
Impact of Altered Gut Microbiota and Its Metabolites in Cystic Fibrosis


Abstract
:1. Introduction
2. Normal Gastrointestinal Microbiota
2.1. Normal Gut Microbiota Composition
2.2. Factors Affecting Gut Microbiota
2.3. Mutualistic Functions of Gut Microbiota
3. Gut Dysbiosis in Cystic Fibrosis
3.1. Mechanisms of Gut Dysbiosis in CF
3.2. Patterns of Gut Dysbiosis in CF
3.2.1. Decrease in Microbial Diversity
3.2.2. Alteration of Gut Microbial Composition
3.3. Gut Dysbiosis with Altered Proteomics and Metabolomics in CF
4. Impact of Gut Dysbiosis on Cystic Fibrosis Manifestations
4.1. Impact on the Gastrointestinal Tract
4.1.1. Increase Intestinal Inflammation and Barrier Permeability
4.1.2. Alteration of Fat Metabolism
4.1.3. Gut Dysbiosis and Colon Cancer in CF
4.2. Extraintestinal Implications of Gut Dysbiosis in CF
4.2.1. Gut Dysbiosis and Liver Involvement in CF
4.2.2. Effect of Gut Dysbiosis on Growth Failure and Glucose Metabolism in CF Patients
4.2.3. Gut Dysbiosis and Respiratory Microbiome Interactions
5. Methods to Modulate the Dysbiosis in Cystic Fibrosis
5.1. Probiotics in Modulation of Gut Microbiome in CF
5.2. Prebiotics in Modulation of Gut Microbiome in CF
5.3. Effect of Vitamins and Dietary Nutrients on the Modulation of Gut Microbiome in CF
5.4. Effect of Targeted Molecular Therapies on the Modulation of Gut Microbiome in CF
6. Conclusions and Future Directions
Author Contributions
Funding
Conflicts of Interest
References
- Cutting, G.R. Modifier Genetics: Cystic Fibrosis. Annu. Rev. Genom. Hum. Genet. 2005, 6, 237–260. [Google Scholar] [CrossRef] [PubMed]
- De Lisle, R.C.; Borowitz, D. The Cystic Fibrosis Intestine. Cold Spring Harb. Perspect. Med. 2013, 3, a009753. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Debray, D.; El Mourabit, H.; Merabtene, F.; Brot, L.; Ulveling, D.; Chrétien, Y.; Rainteau, D.; Moszer, I.; Wendum, D.; Sokol, H.; et al. Diet-Induced dysbiosis and genetic background synergize with cystic fibrosis transmembrane conductance regulator deficiency to promote cholangiopathy in mice. Hepatol. Commun. 2018, 2, 1533–1549. [Google Scholar] [CrossRef]
- Liessi, N.; Pedemonte, N.; Armirotti, A.; Braccia, C. Proteomics and Metabolomics for Cystic Fibrosis Research. Int. J. Mol. Sci. 2020, 21, 5439. [Google Scholar] [CrossRef] [PubMed]
- Lee-Sarwar, K.A.; Lasky-Su, J.; Kelly, R.S.; Litonjua, A.A.; Weiss, S.T. Metabolome–Microbiome Crosstalk and Human Disease. Metabolites 2020, 10, 181. [Google Scholar] [CrossRef]
- Zhang, D.; Li, S.; Wang, N.; Tan, H.-Y.; Zhang, Z.; Feng, Y. The Cross-Talk Between Gut Microbiota and Lungs in Common Lung Diseases. Front. Microbiol. 2020, 11, 301. [Google Scholar] [CrossRef]
- Albenberg, L.; Kelsen, J. Advances in Gut Microbiome Research and Relevance to Pediatric Diseases. J. Pediatr. 2016, 178, 16–23. [Google Scholar] [CrossRef] [Green Version]
- Rinninella, E.; Raoul, P.; Cintoni, M.; Franceschi, F.; Miggiano, G.A.D.; Gasbarrini, A.; Mele, M.C. What is the healthy gut microbiota composition? A changing ecosystem across age, environment, diet, and diseases. Microorganisms 2019, 7, 14. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Coffey, M.J.; Nielsen, S.; Wemheuer, B.; Kaakoush, N.O.; Garg, M.; Needham, B.; Pickford, R.; Jaffe, A.; Thomas, T.; Ooi, C.Y. Gut Microbiota in Children with Cystic Fibrosis: A Taxonomic and Functional Dysbiosis. Sci. Rep. 2019, 9, 1–14. [Google Scholar] [CrossRef]
- Sommer, F.; Bäckhed, F. The gut microbiota—Masters of host development and physiology. Nat. Rev. Microbiol. 2013, 11, 227–238. [Google Scholar] [CrossRef] [PubMed]
- Françoise, A.; Héry-Arnaud, G. The Microbiome in Cystic Fibrosis Pulmonary Disease. Genes 2020, 11, 536. [Google Scholar] [CrossRef] [PubMed]
- Wong, A.C.; Levy, M. New Approaches to Microbiome-Based Therapies. mSystems 2019, 4, e00122-19. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Budden, K.F.; Gellatly, S.L.; Wood, D.L.A.; Cooper, M.A.; Morrison, M.; Hugenholtz, P.; Hansbro, P.M. Emerging pathogenic links between microbiota and the gut–lung axis. Nat. Rev. Microbiol. 2017, 15, 55–63. [Google Scholar] [CrossRef]
- Clemente, J.C.; Manasson, J.; Scher, J.U. The role of the gut microbiome in systemic inflammatory disease. BMJ 2018, 360, j5145. [Google Scholar] [CrossRef] [PubMed]
- Staley, C.; Kaiser, T.; Khoruts, A. Clinician Guide to Microbiome Testing. Dig. Dis. Sci. 2018, 63, 3167–3177. [Google Scholar] [CrossRef] [PubMed]
- Sharon, G.; Garg, N.; Debelius, J.; Knight, R.; Dorrestein, P.C.; Mazmanian, S.K. Specialized Metabolites from the Microbiome in Health and Disease. Cell Metab. 2014, 20, 719–730. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Allaband, C.; McDonald, D.; Vázquez-Baeza, Y.; Minich, J.J.; Tripathi, A.; Brenner, D.A.; Loomba, R.; Smarr, L.; Sandborn, W.J.; Schnabl, B.; et al. Microbiome 101: Studying, Analyzing, and Interpreting Gut Microbiome Data for Clinicians. Clin. Gastroenterol. Hepatol. 2019, 17, 218–230. [Google Scholar] [CrossRef]
- Nielsen, S.; Needham, B.; Leach, S.T.; Day, A.S.; Jaffe, A.; Thomas, T.; Ooi, C.Y. Disrupted progression of the intestinal microbiota with age in children with cystic fibrosis. Sci. Rep. 2016, 6, 24857. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Loman, B.R.; Shrestha, C.L.; Thompson, R.; Groner, J.A.; Mejias, A.; Ruoff, K.L.; O’Toole, G.A.; Bailey, M.T.; Kopp, B.T. Age and environmental exposures influence the fecal bacteriome of young children with cystic fibrosis. Pediatr. Pulmonol. 2020, 55, 1661–1670. [Google Scholar] [CrossRef]
- Madan, J.C.; Koestler, D.C.; Stanton, B.A.; Davidson, L.; Moulton, L.A.; Housman, M.L.; Moore, J.H.; Guill, M.F.; Morrison, H.G.; Sogin, M.L.; et al. Serial analysis of the gut and respiratory microbiome in cystic fibrosis in infancy: Interaction between in-testinal and respiratory tracts and impact of nutritional exposures. MBio 2012, 3, e00251-12. [Google Scholar] [CrossRef] [Green Version]
- Yatsunenko, T.; Rey, F.E.; Manary, M.J.; Trehan, I.; Dominguez-Bello, M.G.; Contreras, M.; Magris, M.; Hidalgo, G.; Baldassano, R.N.; Anokhin, A.P.; et al. Human gut microbiome viewed across age and geography. Nature 2012, 486, 222–227. [Google Scholar] [CrossRef]
- David, L.A.; Maurice, C.F.; Carmody, R.N.; Gootenberg, D.B.; Button, J.E.; Wolfe, B.E.; Ling, A.V.; Devlin, A.S.; Varma, Y.; Fischbach, M.A.; et al. Diet rapidly and reproducibly alters the human gut microbiome. Nat. Cell Biol. 2014, 505, 559–563. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Carmody, R.N.; Gerber, G.K.; Luevano, J.M.; Gatti, D.M.; Somes, L.; Svenson, K.L.; Turnbaugh, P.J. Diet Dominates Host Genotype in Shaping the Murine Gut Microbiota. Cell Host Microbe 2015, 17, 72–84. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- De Filippo, C.; Cavalieri, D.; Di Paola, M.; Ramazzotti, M.; Poullet, J.B.; Massart, S.; Collini, S.; Pieraccini, G.; Lionetti, P. Impact of diet in shaping gut microbiota revealed by a comparative study in children from Europe and rural Africa. Proc. Natl. Acad. Sci. USA 2010, 107, 14691–14696. [Google Scholar] [CrossRef] [Green Version]
- Rogers, G.B.; Narkewicz, M.R.; Hoffman, L.R. The CF gastrointestinal microbiome: Structure and clinical impact. Pediatr. Pulmonol. 2016, 51, S35–S44. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Hayden, H.S.; Eng, A.; Pope, C.E.; Brittnacher, M.J.; Vo, A.T.; Weiss, E.J.; Hager, K.R.; Martin, B.D.; Leung, D.H.; Heltshe, S.L.; et al. Fecal dysbiosis in infants with cystic fibrosis is associated with early linear growth failure. Nat. Med. 2020, 26, 215–221. [Google Scholar] [CrossRef]
- Wang, B.; Jiang, X.; Cao, M.; Ge, J.; Bao, Q.; Tang, L.; Chen, Y.; Li, L. Altered Fecal Microbiota Correlates with Liver Biochemistry in Nonobese Patients with Non-alcoholic Fatty Liver Disease. Sci. Rep. 2016, 6, 32002. [Google Scholar] [CrossRef] [PubMed]
- Louis, P.; Young, P.; Holtrop, G.; Flint, H.J. Diversity of human colonic butyrate-producing bacteria revealed by analysis of the butyryl-CoA: Acetate CoA-transferase gene. Environ. Microbiol. 2010, 12, 304–314. [Google Scholar] [CrossRef]
- Venegas, D.P.; de la Fuente, M.K.; Landskron, G.; González, M.J.; Quera, R.; Dijkstra, G.; Harmsen, H.J.M.; Faber, K.N.; Hermoso, M.A. Short chain fatty acids (SCFAs)-mediated gut epithelial and immune regulation and its relevance for inflammatory bowel diseases. Front. Immunol. 2019, 10, 277. [Google Scholar] [CrossRef] [Green Version]
- Manor, O.; Levy, R.; Pope, C.E.; Hayden, H.S.; Brittnacher, M.J.; Carr, R.; Radey, M.C.; Hager, K.R.; Heltshe, S.L.; Ramsey, B.W.; et al. Metagenomic evidence for taxonomic dysbiosis and functional imbalance in the gastrointestinal tracts of children with cystic fibrosis. Sci. Rep. 2016, 6, 22493. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Da Silva, H.E.; Teterina, A.; Comelli, E.M.; Taibi, A.; Arendt, B.M.; Fischer, S.E.; Lou, W.; Allard, J.P. Nonalcoholic fatty liver disease is associated with dysbiosis independent of body mass index and insulin resistance. Sci. Rep. 2018, 8, 1–12. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Imrie, J.R.; Fagan, D.G.; Sturgess, J.M. Quantitative evaluation of the development of the exocrine pancreas in cystic fibrosis and control infants. Am. J. Pathol. 1979, 95, 697–707. [Google Scholar]
- Meeker, S.M.; Mears, K.S.; Sangwan, N.; Brittnacher, M.J.; Weiss, E.J.; Treuting, P.M.; Tolley, N.; Pope, C.E.; Hager, K.R.; Vo, A.T.; et al. CFTR dysregulation drives active selection of the gut microbiome. PLoS Pathog. 2020, 16, e1008251. [Google Scholar] [CrossRef]
- Ooi, C.Y.; Pang, T.; Leach, S.T.; Katz, T.; Day, A.S.; Jaffe, A. Fecal Human β-Defensin 2 in Children with Cystic Fibrosis: Is There a Diminished Intestinal Innate Immune Response? Dig. Dis. Sci. 2015, 60, 2946–2952. [Google Scholar] [CrossRef]
- Gelfond, D.; Ma, C.; Semler, J.; Borowitz, D. Intestinal pH and Gastrointestinal Transit Profiles in Cystic Fibrosis Patients Measured by Wireless Motility Capsule. Dig. Dis. Sci. 2012, 58, 2275–2281. [Google Scholar] [CrossRef] [PubMed]
- Wang, Y.; Leong, L.E.; Keating, R.L.; Kanno, T.; Abell, G.C.; Mobegi, F.M.; Choo, J.M.; Wesselingh, S.L.; Mason, A.J.; Burr, L.D.; et al. Opportunistic bacteria confer the ability to ferment prebiotic starch in the adult cystic fibrosis gut. Gut Microbes 2019, 10, 367–381. [Google Scholar] [CrossRef] [Green Version]
- Kristensen, M.; Prevaes, S.M.; Kalkman, G.; Tramper-Stranders, G.A.; Hasrat, R.; Groot, K.M.D.W.-D.; Janssens, H.M.; Tiddens, H.A.; Van Westreenen, M.; Sanders, E.A.; et al. Development of the gut microbiota in early life: The impact of cystic fibrosis and antibiotic treatment. J. Cyst. Fibros. 2020, 19, 553–561. [Google Scholar] [CrossRef]
- Debyser, G.; Mesuere, B.; Clement, L.; Van De Weygaert, J.; Van Hecke, P.; Duytschaever, G.; Aerts, M.; Dawyndt, P.; De Boeck, K.; Vandamme, P.; et al. Faecal proteomics: A tool to investigate dysbiosis and inflammation in patients with cystic fibrosis. J. Cyst. Fibros. 2016, 15, 242–250. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Vernocchi, P.; del Chierico, F.; Russo, A.; Majo, F.; Rossitto, M.; Valerio, M.; Casadei, L.; la Storia, A.; de Filippis, F.; Rizzo, C.; et al. Gut microbiota signatures in cystic fibrosis: Loss of host CFTR function drives the microbiota entero-phenotype. PLoS ONE 2018, 13, e0208171. [Google Scholar] [CrossRef]
- Hoffman, L.R.; Pope, C.E.; Hayden, H.S.; Heltshe, S.; Levy, R.; McNamara, S.; Jacobs, M.A.; Rohmer, L.; Radey, M.; Ramsey, B.W.; et al. Escherichia coli dysbiosis correlates with gastrointestinal dysfunction in children with cystic fibrosis. Clin. Infect. Dis. 2013, 58, 396–399. [Google Scholar] [CrossRef] [Green Version]
- Garg, M.; Ooi, C.Y. The Enigmatic Gut in Cystic Fibrosis: Linking Inflammation, Dysbiosis, and the Increased Risk of Malignancy. Curr. Gastroenterol. Rep. 2017, 19, 6. [Google Scholar] [CrossRef]
- Héry-Arnaud, G.; Boutin, S.; Cuthbertson, L.; Elborn, S.J.; Tunney, M.M. The lung and gut microbiome: What has to be taken into consideration for cystic fibrosis? J. Cyst. Fibros. 2019, 18, 13–21. [Google Scholar] [CrossRef]
- Van Biervliet, S.; Declercq, D.; Somerset, S. Clinical effects of probiotics in cystic fibrosis patients: A systematic review. Clin. Nutr. ESPEN 2017, 18, 37–43. [Google Scholar] [CrossRef] [PubMed]
- Schippa, S.; Iebba, V.; Santangelo, F.; Gagliardi, A.; de Biase, R.V.; Stamato, A.; Bertasi, S.; Lucarelli, M.; Conte, M.P.; Quattrucci, S. Cystic fibrosis transmembrane conductance regulator (CFTR) allelic variants relate to shifts in faecal mi-crobiota of cystic fibrosis patients. PLoS ONE 2013, 8, e61176. [Google Scholar] [CrossRef]
- Burke, D.; Fouhy, F.; Harrison, M.J.; Rea, M.C.; Cotter, P.D.; O’Sullivan, O.; Stanton, C.; Hill, C.; Shanahan, F.; Plant, B.J.; et al. The altered gut microbiota in adults with cystic fibrosis. BMC Microbiol. 2017, 17, 1–11. [Google Scholar] [CrossRef] [Green Version]
- Frost, F.; Kacprowski, T.; Rühlemann, M.; Bülow, R.; Kühn, J.; Franke, A.; Heinsen, F.; Pietzner, M.; Nauck, M.; Völker, U.; et al. Impaired exocrine pancreatic function associates with changes in intestinal microbiota composition and di-versity. Gastroenterology 2019, 156, 1010–1015. [Google Scholar] [CrossRef] [PubMed]
- Anitha, M.; Reichardt, F.; Tabatabavakili, S.; Nezami, B.G.; Chassaing, B.; Mwangi, S.; Vijay-Kumar, M.; Gewirtz, A.; Srinivasan, S. Intestinal Dysbiosis Contributes to the Delayed Gastrointestinal Transit in High-Fat Diet Fed Mice. Cell. Mol. Gastroenterol. Hepatol. 2016, 2, 328–339. [Google Scholar] [CrossRef] [Green Version]
- Matamouros, S.; Hayden, H.S.; Hager, K.R.; Brittnacher, M.J.; Lachance, K.; Weiss, E.J.; Pope, C.E.; Imhaus, A.-F.; McNally, C.P.; Borenstein, E.; et al. Adaptation of commensal proliferating Escherichia coli to the intestinal tract of young children with cystic fibrosis. Proc. Natl. Acad. Sci. USA 2018, 115, 1605–1610. [Google Scholar] [CrossRef] [Green Version]
- Li, L.; Somerset, S. The clinical significance of the gut microbiota in cystic fibrosis and the potential for dietary therapies. Clin. Nutr. 2014, 33, 571–580. [Google Scholar] [CrossRef]
- Duytschaever, G.; Huys, G.; Bekaert, M.; Boulanger, L.; De Boeck, K.; Vandamme, P. Dysbiosis of bifidobacteria and Clostridium cluster XIVa in the cystic fibrosis fecal microbiota. J. Cyst. Fibros. 2013, 12, 206–215. [Google Scholar] [CrossRef] [Green Version]
- Dayama, G.; Priya, S.; Niccum, D.E.; Khoruts, A.; Blekhman, R. Interactions between the gut microbiome and host gene regulation in cystic fibrosis. Genome Med. 2020, 12, 1–15. [Google Scholar] [CrossRef] [Green Version]
- Lee, A.J.; Einarsson, G.G.; Gilpin, D.F.; Tunney, M.M. Multi-Omics Approaches: The Key to Improving Respiratory Health in People with Cystic Fibrosis? Front. Pharmacol. 2020, 11, 1382. [Google Scholar] [CrossRef]
- Hoen, A.G.; Li, J.; Moulton, L.A.; O’Toole, G.A.; Housman, M.L.; Koestler, D.C.; Guill, M.F.; Moore, J.H.; Hibberd, P.L.; Morrison, H.G.; et al. Associations between Gut Microbial Colonization in Early Life and Respiratory Outcomes in Cystic Fibrosis. J. Pediatr. 2015, 167, 138–147.e3. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Miragoli, F.; Federici, S.; Ferrari, S.; Minuti, A.; Rebecchi, A.; Bruzzese, E.; Buccigrossi, V.; Guarino, A.; Callegari, M.L. Impact of cystic fibrosis disease on archaea and bacteria composition of gut microbiota. FEMS Microbiol. Ecol. 2016, 93, 230. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Antosca, K.M.; Chernikova, D.A.; Price, C.E.; Ruoff, K.L.; Li, K.; Guill, M.F.; Sontag, N.R.; Morrison, H.G.; Hao, S.; Drumm, M.L.; et al. Altered Stool Microbiota of Infants with Cystic Fibrosis Shows a Reduction in Genera Associated with Immune Programming from Birth. J. Bacteriol. 2019, 201, 00274-19. [Google Scholar] [CrossRef] [Green Version]
- Del Campo, R.; Garriga, M.; Pérez-Aragón, A.; Guallarte, P.; Lamas, A.; Máiz, L.; Bayón, C.; Roy, G.; Cantón, R.; Zamora, J.; et al. Improvement of digestive health and reduction in proteobacterial populations in the gut microbiota of cystic fibrosis patients using a Lactobacillus reuteri probiotic preparation: A double blind prospective study. J. Cyst. Fibros. 2014, 13, 716–722. [Google Scholar] [CrossRef] [Green Version]
- Duytschaever, G.; Huys, G.; Bekaert, M.; Boulanger, L.; De Boeck, K.; Vandamme, P. Cross-Sectional and Longitudinal Comparisons of the Predominant Fecal Microbiota Compositions of a Group of Pediatric Patients with Cystic Fibrosis and Their Healthy Siblings. Appl. Environ. Microbiol. 2011, 77, 8015–8024. [Google Scholar] [CrossRef] [Green Version]
- Scanlan, P.D.; Buckling, A.; Kong, W.; Wild, Y.; Lynch, S.V.; Harrison, F. Gut dysbiosis in cystic fibrosis. J. Cyst. Fibros. 2012, 11, 454–455. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Enaud, R.; Hooks, K.B.; Barre, A.; Barnetche, T.; Hubert, C.; Massot, M.; Bazin, T.; Clouzeau, H.; Bui, S.; Fayon, M.; et al. Intestinal inflammation in children with cystic fibrosis is associated with Crohn’s-like microbiota disturb-ances. J. Clin. Med. 2019, 8, 645. [Google Scholar] [CrossRef] [Green Version]
- Kaakoush, N.O.; Pickford, R.; Jaffe, A.; Ooi, C.Y. Is there a role for stool metabolomics in cystic fibrosis? Pediatr. Int. 2016, 58, 808–811. [Google Scholar] [CrossRef] [PubMed]
- Bruzzese, E.; Callegari, M.L.; Raia, V.; Viscovo, S.; Scotto, R.; Ferrari, S.; Morelli, L.; Buccigrossi, V.; Vecchio, A.L.; Ruberto, E.; et al. Disrupted intestinal microbiota and intestinal inflammation in children with cystic fibrosis and its res-toration with Lactobacillus GG: A randomised clinical trial. PLoS ONE 2014, 9, e87796. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- de Freitas, M.B.; Moreira, E.A.M.; Tomio, C.; Moreno, Y.M.F.; Daltoe, F.P.; Barbosa, E.; Neto, N.L.; Buccigrossi, V.; Guarino, A. Altered intestinal microbiota composition, antibiotic therapy and intestinal inflammation in children and adolescents with cystic fibrosis. PLoS ONE 2018, 13, e0198457. [Google Scholar] [CrossRef] [PubMed]
- Van Biervliet, S.; Eggermont, E.; Carchon, H.; Veereman, G.; DeBoeck, K. Small intestinal brush border enzymes in cystic fibrosis. Acta Gastro-Enterol. Belg. 1999, 62, 267–271. [Google Scholar]
- Van Biervliet, S.; Hauser, B.; Verhulst, S.; Stepman, H.; Delanghe, J.; Warzee, J.; Pot, B.; Vandewiele, T.; Wilschanski, M. Probiotics in cystic fibrosis patients: A double blind crossover placebo controlled study: Pilot study from the ESPGHAN Working Group on Pancreas/CF. Clin. Nutr. ESPEN 2018, 27, 59–65. [Google Scholar] [CrossRef] [PubMed]
- Garg, M.; Leach, S.T.; Coffey, M.J.; Katz, T.; Strachan, R.; Pang, T.; Needham, B.; Lui, K.; Ali, F.; Day, A.S.; et al. Age-dependent variation of fecal calprotectin in cystic fibrosis and healthy children. J. Cyst. Fibros. 2017, 16, 631–636. [Google Scholar] [CrossRef] [Green Version]
- Garg, M.; Leach, S.T.; Pang, T.; Needham, B.; Coffey, M.J.; Katz, T.; Strachan, R.; Widger, J.; Field, P.; Belessis, Y.; et al. Age-related levels of fecal M2-pyruvate kinase in children with cystic fibrosis and healthy children 0 to 10 years old. J. Cyst. Fibros. 2018, 17, 109–113. [Google Scholar] [CrossRef] [Green Version]
- Werlin, S.L.; Benuri-Silbiger, I.; Kerem, E.; Adler, S.N.; Goldin, E.; Zimmerman, J.; Malka, N.; Cohen, L.; Armoni, S.; Yatzkan-Israelit, Y.; et al. Evidence of Intestinal Inflammation in Patients with Cystic Fibrosis. J. Pediatr. Gastroenterol. Nutr. 2010, 51, 304–308. [Google Scholar] [CrossRef] [PubMed]
- Dhaliwal, J.; Leach, S.; Katz, T.; Nahidi, L.; Pang, T.; Lee, J.; Strachan, R.; Day, A.S.; Jaffe, A.; Ooi, C.Y. Intestinal Inflammation and Impact on Growth in Children with Cystic Fibrosis. J. Pediatr. Gastroenterol. Nutr. 2015, 60, 521–526. [Google Scholar] [CrossRef] [Green Version]
- Lee, J.M.; Leach, S.T.; Katz, T.; Day, A.S.; Jaffe, A.; Ooi, C.Y. Update of Faecal Markers of Inflammation in Children with Cystic Fibrosis. Mediat. Inflamm. 2012, 2012, 1–6. [Google Scholar] [CrossRef]
- De Lisle, R.C.; Mueller, R.; Boyd, M. Impaired Mucosal Barrier Function in the Small Intestine of the Cystic Fibrosis Mouse. J. Pediatr. Gastroenterol. Nutr. 2011, 53, 371–379. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Hallberg, K.; Grzegorczyk, A.; Larson, G.; Strandvik, B. Intestinal Permeability in Cystic Fibrosis in Relation to Genotype. J. Pediatr. Gastroenterol. Nutr. 1997, 25, 290–295. [Google Scholar] [CrossRef]
- Peretti, N.; Marcil, V.; Drouin, E.; Levy, E. Mechanisms of lipid malabsorption in Cystic Fibrosis: The impact of essential fatty acids deficiency. Nutr. Metab. 2005, 2, 11. [Google Scholar] [CrossRef] [Green Version]
- De Lisle, R.C. Disrupted tight junctions in the small intestine of cystic fibrosis mice. Cell Tissue Res. 2014, 355, 131–142. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Van Biervliet, S.; Eggermont, E.; Mariën, P.; Hoffman, I.; Veereman, G. Combined impact of mucosal damage and of cystic fibrosis on the small intestinal brush border enzyme activities. Acta Clin. Belg. 2003, 58, 220–224. [Google Scholar] [CrossRef]
- Schubert, A.M.; Sinani, H.; Schloss, P.D. Antibiotic-Induced Alterations of the Murine Gut Microbiota and Subsequent Effects on Colonization Resistance against Clostridium difficile. mBio 2015, 6, e00974-15. [Google Scholar] [CrossRef] [Green Version]
- Fouhy, F.; Ronan, N.J.; O’Sullivan, O.; McCarthy, Y.; Walsh, A.M.; Murphy, D.M.; Daly, M.; Flanagan, E.T.; Fleming, C.; McCarthy, M.; et al. A pilot study demonstrating the altered gut microbiota functionality in stable adults with Cystic Fibrosis. Sci. Rep. 2017, 7, 6685. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Scott, P.; Anderson, K.; Singhania, M.; Cormier, R. Cystic Fibrosis, CFTR, and Colorectal Cancer. Int. J. Mol. Sci. 2020, 21, 2891. [Google Scholar] [CrossRef] [Green Version]
- Yamada, A.; Komaki, Y.; Komaki, F.; Micic, D.; Zullow, S.; Sakuraba, A. Risk of gastrointestinal cancers in patients with cystic fibrosis: A systematic review and meta-analysis. Lancet Oncol. 2018, 19, 758–767. [Google Scholar] [CrossRef]
- Hussan, H.; Clinton, S.K.; Roberts, K.; Bailey, M.T. Fusobacterium’s link to colorectal neoplasia sequenced: A systematic review and future insights. World J. Gastroenterol. 2017, 23, 8626–8650. [Google Scholar] [CrossRef]
- Leung, D.H.; Yimlamai, D. The intestinal microbiome and paediatric liver disease. Lancet Gastroenterol. Hepatol. 2017, 2, 446–455. [Google Scholar] [CrossRef]
- Wu, H.; Vu, M.; Dhingra, S.; Ackah, R.; Goss, J.A.; Rana, A.; Quintanilla, N.; Patel, K.; Leung, D.H. Obliterative Portal Venopathy Without Cirrhosis Is Prevalent in Pediatric Cystic Fibrosis Liver Disease with Portal Hypertension. Clin. Gastroenterol. Hepatol. 2019, 17, 2134–2136. [Google Scholar] [CrossRef]
- Flass, T.; Tong, S.; Frank, D.N.; Wagner, B.D.; Robertson, C.E.; Kotter, C.V.; Sokol, R.J.; Zemanick, E.; Accurso, F.; Hoffenberg, E.J.; et al. Intestinal Lesions Are Associated with Altered Intestinal Microbiome and Are More Frequent in Children and Young Adults with Cystic Fibrosis and Cirrhosis. PLoS ONE 2015, 10, e0116967. [Google Scholar] [CrossRef] [Green Version]
- Fiorotto, R.; Scirpo, R.; Trauner, M.; Fabris, L.; Hoque, R.; Spirli, C.; Strazzabosco, M. Loss of CFTR affects biliary epithelium innate immunity and causes TLR4–NF-κB—mediated inflammatory response in mice. Gastroenterology 2011, 141, 1498–1508.e5. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Rath, T.; Hage, L.; Kügler, M.; Menendez, K.M.; Zachoval, R.; Naehrlich, L.; Schulz, R.; Roderfeld, M.; Roeb, E. Serum Proteome Profiling Identifies Novel and Powerful Markers of Cystic Fibrosis Liver Disease. PLoS ONE 2013, 8, e58955. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Vallianou, N.G.; Stratigou, T.; Tsagarakis, S. Microbiome and diabetes: Where are we now? Diabetes Res. Clin. Pr. 2018, 146, 111–118. [Google Scholar] [CrossRef] [PubMed]
- Charlson, E.S.; Bittinger, K.; Haas, A.R.; Fitzgerald, A.S.; Frank, I.; Yadav, A.; Bushman, F.D.; Collman, R.G. Topographical Continuity of Bacterial Populations in the Healthy Human Respiratory Tract. Am. J. Respir. Crit. Care Med. 2011, 184, 957–963. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Boutin, S.; Graeber, S.Y.; Weitnauer, M.; Panitz, J.; Stahl, M.; Clausznitzer, D.; Kaderali, L.; Einarsson, G.; Tunney, M.M.; Elborn, J.S.; et al. Comparison of Microbiomes from Different Niches of Upper and Lower Airways in Children and Adolescents with Cystic Fibrosis. PLoS ONE 2015, 10, e0116029. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Prevaes, S.M.; Piters, W.A.D.S.; Groot, K.M.D.W.-D.; Janssens, H.M.; Tramper-Stranders, G.A.; Chu, M.L.J.; Tiddens, H.A.; Van Westreenen, M.; Van Der Ent, C.K.; Sanders, E.A.; et al. Concordance between upper and lower airway microbiota in infants with cystic fibrosis. Eur. Respir. J. 2017, 49, 1602235. [Google Scholar] [CrossRef] [Green Version]
- Madan, J.C. Neonatal Gastrointestinal and Respiratory Microbiome in Cystic Fibrosis: Potential Interactions and Implications for Systemic Health. Clin. Ther. 2016, 38, 740–746. [Google Scholar] [CrossRef] [Green Version]
- Marsland, B.J.; Gollwitzer, E.S. Host–microorganism interactions in lung diseases. Nat. Rev. Immunol. 2014, 14, 827–835. [Google Scholar] [CrossRef]
- Bazett, M.; Bergeron, M.-E.; Haston, C.K. Streptomycin treatment alters the intestinal microbiome, pulmonary T cell profile and airway hyperresponsiveness in a cystic fibrosis mouse model. Sci. Rep. 2016, 6, srep19189. [Google Scholar] [CrossRef] [Green Version]
- Huang, Y.J.; LiPuma, J.J. The Microbiome in Cystic Fibrosis. Clin. Chest Med. 2016, 37, 59–67. [Google Scholar] [CrossRef]
- Bilski, J.; Mazur-Bialy, A.; Wojcik, D.; Zahradnik-Bilska, J.; Brzozowski, B.; Magierowski, M.; Mach, T.; Magierowska, K.; Brzozowski, T. The Role of Intestinal Alkaline Phosphatase in Inflammatory Disorders of Gastrointestinal Tract. Mediat. Inflamm. 2017, 2017, 1–9. [Google Scholar] [CrossRef] [PubMed]
- Li, L.; Somerset, S. Associations between Flavonoid Intakes and Gut Microbiota in a Group of Adults with Cystic Fibrosis. Nutrients 2018, 10, 1264. [Google Scholar] [CrossRef] [Green Version]
- Di Nardo, G.; Oliva, S.; Menichella, A.; Pistelli, R.; De Biase, R.V.; Patriarchi, F.; Cucchiara, S.; Stronati, L. Lactobacillus reuteri ATCC55730 in Cystic Fibrosis. J. Pediatr. Gastroenterol. Nutr. 2014, 58, 81–86. [Google Scholar] [CrossRef]
- Bruzzese, E.; Raia, V.; Spagnuolo, M.I.; Volpicelli, M.; De Marco, G.; Maiuri, L.; Guarino, A. Effect of Lactobacillus GG supplementation on pulmonary exacerbations in patients with cystic fibrosis: A pilot study. Clin. Nutr. 2007, 26, 322–328. [Google Scholar] [CrossRef] [PubMed]
- Jafari, S.-A.; Mehdizadeh-Hakkak, A.; Kianifar, H.-R.; Hebrani, P.; Ahanchian, H.; Abbasnejad, E. Effects of Probiotics on Quality of Life in Children with Cystic Fibrosis; A Randomized Controlled Trial. Iran. J. Pediatr. 2013, 23, 669–674. [Google Scholar]
- Fallahi, G.; Motamed, F.; Yousefi, A.; Shafieyoun, A.; Najafi, M.; Khodadad, A.; Farhmand, F.; Ahmadvand, A.; Rezaei, N. The effect of probiotics on fecal calprotectin in patients with cystic fibrosis. Turk. J. Pediatr. 2014, 55, 475–478. [Google Scholar]
- Nikniaz, Z.; Nikniaz, L.; Bilan, N.; Somi, M.H.; Faramarzi, E. Does probiotic supplementation affect pulmonary exacerbation and intestinal inflammation in cystic fibrosis: A systematic review of randomized clinical trials. World J. Pediatr. 2017, 13, 307–313. [Google Scholar] [CrossRef] [PubMed]
- Fukuda, S.; Toh, H.; Hase, K.; Oshima, K.; Nakanishi, Y.; Yoshimura, K.; Tobe, T.; Clarke, J.M.; Topping, D.L.; Suzuki, T.; et al. Bifidobacteria can protect from enteropathogenic infection through production of acetate. Nature 2011, 469, 543–547. [Google Scholar] [CrossRef]
- Li, L.; Krause, L.; Somerset, S. Associations between micronutrient intakes and gut microbiota in a group of adults with cystic fibrosis. Clin. Nutr. 2017, 36, 1097–1104. [Google Scholar] [CrossRef] [PubMed]
- Alvarez, J.A.; Chong, E.Y.; Walker, D.I.; Chandler, J.D.; Michalski, E.S.; Grossmann, R.E.; Uppal, K.; Li, S.; Frediani, J.K.; Tirouvanziam, R.; et al. Plasma metabolomics in adults with cystic fibrosis during a pulmonary exacerbation: A pilot randomized study of high-dose vitamin D3 administration. Metabolism 2017, 70, 31–41. [Google Scholar] [CrossRef] [Green Version]
- Portal, C.; Gouyer, V.; Léonard, R.; Husson, M.-O.; Gottrand, F.; Desseyn, J.-L. Long-term dietary (n-3) polyunsaturated fatty acids show benefits to the lungs of Cftr F508del mice. PLoS ONE 2018, 13, e0197808. [Google Scholar] [CrossRef]
- Argo, K.B. Gut Microbiome Diversity and Community Structure Following Dietary Genistein Treatment in a Murine Model of Cystic Fibrosis. Master’s Thesis, Arizona State University, Tempe, AZ, USA, 2019. [Google Scholar]
- Ooi, C.Y.; Syed, S.A.; Rossi, L.; Garg, M.; Needham, B.; Avolio, J.; Young, K.; Surette, M.G.; Gonska, T. Impact of CFTR modulation with Ivacaftor on Gut Microbiota and Intestinal Inflammation. Sci. Rep. 2018, 8, 1–8. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Kopp, B.T.; McCulloch, S.; Shrestha, C.L.; Zhang, S.; Sarzynski, L.; Woodley, F.W.; Hayes, D. Metabolomic responses to lumacaftor/ivacaftor in cystic fibrosis. Pediatr. Pulmonol. 2018, 53, 583–591. [Google Scholar] [CrossRef]
- Kashyap, P.C.; Chia, N.; Nelson, H.; Segal, E.; Elinav, E. Microbiome at the frontier of personalized medicine. In Mayo Clinic Proceedings; Elsevier: Amsterdam, The Netherlands, 2017. [Google Scholar]
- Rezasoltani, S.; Bashirzadeh, D.A.; Mojarad, E.N.; Aghdaei, H.A.; Norouzinia, M.; Shahrokh, S. Signature of gut microbiome by conventional and advanced analysis techniques: Advantages and dis-advantages. Middle East J. Dig. Dis. 2020, 12, 5. [Google Scholar] [CrossRef] [Green Version]
- Virkud, Y.V.; Kelly, R.S.; Wood, C.; Lasky-Su, J.A. The nuts and bolts of omics for the clinical allergist. Ann. Allergy Asthma Immunol. 2019, 123, 558–563. [Google Scholar] [CrossRef] [Green Version]
- Galloway-Peña, J.; Hanson, B. Tools for Analysis of the Microbiome. Dig. Dis. Sci. 2020, 65, 674–685. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Güemes, A.G.C.; Lim, Y.W.; Quinn, R.A.; Conrad, D.J.; Benler, S.; Maughan, H.; Edwards, R.; Brettin, T.; Cantú, V.A.; Cuevas, D.; et al. Cystic Fibrosis Rapid Response: Translating Multi-omics Data into Clinically Relevant Information. mBio 2019, 10, e00431-19. [Google Scholar] [CrossRef] [Green Version]
- Coffey, M.J.; Low, I.; Stelzer-Braid, S.; Wemheuer, B.; Garg, M.; Thomas, T.; Jaffe, A.; Rawlinson, W.D.; Ooi, C.Y. The intestinal virome in children with cystic fibrosis differs from healthy controls. PLoS ONE 2020, 15, e0233557. [Google Scholar] [CrossRef] [PubMed]




{kind=link}
{kind=link}
{kind=link}
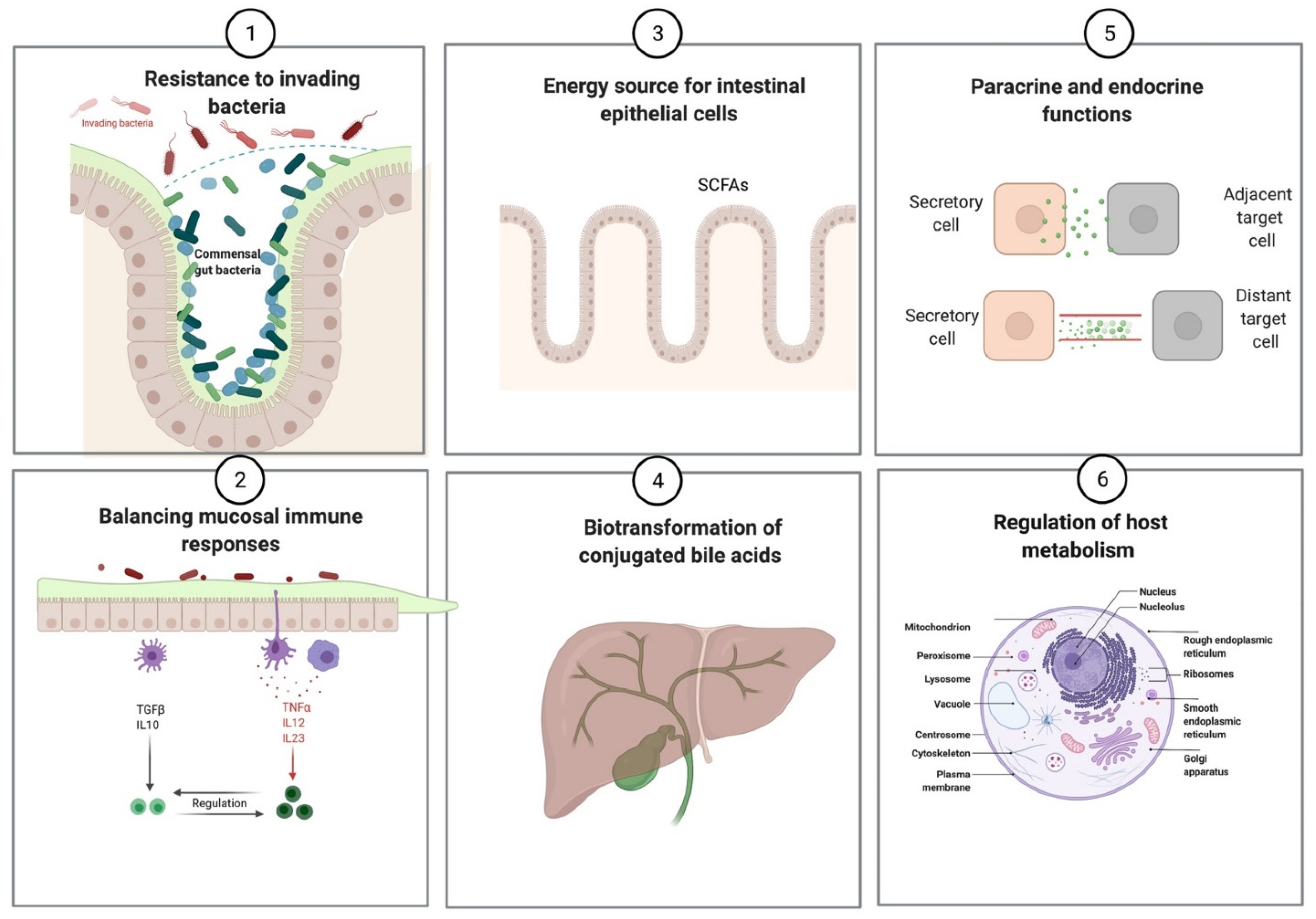
| Prevention of pathogenic infections |
| Synthesis of vitamins (vitamin K and vitamin B complex) |
Production of short-chain fatty acids
|
| Regulation of mucosal immune responses |
| Influence host metabolism and behavior via various endocrine and paracrine functions |
| Biotransformation of conjugated bile acids |
| Xenobiotic metabolism |
| CFTR-related mechanisms |
| Thick and inspissated mucus due to chloride channel dysfunction Defective bicarbonate secretion altering the intestinal milieu pH Malabsorption due to pancreatic insufficiency Intestinal dysmotility with prolonged intestinal transit Altered mucosal immune mechanisms Enhanced intestinal inflammation Epithelial barrier disruption |
| Acquired factors |
| Frequent use of antibiotics for recurrent pulmonary infections High-fat, high-calorie diet Other medications used in cystic fibrosis patients such as acid-suppressive medications, opioids, anticholinergic agents, and immunosuppressive mediations, etc. |
| Study Population | Key Findings in CF | Conclusions | References |
|---|---|---|---|
| 7 infants with CF enrolled at birth | High degree of concordance between gut and respiratory microbial samples. For seven genera, gut colonization predicted their appearance in the lungs | Nutritional factors and gut colonization patterns could determine respiratory microbiome in CF | Madan et al., 2012 [20] |
| 21 family units (one patient with CF and one to two healthy siblings) | ↓ Abundance and temporal stability of Bifidobacteria and Clostridium cluster XIVa | Dysbiosis in CF could be due to disease-related impairment of essential gastrointestinal tract functions or a side effect of antibiotic usage | Duytschaever et al., 2013 [50] |
| 12 patients with CF and 12 HC aged several weeks to 5 years | ↑ E. coli correlated with nutrient malabsorption and intestinal inflammation | E. coli contributed to CF-related gastrointestinal dysfunction | Hoffman et al., 2014 [40] |
| 13 patients with CF aged 0–34 months | Specific clustering of bacteria in fecal samples, but not respiratory samples, were associated with pulmonary exacerbations | Specific bacterial communities colonized the gut before the lungs in CF patients | Hoen et al., 2015 [53] |
| 14 patients with CF and 12 HC aged < 3 years | ↑ E. coli, E. faecalis, Veillonella, C. difficile ↓ Beneficial Clostridiales Dysbiosis significantly altered lipid metabolism (↓ FFA biosynthesis and ↑ anti-inflammatory SCFAs degradation) | Taxonomic and functional microbial shifts in young children with CF decreased with age Gut dysbiosis in CF correlated with fat malabsorption & inflammation | Manor et al., 2016 [30] |
| 23 HC and 35 patients with CF (age range 0–18 years) | Progressive ↓ and alteration in richness and diversity of gut bacteria that was associated with CF from early childhood until late adolescence independent of pancreatic function | ↑ Deviation in the number and diversity of intestinal microbiome with age in CF Efforts to rectify loss of bacterial diversity should be conducted no later than early childhood | Nielsen et al., 2016 [18] |
| 43 patients with CF aged 21–38 years and 69 HC aged 24–40 years | ↓ Microbial diversity ↑ Firmicutes ↓ Bacteroidetes | Gut dysbiosis in CF positively correlated with lung dysfunction and intravenous antibiotic use | Burke et al., 2017 [45] |
| 30 patients with CF (14 were homozygous for delF508 and 14 were heterozygous, and 2 had mild genotype) age range 10–22 years and 8 HC (mean age 14.3 years) | ↓ Clostridium coccoides ↓ Bacteroides-Proveotella ↓ Bifidobacterium genera ↓ Key butyrate producers | Low frequency of sulfate reducing bacteria in CF Significant reduction in hydrogen-consuming microbes in CF | Miragoli et al., 2017 [54] |
| 31 patients with CF between 1–6 years and age-matched 1:1 HC | ↑ Propionibacterium, Staphylococcus, C. difficile ↓ Eggerthella, Eubacterium, Ruminococcus, F. prausnitzii, Lachnospiraceae ↑ GABA, choline, propylbutyrate, and pyridine↓ Sarcosine, methylphenol, uracil, glucose, acetate, phenol, and benzaldehyde | CF gut microbiota revealed an enterophenotype that was correlated with disease status regardless of age and pancreatic status. This distinct dysbiosis was partially related to pulmonary infections and oral antibiotic use | Vernocchi et al., 2018 [39] |
| 27 patients with CF and age/gender matched HC (age range 0.8–18 years) | Prominent taxonomic and functional dysbiosis in CF compared to HC ↓ richness and diversity of gut microbiota in CF | Enrichment of genes involved in SCFAs, antioxidant and nutrient metabolisms in CF | Coffey et al., 2019 [9] |
| 21 patients with CF and 409 healthy infant controls | Unlike the healthy infants, the alpha diversity did not increase in CF infants ↓ Bacteroides ↓ Roseburia ↑ Veillonella | The distinct CF gut microbiota in infants was associated with pulmonary exacerbations. In vitro models suggested the role of Bacteroides in reduction of IL-8 linking the gut dysbiosis in CF-related inflammation | Antosca et al., 2019 [55] |
| 20 patients with CF and 45 HC, fecal samples collected over the first 18 months of life | ↓ Akkermansia, Bifidobacterium, Bacteroides and Anaerostipes ↑ Streptococci, Enterococcus and E. coli ↓ Alpha diversity | Antibiotic use in infants with CF was associated with a lower alpha diversity and altered microbial composition | Kristensen et al., 2020 [37] |
| 207 infants with CF and 25 HC | ↓ Bacteroidetes ↑ Proteobacteria | CF infants with low length had pronounced dysbiosis than HC and CF infants with normal length | Hayden et al, 2020 [26] |
| Increase | Decrease | |
|---|---|---|
| Phylum level | Firmicutes/Bacteroidetes ratio γ-Proteobacteria | Bacteroidetes Firmicutes Actinobacteria |
| Genus/Species level | Pro-inflammatory microbiota Enterobacteriaceae Streptococcus Veillonella Staphylococcus Propionibacterium (promoted by low pH and anaerobic milieu) Colorectal cancer-related microbiota Fusobacterium Veillonella Escherichia coli (growth–promotional transcription profile) Antibiotics-resistant species Enterococcus faecalis Enterococcus faecium Pro-inflammatory species Veillonella dispar Clostridiales difficile | Beneficial microbiota Bifidobacterium Clostridium Akkermansia Eggerthella Immune modulatory microbiota Bacteroides species Butyrate producers Anaerostipes Butyricicoccus Ruminococcus Faecalibacterium prausnitzii Eubacterium rectale Blautia species Anti-inflammatory species Bifidobacterium adolescentis |
| Metabolite level | Gamma aminobutyric acid Choline Ethanol Propylbutyrate Pyridine | Butyric acid Pantetheine Sarcosine Methylphenol Uracil Glucose Acetate Phenol Benzaldehyde Methylacetate |
| Specific antibiotic therapy |
| Dietary interventions (modification of macronutrient composition) |
| Probiotics |
| Prebiotics (indigestible carbohydrates) |
| Probiotics and prebiotics (synbiotics) |
| Vitamins and supplements |
| Fecal microbiome transplantation |
| Targeted molecular therapies (e.g., ivacaftor) |
| Metabolite-based therapies (postbiotics) |
| Study Population | Probiotic Strains | Clinical Responses | Proposed Mechanisms | References |
|---|---|---|---|---|
| 19 patients | LGG* | ↓ Pulmonary exacerbations and hospital admissions | ↓ DCs* maturation resulting in induction of Treg*-cells | Bruzzese et al., 2007 [96] |
| 37 patients (20 received probiotics and 17 took placebo capsules) | L. casei*, L. rhamnosus*, S. thermophiles*, B. breve*, L. acidophilus*, B. infantis*, L. bulgaricus* | ↓ Pulmonary exacerbations and improving quality of life | Preventing deleterious effects of inflammatory cytokines (TNF-α * IFN-γ*) on epithelial function leading to a less-disrupted intestinal barrier | Jafari et al., 2013 [97] |
| 61 Patients with mild to moderate pulmonary disease | L. reuteri * ATCC55730 | ↓ Pulmonary exacerbations and URTI* | Improvement of intestinal barrier function and modulation of immune response | Di Nardo et al., 2014 [95] |
| 22 patients aged 2–9 years | LGG* | ↓ Fecal calprotectin (↓ intestinal inflammation) | Partial restoration of healthy intestinal microbiota that limit intestinal inflammation | Bruzzese et al., 2014 [61] |
| 30 patients in two groups (probiotic and placebo group) | L. reuteri* | ↓ Fecal calprotectin (↓ intestinal inflammation) and ↑ digestive comfort | ↑ Microbial diversity with ↑ representation of Firmicutes. ↓ γ-Proteobacteria genera Enterobacteriaceae | Del Campo et al., 2014 [56] |
| 25 patients aged 7–12 years (crossover study) | L. rhamnosus* SP1 (DSM 21690) & B. animalis spp.BLC1 (LGM23512) | Normalization of gut permeability in 13% of patients. No change in BMI*, FEV1*%, abdominal pain, and pulmonary exacerbations | Probiotic supplementation did not change the microbiota (both at phylum or phylogenetic levels) | Van Biervliet, 2018 [64] |
Publisher’s Note: MDPI stays neutral with regard to jurisdictional claims in published maps and institutional affiliations. |
© 2021 by the authors. Licensee MDPI, Basel, Switzerland. This article is an open access article distributed under the terms and conditions of the Creative Commons Attribution (CC BY) license (http://creativecommons.org/licenses/by/4.0/).
Share and Cite
Thavamani, A.; Salem, I.; Sferra, T.J.; Sankararaman, S. Impact of Altered Gut Microbiota and Its Metabolites in Cystic Fibrosis. Metabolites 2021, 11, 123. https://doi.org/10.3390/metabo11020123
Thavamani A, Salem I, Sferra TJ, Sankararaman S. Impact of Altered Gut Microbiota and Its Metabolites in Cystic Fibrosis. Metabolites. 2021; 11(2):123. https://doi.org/10.3390/metabo11020123
Chicago/Turabian StyleThavamani, Aravind, Iman Salem, Thomas J. Sferra, and Senthilkumar Sankararaman. 2021. "Impact of Altered Gut Microbiota and Its Metabolites in Cystic Fibrosis" Metabolites 11, no. 2: 123. https://doi.org/10.3390/metabo11020123
APA StyleThavamani, A., Salem, I., Sferra, T. J., & Sankararaman, S. (2021). Impact of Altered Gut Microbiota and Its Metabolites in Cystic Fibrosis. Metabolites, 11(2), 123. https://doi.org/10.3390/metabo11020123