniels, M.A.; Willmes, D.M.; Bhanot, S.; Bornstein, S.R.; et al. Prevention of diet-induced hepatic steatosis and hepatic insulin resistance by second generation antisense oligonucleotides targeted to the longevity gene mIndy (Slc13a5). Aging 2015, 7, 1086–1093. [Google Scholar] [CrossRef] [Green Version]
- Neuschafer-Rube, F.; Schraplau, A.; Schewe, B.; Lieske, S.; Krutzfeldt, J.M.; Ringel, S.; Henkel, J.; Birkenfeld, A.L.; Puschel, G.P. Arylhydrocarbon receptor-dependent mIndy (Slc13a5) induction as possible contributor to benzo[a]pyrene-induced lipid accumulation in hepatocytes. Toxicology 2015, 337, 1–9. [Google Scholar] [CrossRef]
- Li, L.; Li, H.; Garzel, B.; Yang, H.; Sueyoshi, T.; Li, Q.; Shu, Y.; Zhang, J.; Hu, B.; Heyward, S.; et al. SLC13A5 is a novel transcriptional target of the pregnane X receptor and sensitizes drug-induced steatosis in human liver. Mol. Pharmacol. 2015, 87, 674–682. [Google Scholar] [CrossRef] [Green Version]
- Von Loeffelholz, C.; Lieske, S.; Neuschafer-Rube, F.; Willmes, D.M.; Raschzok, N.; Sauer, I.M.; Konig, J.; Fromm, M.F.; Horn, P.; Chatzigeorgiou, A.; et al. The human longevity gene homolog INDY and interleukin-6 interact in hepatic lipid metabolism. Hepatology 2017, 66, 616–630. [Google Scholar] [CrossRef] [Green Version]
- Sauer, D.B.; Song, J.; Wang, B.; Hilton, J.K.; Karpowich, N.K.; Mindell, J.A.; Rice, W.J.; Wang, D.N. Structure and inhibition mechanism of the human citrate transporter NaCT. Nature 2021, 591, 157–161. [Google Scholar] [CrossRef] [PubMed]
- Hardies, K.; de Kovel, C.G.; Weckhuysen, S.; Asselbergh, B.; Geuens, T.; Deconinck, T.; Azmi, A.; May, P.; Brilstra, E.; Becker, F.; et al. Recessive mutations in SLC13A5 result in a loss of citrate transport and cause neonatal epilepsy, developmental delay and teeth hypoplasia. Brain A J. Neurol. 2015, 138, 3238–3250. [Google Scholar] [CrossRef] [Green Version]
- Klotz, J.; Porter, B.E.; Colas, C.; Schlessinger, A.; Pajor, A.M. Mutations in the Na(+)/citrate cotransporter NaCT (SLC13A5) in pediatric patients with epilepsy and developmental delay. Mol. Med. 2016, 22. [Google Scholar] [CrossRef] [PubMed]
- Selch, S.; Chafai, A.; Sticht, H.; Birkenfeld, A.L.; Fromm, M.F.; Konig, J. Analysis of naturally occurring mutations in the human uptake transporter NaCT important for bone and brain development and energy metabolism. Sci. Rep. 2018, 8, 11330. [Google Scholar] [CrossRef] [PubMed]
- Sueyoshi, T.; Negishi, M. Phenobarbital response elements of cytochrome P450 genes and nuclear receptors. Annu. Rev. Pharmacol. Toxicol. 2001, 41, 123–143. [Google Scholar] [CrossRef] [PubMed]
- Kliewer, S.A.; Moore, J.T.; Wade, L.; Staudinger, J.L.; Watson, M.A.; Jones, S.A.; McKee, D.D.; Oliver, B.B.; Willson, T.M.; Zetterstrom, R.H.; et al. An orphan nuclear receptor activated by pregnanes defines a novel steroid signaling pathway. Cell 1998, 92, 73–82. [Google Scholar] [CrossRef] [Green Version]
- Lehmann, J.M.; McKee, D.D.; Watson, M.A.; Willson, T.M.; Moore, J.T.; Kliewer, S.A. The human orphan nuclear receptor PXR is activated by compounds that regulate CYP3A4 gene expression and cause drug interactions. J. Clin. Investig. 1998, 102, 1016–1023. [Google Scholar] [CrossRef] [PubMed]
- Kliewer, S.A.; Willson, T.M. Regulation of xenobiotic and bile acid metabolism by the nuclear pregnane X receptor. J. Lipid Res. 2002, 43, 359–364. [Google Scholar] [CrossRef]
- di Masi, A.; De Marinis, E.; Ascenzi, P.; Marino, M. Nuclear receptors CAR and PXR: Molecular, functional, and biomedical aspects. Mol. Asp. Med. 2009, 30, 297–343. [Google Scholar] [CrossRef]
- Tolson, A.H.; Wang, H. Regulation of drug-metabolizing enzymes by xenobiotic receptors: PXR and CAR. Adv. Drug Deliv. Rev. 2010, 62, 1238–1249. [Google Scholar] [CrossRef] [Green Version]
- Zhou, J.; Febbraio, M.; Wada, T.; Zhai, Y.; Kuruba, R.; He, J.; Lee, J.H.; Khadem, S.; Ren, S.; Li, S.; et al. Hepatic fatty acid transporter Cd36 is a common target of LXR, PXR, and PPARgamma in promoting steatosis. Gastroenterology 2008, 134, 556–567. [Google Scholar] [CrossRef] [PubMed]
- Wada, T.; Gao, J.; Xie, W. PXR and CAR in energy metabolism. Trends Endocrinol. Metab. 2009, 20, 273–279. [Google Scholar] [CrossRef]
- Nakamura, K.; Moore, R.; Negishi, M.; Sueyoshi, T. Nuclear pregnane X receptor cross-talk with FoxA2 to mediate drug-induced regulation of lipid metabolism in fasting mouse liver. J. Biol. Chem. 2007, 282, 9768–9776. [Google Scholar] [CrossRef] [Green Version]
- Biswas, A.; Pasquel, D.; Tyagi, R.K.; Mani, S. Acetylation of pregnane X receptor protein determines selective function independent of ligand activation. Biochem. Biophys. Res. Commun. 2011, 406, 371–376. [Google Scholar] [CrossRef] [Green Version]
- Staudinger, J.L.; Xu, C.; Biswas, A.; Mani, S. Post-translational modification of pregnane x receptor. Pharmacol. Res. 2011, 64, 4–10. [Google Scholar] [CrossRef] [Green Version]
- Biswas, A.; Mani, S.; Redinbo, M.R.; Krasowski, M.D.; Li, H.; Ekins, S. Elucidating the ‘Jekyll and Hyde’ nature of PXR: The case for discovering antagonists or allosteric antagonists. Pharm. Res. 2009, 26, 1807–1815. [Google Scholar] [CrossRef] [PubMed]
- He, J.; Gao, J.; Xu, M.; Ren, S.; Stefanovic-Racic, M.; O’Doherty, R.M.; Xie, W. PXR ablation alleviates diet-induced and genetic obesity and insulin resistance in mice. Diabetes 2013, 62, 1876–1887. [Google Scholar] [CrossRef] [Green Version]
- Zhou, J.; Zhai, Y.; Mu, Y.; Gong, H.; Uppal, H.; Toma, D.; Ren, S.; Evans, R.M.; Xie, W. A novel pregnane X receptor-mediated and sterol regulatory element-binding protein-independent lipogenic pathway. J. Biol. Chem. 2006, 281, 15013–15020. [Google Scholar] [CrossRef] [Green Version]
- Bitter, A.; Rummele, P.; Klein, K.; Kandel, B.A.; Rieger, J.K.; Nussler, A.K.; Zanger, U.M.; Trauner, M.; Schwab, M.; Burk, O. Pregnane X receptor activation and silencing promote steatosis of human hepatic cells by distinct lipogenic mechanisms. Arch. Toxicol. 2015, 89, 2089–2103. [Google Scholar] [CrossRef]
- Zhou, C.; Poulton, E.J.; Grun, F.; Bammler, T.K.; Blumberg, B.; Thummel, K.E.; Eaton, D.L. The dietary isothiocyanate sulforaphane is an antagonist of the human steroid and xenobiotic nuclear receptor. Mol. Pharmacol. 2007, 71, 220–229. [Google Scholar] [CrossRef]
- Jiang, Y.; Yao, X.; Fan, S.; Gao, Y.; Zhang, H.; Huang, M.; Bi, H. Lipidomic profiling reveals triacylglycerol accumulation in the liver during pregnane X receptor activation-induced hepatomegaly. J. Pharm. Biomed. Anal. 2021, 195, 113851. [Google Scholar] [CrossRef] [PubMed]
- Li, D.; Mackowiak, B.; Brayman, T.G.; Mitchell, M.; Zhang, L.; Huang, S.M.; Wang, H. Genome-wide analysis of human constitutive androstane receptor (CAR) transcriptome in wild-type and CAR-knockout HepaRG cells. Biochem. Pharmacol. 2015, 98, 190–202. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Li, L.; Welch, M.A.; Li, Z.; Mackowiak, B.; Heyward, S.; Swaan, P.W.; Wang, H. Mechanistic Insights of Phenobarbital-Mediated Activation of Human but Not Mouse Pregnane X Receptor. Mol. Pharmacol. 2019, 96, 345–354. [Google Scholar] [CrossRef] [PubMed]
- Williamson, M.A.; Gasiewicz, T.A.; Opanashuk, L.A. Aryl hydrocarbon receptor expression and activity in cerebellar granule neuroblasts: Implications for development and dioxin neurotoxicity. Toxicol. Sci. 2005, 83, 340–348. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Guyot, E.; Chevallier, A.; Barouki, R.; Coumoul, X. The AhR twist: Ligand-dependent AhR signaling and pharmaco-toxicological implications. Drug Discov. Today 2013, 18, 479–486. [Google Scholar] [CrossRef] [Green Version]
- Kudo, I.; Hosaka, M.; Haga, A.; Tsuji, N.; Nagata, Y.; Okada, H.; Fukuda, K.; Kakizaki, Y.; Okamoto, T.; Grave, E.; et al. The regulation mechanisms of AhR by molecular chaperone complex. J. Biochem. 2018, 163, 223–232. [Google Scholar] [CrossRef]
- Beischlag, T.V.; Luis Morales, J.; Hollingshead, B.D.; Perdew, G.H. The aryl hydrocarbon receptor complex and the control of gene expression. Crit. Rev. Eukaryot Gene Expr. 2008, 18, 207–250. [Google Scholar] [CrossRef] [Green Version]
- Yueh, M.F.; Huang, Y.H.; Hiller, A.; Chen, S.; Nguyen, N.; Tukey, R.H. Involvement of the xenobiotic response element (XRE) in Ah receptor-mediated induction of human UDP-glucuronosyltransferase 1A1. J. Biol. Chem. 2003, 278, 15001–15006. [Google Scholar] [CrossRef] [Green Version]
- Shelby, M.K.; Cherrington, N.J.; Vansell, N.R.; Klaassen, C.D. Tissue mRNA expression of the rat UDP-glucuronosyltransferase gene family. Drug Metab. Dispos. 2003, 31, 326–333. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Hankinson, O.; Brooks, B.A.; Weir-Brown, K.I.; Hoffman, E.C.; Johnson, B.S.; Nanthur, J.; Reyes, H.; Watson, A.J. Genetic and molecular analysis of the Ah receptor and of Cyp1a1 gene expression. Biochimie 1991, 73, 61–66. [Google Scholar] [CrossRef]
- Gu, Y.Z.; Hogenesch, J.B.; Bradfield, C.A. The PAS superfamily: Sensors of environmental and developmental signals. Annu. Rev. Pharmacol. Toxicol. 2000, 40, 519–561. [Google Scholar] [CrossRef] [Green Version]
- Emi, Y.; Ikushiro, S.; Iyanagi, T. Xenobiotic responsive element-mediated transcriptional activation in the UDP-glucuronosyltransferase family 1 gene complex. J. Biol. Chem. 1996, 271, 3952–3958. [Google Scholar] [CrossRef] [Green Version]
- Munzel, P.A.; Schmohl, S.; Buckler, F.; Jaehrling, J.; Raschko, F.T.; Kohle, C.; Bock, K.W. Contribution of the Ah receptor to the phenolic antioxidant-mediated expression of human and rat UDP-glucuronosyltransferase UGT1A6 in Caco-2 and rat hepatoma 5L cells. Biochem. Pharmacol. 2003, 66, 841–847. [Google Scholar] [CrossRef]
- Ebert, B.; Seidel, A.; Lampen, A. Identification of BCRP as transporter of benzo[a]pyrene conjugates metabolically formed in Caco-2 cells and its induction by Ah-receptor agonists. Carcinogenesis 2005, 26, 1754–1763. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Rothhammer, V.; Quintana, F.J. The aryl hydrocarbon receptor: An environmental sensor integrating immune responses in health and disease. Nat. Rev. Immunol. 2019, 19, 184–197. [Google Scholar] [CrossRef]
- Mackowiak, B.; Wang, H. Mechanisms of xenobiotic receptor activation: Direct vs. indirect. Biochim. Biophys. Acta 2016, 1859, 1130–1140. [Google Scholar] [CrossRef] [Green Version]
- Lee, J.H.; Wada, T.; Febbraio, M.; He, J.; Matsubara, T.; Lee, M.J.; Gonzalez, F.J.; Xie, W. A novel role for the dioxin receptor in fatty acid metabolism and hepatic steatosis. Gastroenterology 2010, 139, 653–663. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Cave, M.; Appana, S.; Patel, M.; Falkner, K.C.; McClain, C.J.; Brock, G. Polychlorinated biphenyls, lead, and mercury are associated with liver disease in American adults: NHANES 2003-2004. Environ. Health Perspect. 2010, 118, 1735–1742. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Shan, Q.; Li, H.; Chen, N.; Qu, F.; Guo, J. Understanding the Multiple Effects of PCBs on Lipid Metabolism. Diabetes Metab. Syndr. Obes. 2020, 13, 3691–3702. [Google Scholar] [CrossRef] [PubMed]
- Okino, S.T.; Quattrochi, L.C.; Pookot, D.; Iwahashi, M.; Dahiya, R. A dioxin-responsive enhancer 3’ of the human CYP1A2 gene. Mol. Pharmacol. 2007, 72, 1457–1465. [Google Scholar] [CrossRef] [Green Version]
- Nakata, A.; Urano, D.; Fujii-Kuriyama, Y.; Mizuno, N.; Tago, K.; Itoh, H. G-protein signalling negatively regulates the stability of aryl hydrocarbon receptor. EMBO Rep. 2009, 10, 622–628. [Google Scholar] [CrossRef]
- Schraplau, A.; Schewe, B.; Neuschafer-Rube, F.; Ringel, S.; Neuber, C.; Kleuser, B.; Puschel, G.P. Enhanced thyroid hormone breakdown in hepatocytes by mutual induction of the constitutive androstane receptor (CAR, NR1I3) and arylhydrocarbon receptor by benzo[a]pyrene and phenobarbital. Toxicology 2015, 328, 21–28. [Google Scholar] [CrossRef]
- Li, Q.; Gao, C.; Deng, H.; Song, Q.; Yuan, L. Benzo[a]pyrene induces pyroptotic and autophagic death through inhibiting PI3K/Akt signaling pathway in HL-7702 human normal liver cells. J. Toxicol. Sci. 2019, 44, 121–131. [Google Scholar] [CrossRef] [Green Version]
- Benchoula, K.; Parhar, I.S.; Madhavan, P.; Hwa, W.E. CREB nuclear transcription activity as a targeting factor in the treatment of diabetes and diabetes complications. Biochem. Pharmacol. 2021, 188, 114531. [Google Scholar] [CrossRef]
- Altarejos, J.Y.; Montminy, M. CREB and the CRTC co-activators: Sensors for hormonal and metabolic signals. Nat. Rev. Mol. Cell Biol. 2011, 12, 141–151. [Google Scholar] [CrossRef] [Green Version]
- Neuschafer-Rube, F.; Lieske, S.; Kuna, M.; Henkel, J.; Perry, R.J.; Erion, D.M.; Pesta, D.; Willmes, D.M.; Brachs, S.; von Loeffelholz, C.; et al. The mammalian INDY homolog is induced by CREB in a rat model of type 2 diabetes. Diabetes 2014, 63, 1048–1057. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Jitrapakdee, S. Transcription factors and coactivators controlling nutrient and hormonal regulation of hepatic gluconeogenesis. Int. J. Biochem. Cell Biol. 2012, 44, 33–45. [Google Scholar] [CrossRef]
- Zhang, E.E.; Liu, Y.; Dentin, R.; Pongsawakul, P.Y.; Liu, A.C.; Hirota, T.; Nusinow, D.A.; Sun, X.; Landais, S.; Kodama, Y.; et al. Cryptochrome mediates circadian regulation of cAMP signaling and hepatic gluconeogenesis. Nat. Med. 2010, 16, 1152–1156. [Google Scholar] [CrossRef]
- Kopel, J.; Higuchi, K.; Ristic, B.; Sato, T.; Ramachandran, S.; Ganapathy, V. The Hepatic Plasma Membrane Citrate Transporter NaCT (SLC13A5) as a Molecular Target for Metformin. Sci. Rep. 2020, 10, 8536. [Google Scholar] [CrossRef] [PubMed]
- He, L.; Wondisford, F.E. Metformin action: Concentrations matter. Cell Metab. 2015, 21, 159–162. [Google Scholar] [CrossRef] [Green Version]
- Perry, R.J.; Camporez, J.G.; Kursawe, R.; Titchenell, P.M.; Zhang, D.; Perry, C.J.; Jurczak, M.J.; Abudukadier, A.; Han, M.S.; Zhang, X.M.; et al. Hepatic acetyl CoA links adipose tissue inflammation to hepatic insulin resistance and type 2 diabetes. Cell 2015, 160, 745–758. [Google Scholar] [CrossRef] [Green Version]
- Su, H.; Lei, C.T.; Zhang, C. Interleukin-6 Signaling Pathway and Its Role in Kidney Disease: An Update. Front. Immunol. 2017, 8, 405. [Google Scholar] [CrossRef] [Green Version]
- Sabio, G.; Das, M.; Mora, A.; Zhang, Z.; Jun, J.Y.; Ko, H.J.; Barrett, T.; Kim, J.K.; Davis, R.J. A stress signaling pathway in adipose tissue regulates hepatic insulin resistance. Science 2008, 322, 1539–1543. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Senn, J.J.; Klover, P.J.; Nowak, I.A.; Zimmers, T.A.; Koniaris, L.G.; Furlanetto, R.W.; Mooney, R.A. Suppressor of cytokine signaling-3 (SOCS-3), a potential mediator of interleukin-6-dependent insulin resistance in hepatocytes. J. Biol. Chem. 2003, 278, 13740–13746. [Google Scholar] [CrossRef] [Green Version]
- Mathis, D.; Shoelson, S.E. Immunometabolism: An emerging frontier. Nat. Rev. Immunol. 2011, 11, 81. [Google Scholar] [CrossRef] [Green Version]
- Yang, Q.Z.; Spelbrink, E.M.; Nye, K.L.; Hsu, E.R.; Porter, B.E. Epilepsy and EEG Phenotype of SLC13A5 Citrate Transporter Disorder. Child. Neurol. Open 2020, 7. [Google Scholar] [CrossRef]
- Thevenon, J.; Milh, M.; Feillet, F.; St-Onge, J.; Duffourd, Y.; Juge, C.; Roubertie, A.; Heron, D.; Mignot, C.; Raffo, E.; et al. Mutations in SLC13A5 cause autosomal-recessive epileptic encephalopathy with seizure onset in the first days of life. Am. J. Hum. Genet. 2014, 95, 113–120. [Google Scholar] [CrossRef]
- Matricardi, S.; De Liso, P.; Freri, E.; Costa, P.; Castellotti, B.; Magri, S.; Gellera, C.; Granata, T.; Musante, L.; Lesca, G.; et al. Neonatal developmental and epileptic encephalopathy due to autosomal recessive variants in SLC13A5 gene. Epilepsia 2020, 61, 2474–2485. [Google Scholar] [CrossRef]
- Spielmann, M.; Mundlos, S. Looking beyond the genes: The role of non-coding variants in human disease. Hum. Mol. Genet. 2016, 25, R157–R165. [Google Scholar] [CrossRef] [PubMed]
- Loehlin, D.W.; Ames, J.R.; Vaccaro, K.; Carroll, S.B. A major role for noncoding regulatory mutations in the evolution of enzyme activity. Proc. Natl. Acad. Sci. USA 2019, 116, 12383–12389. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Hedrich, W.D.; Hassan, H.E.; Wang, H. Insights into CYP2B6-mediated drug-drug interactions. Acta Pharm. Sin. B 2016, 6, 413–425. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Lek, M.; Karczewski, K.J.; Minikel, E.V.; Samocha, K.E.; Banks, E.; Fennell, T.; O’Donnell-Luria, A.H.; Ware, J.S.; Hill, A.J.; Cummings, B.B.; et al. Analysis of protein-coding genetic variation in 60,706 humans. Nature 2016, 536, 285–291. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Handy, D.E.; Castro, R.; Loscalzo, J. Epigenetic modifications: Basic mechanisms and role in cardiovascular disease. Circulation 2011, 123, 2145–2156. [Google Scholar] [CrossRef] [Green Version]
- Schiano, C.; Benincasa, G.; Franzese, M.; Della Mura, N.; Pane, K.; Salvatore, M.; Napoli, C. Epigenetic-sensitive pathways in personalized therapy of major cardiovascular diseases. Pharmacol. Ther. 2020, 210, 107514. [Google Scholar] [CrossRef]
- Etcheverry, A.; Aubry, M.; de Tayrac, M.; Vauleon, E.; Boniface, R.; Guenot, F.; Saikali, S.; Hamlat, A.; Riffaud, L.; Menei, P.; et al. DNA methylation in glioblastoma: Impact on gene expression and clinical outcome. BMC Genom. 2010, 11, 701. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Diaz, M.; Garcia, C.; Sebastiani, G.; de Zegher, F.; Lopez-Bermejo, A.; Ibanez, L. Placental and Cord Blood Methylation of Genes Involved in Energy Homeostasis: Association with Fetal Growth and Neonatal Body Composition. Diabetes 2017, 66, 779–784. [Google Scholar] [CrossRef] [Green Version]
- Arai, E.; Chiku, S.; Mori, T.; Gotoh, M.; Nakagawa, T.; Fujimoto, H.; Kanai, Y. Single-CpG-resolution methylome analysis identifies clinicopathologically aggressive CpG island methylator phenotype clear cell renal cell carcinomas. Carcinogenesis 2012, 33, 1487–1493. [Google Scholar] [CrossRef]
- Hu, T.; Huang, W.; Li, Z.; Kane, M.A.; Zhang, L.; Huang, S.M.; Wang, H. Comparative proteomic analysis of SLC13A5 knockdown reveals elevated ketogenesis and enhanced cellular toxic response to chemotherapeutic agents in HepG2 cells. Toxicol. Appl. Pharmacol. 2020, 402, 115117. [Google Scholar] [CrossRef]
- Ilagan, Y.; Mamillapalli, R.; Goetz, T.G.; Kayani, J.; Taylor, H.S. Bisphenol-A exposure in utero programs a sexually dimorphic estrogenic state of hepatic metabolic gene expression. Reprod. Toxicol. 2017, 71, 84–94. [Google Scholar] [CrossRef] [PubMed]
- Tan, M.; Mosaoa, R.; Graham, G.T.; Kasprzyk-Pawelec, A.; Gadre, S.; Parasido, E.; Catalina-Rodriguez, O.; Foley, P.; Giaccone, G.; Cheema, A.; et al. Inhibition of the mitochondrial citrate carrier, Slc25a1, reverts steatosis, glucose intolerance, and inflammation in preclinical models of NAFLD/NASH. Cell Death Differ. 2020, 27, 2143–2157. [Google Scholar] [CrossRef] [PubMed] [Green Version]





{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
| Chromosome | Position | rsIDs b | Reference | Alternate | Annotation | Allele Frequency (%) |
|---|---|---|---|---|---|---|
| 17 | 6616669 | rs62061545 | T | G | 5_prime_UTR_variant | 2.063 |
| 17 | 6616527 | rs62621831 | G | A | intron_variant | 8.467 |
| 17 | 6610536 | rs62061541 | A | G | intron_variant | 1.730 |
| 17 | 6610295 | rs144316437 | AAG | A | intron_variant | 4.250 |
| 17 | 6607435 | rs218678 | G | A | intron_variant | 9.240 |
| 17 | 6607389 | rs218677 | G | A | intron_variant | 2.729 |
| 17 | 6607150 | rs62061540 | G | A | intron_variant | 2.400 |
| 17 | 6599288 | rs77795940 | C | T | intron_variant | 3.760 |
| 17 | 6596497 | rs896810583 | A | AAC | intron_variant | 8.085 |
| 17 | 6594322 | rs4796540 | G | A | intron_variant | 23.338 |
| 17 | 6594291 | rs116929585 | C | T | intron_variant | 1.502 |
| 17 | 6593417 | rs218694 | A | G | intron_variant | 12.780 |
| 17 | 6593414 | rs934718226 | AC | A | intron_variant | 5.223 |
| 17 | 6591041 | rs78203589 | C | T | intron_variant | 1.335 |
| 17 | 6589495 | rs16956120 | A | G | 3_prime_UTR_variant | 1.073 |
| Species | Tissue | Effect a | Reference | ||
|---|---|---|---|---|---|
| PXR | Human | Human primary hepatocytes | + | [25] | |
| AhR | Rat | Rat primary hepatocytes | + | [24] | |
| Transcription Factor | Mouse | Mouse primary hepatocytes | + | ||
| CREB | Rat | Rat primary hepatocytes | + | [72] | |
| STAT3 | Human | Human primary hepatocytes | + | [26] | |
| Glucagon | Rat | Rat primary hepatocytes | + | [72] | |
| Hormone or Cytokine | IL-6 | Human | Human primary hepatocytes | + | [26] |
| Mouse | Mouse liver | + | |||
| RIF | Human | Human primary hepatocytes | + | [25] | |
| TCDD | Rat | Rat primary hepatocytes | + | [24] | |
| 3MC | Rat | Rat primary hepatocytes | + | [24] | |
| BaP | Rat | Rat primary hepatocytes | + | [24] | |
| Mouse | Mouse primary hepatocytes | + | |||
| Chemicals or Others | Metformin | Human | HepG2 cells | − | [75] |
| AICAR | Human | HepG2 cells | − | [75] | |
| Phenobarbital | Rat | Rat primary hepatocytes | + | [24] | |
| Bisphenol-A | Mouse | Mouse liver | − | [95] | |
| CTPI-2 | Mouse | Mouse liver | + | [96] | |
| LPS | Human | Human non-parenchymal cells (including Kupffer cells) were co-cultivated with human primary hepatocytes | + | [26] | |
| 6-Bnz-cAMP | Rat | Rat primary hepatocytes | + | [72] | |
Publisher’s Note: MDPI stays neutral with regard to jurisdictional claims in published maps and institutional affiliations. |
© 2021 by the authors. Licensee MDPI, Basel, Switzerland. This article is an open access article distributed under the terms and conditions of the Creative Commons Attribution (CC BY) license (https://creativecommons.org/licenses/by/4.0/).
Share and Cite
Li, Z.; Wang, H. Molecular Mechanisms of the SLC13A5 Gene Transcription. Metabolites 2021, 11, 706. https://doi.org/10.3390/metabo11100706
Li Z, Wang H. Molecular Mechanisms of the SLC13A5 Gene Transcription. Metabolites. 2021; 11(10):706. https://doi.org/10.3390/metabo11100706
Chicago/Turabian StyleLi, Zhihui, and Hongbing Wang. 2021. "Molecular Mechanisms of the SLC13A5 Gene Transcription" Metabolites 11, no. 10: 706. https://doi.org/10.3390/metabo11100706
APA StyleLi, Z., & Wang, H. (2021). Molecular Mechanisms of the SLC13A5 Gene Transcription. Metabolites, 11(10), 706. https://doi.org/10.3390/metabo11100706