
Worms, Fat, and Death: Caenorhabditis elegans Lipid Metabolites Regulate Cell Death


Abstract
:1. Introduction
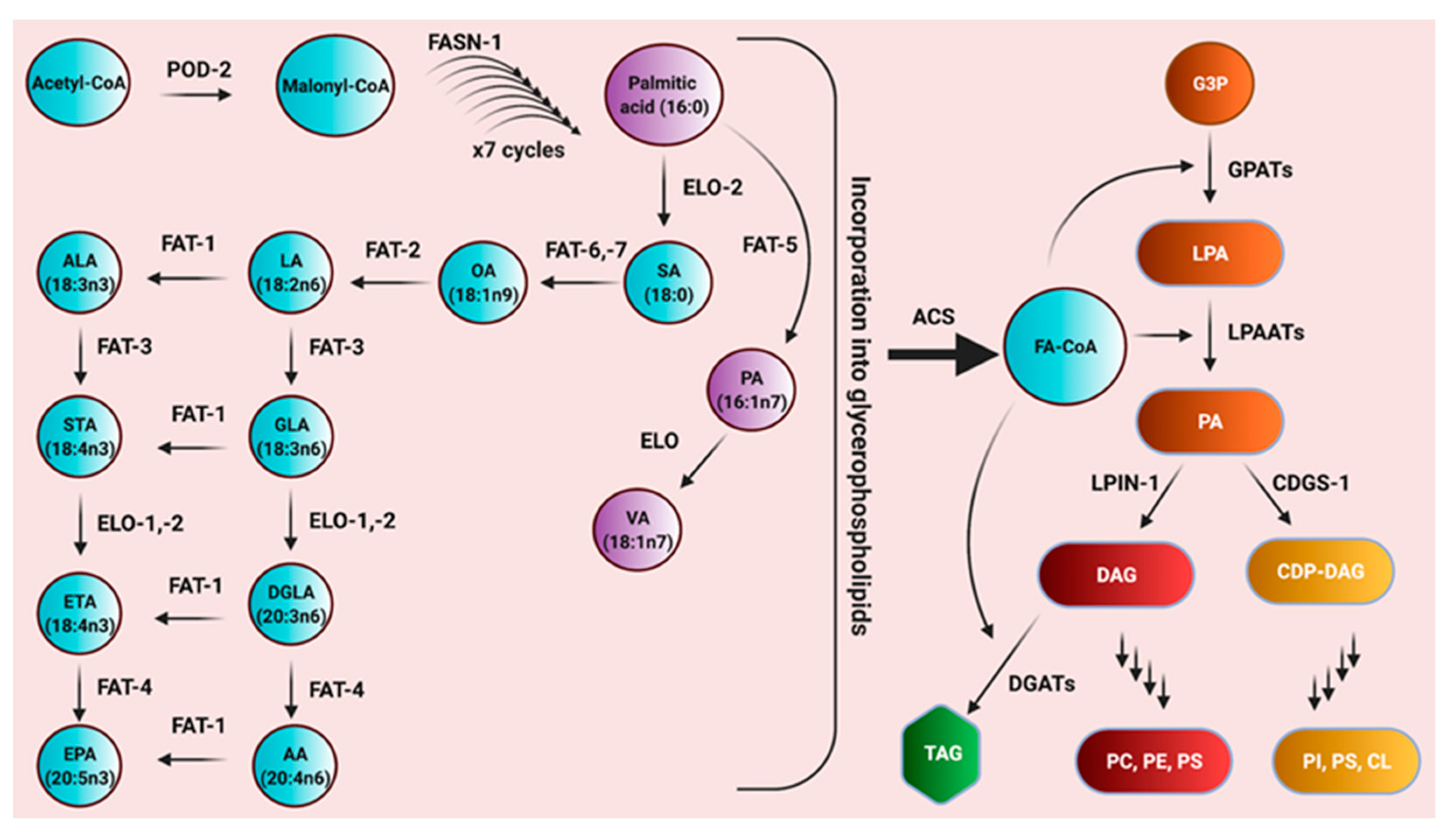
C. elegans Lipid Composition
2. Apoptosis
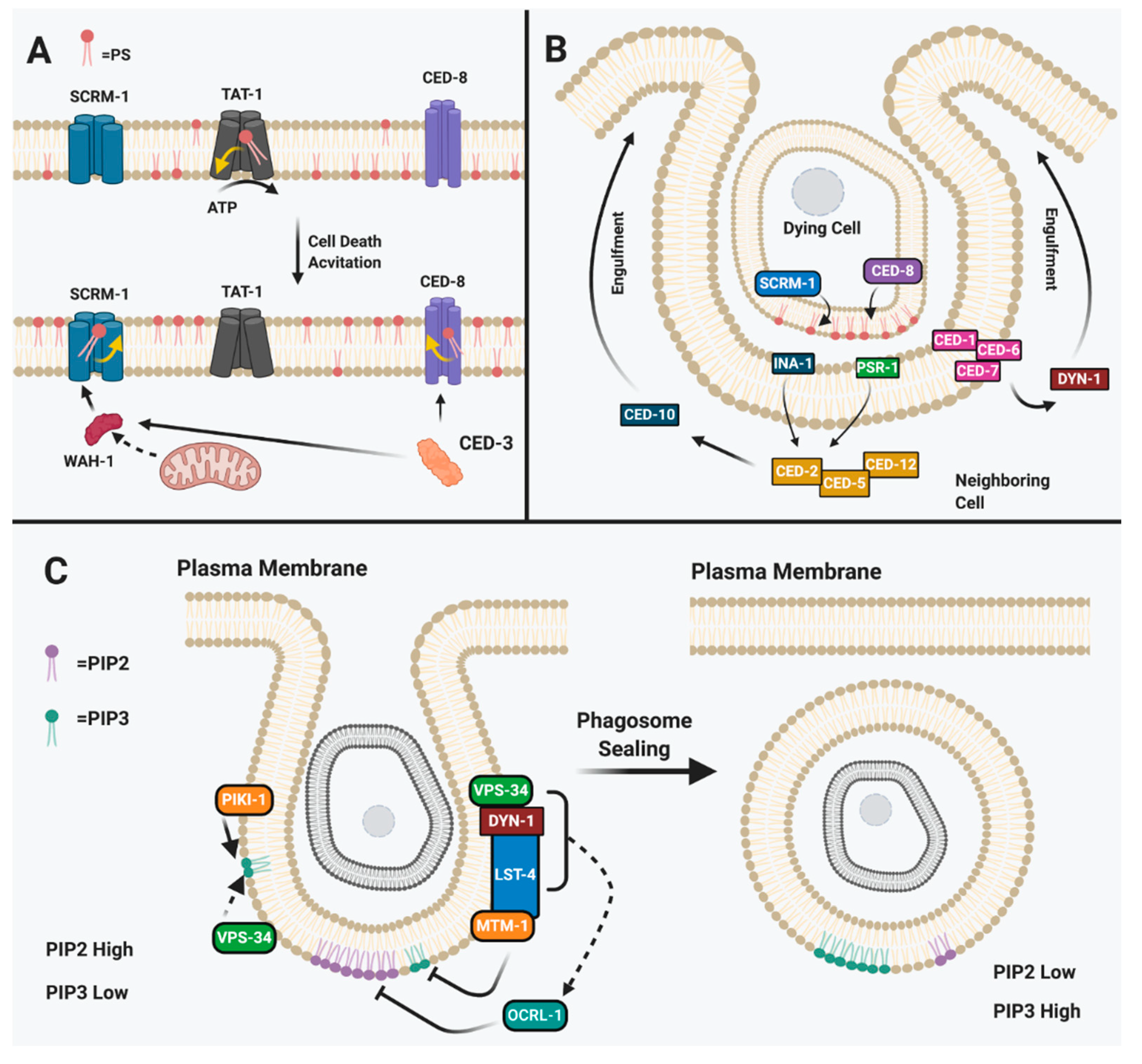
2.1. Phosphatidylserine in Signaling Execution in Apoptotic Cells
2.2. Phosphotidylinositol in Consuming the Apoptotic Corpse
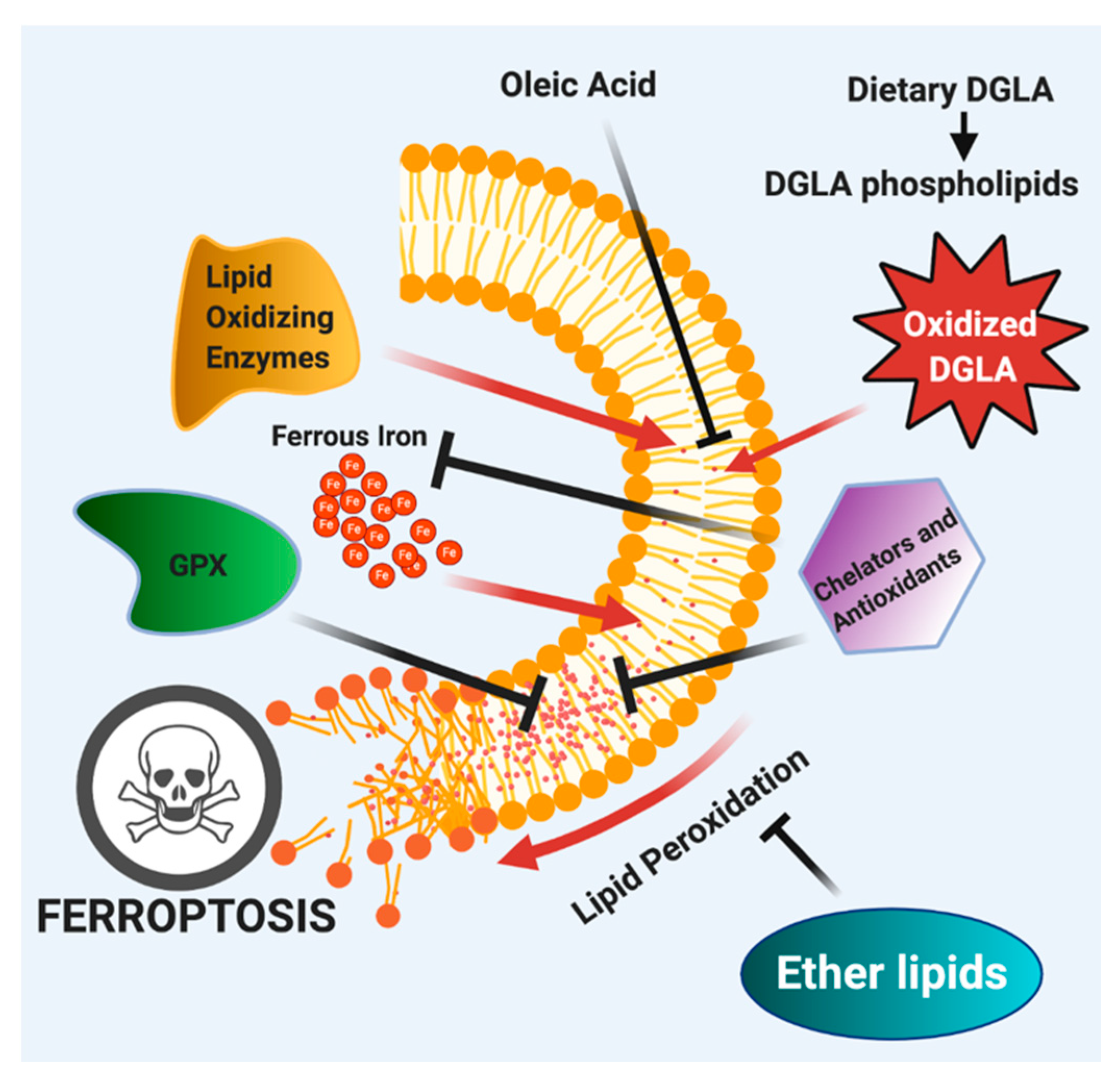
3. Ferroptosis
3.1. Dietary Polyunsaturated Fatty Acid Induction of Ferroptosis in C. elegans Germ Cells and Cancer Cells
3.2. Dietary Monounsaturated Fatty Acids Protect Cells from Ferroptosis
3.3. Iron, Ferroptosis and Aging
3.4. Ether Lipids in Ferroptosis
4. Conclusions and Future Directions
Author Contributions
Funding
Acknowledgments
Conflicts of Interest
References
- Gudipaty, S.A.; Conner, C.M.; Rosenblatt, J.; Montell, D.J. Unconventional Ways to Live and Die: Cell Death and Survival in Development, Homeostasis, and Disease. Annu. Rev. Cell Dev. Biol. 2018, 34, 311–332. [Google Scholar] [CrossRef] [PubMed]
- Tang, D.; Kang, R.; Berghe, T.V.; Vandenabeele, P.; Kroemer, G. The molecular machinery of regulated cell death. Cell Res. 2019, 29, 347–364. [Google Scholar] [CrossRef] [Green Version]
- Labi, V.; Erlacher, M. How cell death shapes cancer. Cell Death Dis. 2015, 6, e1675. [Google Scholar] [CrossRef] [Green Version]
- Pemberton, J.M.; Pogmore, J.P.; Andrews, D.W. Neuronal cell life, death, and axonal degeneration as regulated by the BCL-2 family proteins. Cell Death Differ. 2021, 28, 108–122. [Google Scholar] [CrossRef] [PubMed]
- Ellis, H.M.; Horvitz, H.R. Genetic control of programmed cell death in the nematode C. elegans. Cell 1986, 44, 817–829. [Google Scholar] [CrossRef]
- Ellis, R.E.; Yuan, J.; Horvitz, H.R. Mechanisms and Functions of Cell Death. Annu. Rev. Cell Biol. 1991, 7, 663–698. [Google Scholar] [CrossRef]
- Taylor, R.C.; Cullen, S.P.; Martin, S.J. Apoptosis: Controlled demolition at the cellular level. Nat. Rev. Mol. Cell Biol. 2008, 9, 231–241. [Google Scholar] [CrossRef]
- Galluzzi, L.; Vitale, I.; Aaronson, S.A.; Abrams, J.M.; Adam, D.; Agostinis, P.; Alnemri, E.S.; Altucci, L.; Amelio, I.; Andrews, D.W.; et al. Molecular mechanisms of cell death: Recommendations of the Nomenclature Committee on Cell Death 2018. Cell Death Differ. 2018, 25, 486–541. [Google Scholar] [CrossRef] [PubMed]
- Vanlangenakker, N.; Vanden Berghe, T.; Vandenabeele, P. Many stimuli pull the necrotic trigger, an overview. Cell Death Differ. 2012, 19, 75–86. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Cookson, B.T.; Brennan, M.A. Pro-inflammatory programmed cell death. Trends Microbiol. 2001, 3, 113–114. [Google Scholar] [CrossRef]
- Man, S.M.; Karki, R.; Kanneganti, T.D. Molecular mechanisms and functions of pyroptosis, inflammatory caspases and inflammasomes in infectious diseases. Immunol. Rev. 2017, 277, 61–75. [Google Scholar] [CrossRef] [Green Version]
- Cho, Y.; Challa, S.; Moquin, D.; Genga, R.; Ray, T.D.; Guildford, M.; Chan, F.K.-M. Phosphorylation-driven assembly of the RIP1-RIP3 complex regulates programmed necrosis and virus-induced inflammation. Cell 2009, 137, 1112–1123. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Sun, L.; Wang, H.; Wang, Z.; He, S.; Chen, S.; Liao, D.; Wang, L.; Yan, J.; Liu, W.; Lei, X. Mixed lineage kinase domain-like protein mediates necrosis signaling downstream of RIP3 kinase. Cell 2012, 148, 213–227. [Google Scholar] [CrossRef] [Green Version]
- Cai, Z.; Jitkaew, S.; Zhao, J.; Chiang, H.-C.; Choksi, S.; Liu, J.; Ward, Y.; Wu, L.-G.; Liu, Z.-G. Plasma membrane translocation of trimerized MLKL protein is required for TNF-induced necroptosis. Nat. Cell Biol. 2014, 16, 55–65. [Google Scholar] [CrossRef]
- Choi, M.E.; Price, D.R.; Ryter, S.W.; Choi, A.M.K. Necroptosis: A crucial pathogenic mediator of human disease. JCI Insight 2019, 4. [Google Scholar] [CrossRef]
- Brinkmann, V.; Reichard, U.; Goosmann, C.; Fauler, B.; Uhlemann, Y.; Weiss, D.S.; Weinrauch, Y.; Zychlinsky, A. Neutrophil extracellular traps kill bacteria. Science 2004, 303, 1532–1535. [Google Scholar] [CrossRef] [PubMed]
- Thiam, H.R.; Wong, S.L.; Wagner, D.D.; Waterman, C.M. Cellular Mechanisms of NETosis. Annu. Rev. Cell Dev. Biol. 2020, 36, 191–218. [Google Scholar] [CrossRef] [PubMed]
- Frisch, S.M.; Francis, H. Disruption of epithelial cell-matrix interactions induces apoptosis. J. Cell Biol. 1994, 124, 619–626. [Google Scholar] [CrossRef] [Green Version]
- Paoli, P.; Giannoni, E.; Chiarugi, P. Anoikis molecular pathways and its role in cancer progression. Biochim. Biophys. Acta (BBA) Mol. Cell Res. 2013, 1833, 3481–3498. [Google Scholar] [CrossRef] [Green Version]
- Dixon, S.J.; Lemberg, K.M.; Lamprecht, M.R.; Skouta, R.; Zaitsev, E.M.; Gleason, C.E.; Patel, D.N.; Bauer, A.J.; Cantley, A.M.; Yang, W.S.; et al. Ferroptosis: An iron-dependent form of nonapoptotic cell death. Cell 2012, 149, 1060–1072. [Google Scholar] [CrossRef] [Green Version]
- Dixon, S.J.; Stockwell, B.R. The Hallmarks of Ferroptosis. Annu. Rev. Cancer Biol. 2019, 3, 35–54. [Google Scholar] [CrossRef]
- Brown, G.C.; Neher, J.J. Eaten alive! Cell death by primary phagocytosis:‘phagoptosis’. Trends Biochem. Sci. 2012, 37, 325–332. [Google Scholar] [CrossRef] [PubMed]
- Brown, G.C.; Vilalta, A.; Fricker, M. Phagoptosis—Cell Death By Phagocytosis—Plays Central Roles in Physiology, Host Defense and Pathology. Curr. Mol. Med. 2015, 15, 842–851. [Google Scholar] [CrossRef] [PubMed]
- Overholtzer, M.; Mailleux, A.A.; Mouneimne, G.; Normand, G.; Schnitt, S.J.; King, R.W.; Cibas, E.S.; Brugge, J.S. A nonapoptotic cell death process, entosis, that occurs by cell-in-cell invasion. Cell 2007, 131, 966–979. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Abraham, M.C.; Lu, Y.; Shaham, S. A morphologically conserved nonapoptotic program promotes linker cell death in Caenorhabditis elegans. Dev. Cell 2007, 12, 73–86. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Malin, J.A.; Kinet, M.J.; Abraham, M.C.; Blum, E.S.; Shaham, S. Transcriptional control of non-apoptotic developmental cell death in C. elegans. Cell Death Differ. 2016, 23, 1985–1994. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Yu, S.-W.; Wang, H.; Poitras, M.F.; Coombs, C.; Bowers, W.J.; Federoff, H.J.; Poirier, G.G.; Dawson, T.M.; Dawson, V.L. Mediation of Poly(ADP-Ribose) Polymerase-1-Dependent Cell Death by Apoptosis-Inducing Factor. Science 2002, 297, 259–263. [Google Scholar] [CrossRef] [PubMed]
- Wang, X.; Ge, P. Parthanatos in the pathogenesis of nervous system diseases. Neuroscience 2020, 449, 241–250. [Google Scholar] [CrossRef]
- Fuchslocher Chico, J.; Saggau, C.; Adam, D. Proteolytic control of regulated necrosis. Biochim. Biophys. Acta Mol. Cell Res. 2017, 1864, 2147–2161. [Google Scholar] [CrossRef] [PubMed]
- Takeshige, K.; Baba, M.; Tsuboi, S.; Noda, T.; Ohsumi, Y. Autophagy in yeast demonstrated with proteinase-deficient mutants and conditions for its induction. J. Cell Biol. 1992, 119, 301–311. [Google Scholar] [CrossRef] [Green Version]
- Hansen, M.; Chandra, A.; Mitic, L.L.; Onken, B.; Driscoll, M.; Kenyon, C. A role for autophagy in the extension of lifespan by dietary restriction in C. elegans. PLoS Genet. 2008, 4, e24. [Google Scholar] [CrossRef] [Green Version]
- Bursch, W. The autophagosomal–lysosomal compartment in programmed cell death. Cell Death Differ. 2001, 8, 569–581. [Google Scholar] [CrossRef]
- Wu, C.; Zhou, L.; Yuan, H.; Wu, S. Interconnections among major forms of regulated cell death. Apoptosis 2020, 25, 616–624. [Google Scholar] [CrossRef] [PubMed]
- Conradt, B.; Wu, Y.-C.; Xue, D. Programmed Cell Death During Caenorhabditis elegans Development. Genetics 2016, 203, 1533–1562. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Blackwell, T.K.; Sewell, A.K.; Wu, Z.; Han, M. TOR Signaling in Caenorhabditis elegans Development, Metabolism, and Aging. Genetics 2019, 213, 329–360. [Google Scholar] [CrossRef] [PubMed]
- Lapierre, L.R.; Gelino, S.; Meléndez, A.; Hansen, M. Autophagy and lipid metabolism coordinately modulate life span in germline-less C. elegans. Curr. Biol. 2011, 21, 1507–1514. [Google Scholar] [CrossRef] [Green Version]
- Johnson, A.A.; Stolzing, A. The role of lipid metabolism in aging, lifespan regulation, and age-related disease. Aging Cell 2019, 18, e13048. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Chalfie, M.; Wolinsky, E. The identification and suppression of inherited neurodegeneration in Caenorhabditis elegans. Nature 1990, 345, 410–416. [Google Scholar] [CrossRef] [PubMed]
- Kourtis, N.; Nikoletopoulou, V.; Tavernarakis, N. Small heat-shock proteins protect from heat-stroke-associated neurodegeneration. Nature 2012, 490, 213–218. [Google Scholar] [CrossRef]
- Nikoletopoulou, V.; Tavernarakis, N. Chapter Six—Necrotic Cell Death in Caenorhabditis elegans. In Methods in Enzymology; Ashkenazi, A., Wells, J.A., Yuan, J., Eds.; Academic Press: Cambridge, MA, USA, 2014; Volume 545, pp. 127–155. [Google Scholar]
- Wong, D.; Bazopoulou, D.; Pujol, N.; Tavernarakis, N.; Ewbank, J.J. Genome-wide investigation reveals pathogen-specific and shared signatures in the response of Caenorhabditis elegans to infection. Genome Biol. 2007, 8, R194. [Google Scholar] [CrossRef] [Green Version]
- Lee, Y.; Hamann, J.C.; Pellegrino, M.; Durgan, J.; Domart, M.-C.; Collinson, L.M.; Haynes, C.M.; Florey, O.; Overholtzer, M. Entosis Controls a Developmental Cell Clearance in C. elegans. Cell Rep. 2019, 26, 3212–3220.e4. [Google Scholar] [CrossRef] [Green Version]
- Perez, M.A.; Magtanong, L.; Dixon, S.J.; Watts, J.L. Dietary Lipids Induce Ferroptosis in Caenorhabditiselegans and Human Cancer Cells. Dev. Cell 2020. [Google Scholar] [CrossRef] [PubMed]
- Jenkins, N.L.; James, S.A.; Salim, A.; Sumardy, F.; Speed, T.P.; Conrad, M.; Richardson, D.R.; Bush, A.I.; McColl, G. Changes in ferrous iron and glutathione promote ferroptosis and frailty in aging Caenorhabditis elegans. eLife 2020, 9, e56580. [Google Scholar] [CrossRef] [PubMed]
- Flores-Romero, H.; Ros, U.; García-Sáez, A.J. A lipid perspective on regulated cell death. Int. Rev. Cell Mol. Biol. 2020, 351, 197–236. [Google Scholar] [CrossRef] [PubMed]
- Han, S.; Schroeder, E.A.; Silva-García, C.G.; Hebestreit, K.; Mair, W.B.; Brunet, A. Mono-unsaturated fatty acids link H3K4me3 modifiers to C. elegans lifespan. Nature 2017, 544, 185–190. [Google Scholar] [CrossRef] [PubMed]
- Littlejohn, N.K.; Seban, N.; Liu, C.C.; Srinivasan, S. A feedback loop governs the relationship between lipid metabolism and longevity. eLife 2020, 9. [Google Scholar] [CrossRef] [PubMed]
- Dixit, A.; Sandhu, A.; Modi, S.; Shashikanth, M.; Koushika, S.P.; Watts, J.L.; Singh, V. Neuronal control of lipid metabolism by STR-2 G protein-coupled receptor promotes longevity in Caenorhabditis elegans. Aging Cell 2020, 19, e13160. [Google Scholar] [CrossRef] [PubMed]
- Wang, X.; Zhang, L.; Zhang, L.; Wang, W.; Wei, S.; Wang, J.; Che, H.; Zhang, Y. Effects of excess sugars and lipids on the growth and development of Caenorhabditis elegans. Genes Nutr. 2020, 15, 1. [Google Scholar] [CrossRef]
- Brock, T.J.; Browse, J.; Watts, J.L. Genetic regulation of unsaturated fatty acid composition in C. elegans. PLoS Genet. 2006, 2, e108. [Google Scholar] [CrossRef] [Green Version]
- Watts, J.L.; Browse, J. Genetic dissection of polyunsaturated fatty acid synthesis in Caenorhabditis elegans. Proc. Natl. Acad. Sci. USA 2002, 99, 5854–5859. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Watts, J.L.; Ristow, M. Lipid and Carbohydrate Metabolism in Caenorhabditis elegans. Genetics 2017, 207, 413–446. [Google Scholar] [CrossRef]
- Watts, J.L.; Phillips, E.; Griffing, K.R.; Browse, J. Deficiencies in C20 polyunsaturated fatty acids cause behavioral and developmental defects in Caenorhabditis elegans fat-3 mutants. Genetics 2003, 163, 581–589. [Google Scholar] [PubMed] [Green Version]
- Kubagawa, H.M.; Watts, J.L.; Corrigan, C.; Edmonds, J.W.; Sztul, E.; Browse, J.; Miller, M.A. Oocyte signals derived from polyunsaturated fatty acids control sperm recruitment in vivo. Nat. Cell Biol. 2006, 8, 1143–1148. [Google Scholar] [CrossRef] [PubMed]
- Watts, J.L.; Browse, J. Dietary manipulation implicates lipid signaling in the regulation of germ cell maintenance in C. elegans. Dev. Biol. 2006, 292, 381–392. [Google Scholar] [CrossRef] [Green Version]
- Hoang, H.D.; Prasain, J.K.; Dorand, D.; Miller, M.A. A heterogeneous mixture of F-series prostaglandins promotes sperm guidance in the Caenorhabditis elegans reproductive tract. PLoS Genet. 2013, 9, e1003271. [Google Scholar] [CrossRef] [Green Version]
- Chen, W.W.; Yi, Y.H.; Chien, C.H.; Hsiung, K.C.; Ma, T.H.; Lin, Y.C.; Lo, S.J.; Chang, T.C. Specific polyunsaturated fatty acids modulate lipid delivery and oocyte development in C. elegans revealed by molecular-selective label-free imaging. Sci. Rep. 2016, 6, 32021. [Google Scholar] [CrossRef] [Green Version]
- Watts, J.S.; Morton, D.G.; Kemphues, K.J.; Watts, J.L. The biotin-ligating protein BPL-1 is critical for lipid biosynthesis and polarization of the Caenorhabditis elegans embryo. J. Biol. Chem. 2018, 293, 610–622. [Google Scholar] [CrossRef] [Green Version]
- Webster, C.M.; Deline, M.L.; Watts, J.L. Stress response pathways protect germ cells from omega-6 polyunsaturated fatty acid-mediated toxicity in Caenorhabditis elegans. Dev. Biol. 2013, 373, 14–25. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Deline, M.; Keller, J.; Rothe, M.; Schunck, W.H.; Menzel, R.; Watts, J.L. Epoxides Derived from Dietary Dihomo-Gamma-Linolenic Acid Induce Germ Cell Death in C. elegans. Sci. Rep. 2015, 5, 15417. [Google Scholar] [CrossRef] [PubMed]
- Hou, N.S.; Gutschmidt, A.; Choi, D.Y.; Pather, K.; Shi, X.; Watts, J.L.; Hoppe, T.; Taubert, S. Activation of the endoplasmic reticulum unfolded protein response by lipid disequilibrium without disturbed proteostasis in vivo. Proc. Natl. Acad. Sci. USA 2014, 111, E2271–E2280. [Google Scholar] [CrossRef] [Green Version]
- Chamoli, M.; Goyala, A.; Tabrez, S.S.; Siddiqui, A.A.; Singh, A.; Antebi, A.; Lithgow, G.J.; Watts, J.L.; Mukhopadhyay, A. Polyunsaturated fatty acids and p38-MAPK link metabolic reprogramming to cytoprotective gene expression during dietary restriction. Nat. Commun. 2020, 11, 4865. [Google Scholar] [CrossRef]
- Jung, Y.; Kwon, S.; Ham, S.; Lee, D.; Park, H.H.; Yamaoka, Y.; Jeong, D.E.; Artan, M.; Altintas, O.; Park, S.; et al. Caenorhabditis elegans Lipin 1 moderates the lifespan-shortening effects of dietary glucose by maintaining ω-6 polyunsaturated fatty acids. Aging Cell 2020, 19, e13150. [Google Scholar] [CrossRef]
- Watts, J.L. Using Caenorhabditis elegans to Uncover Conserved Functions of Omega-3 and Omega-6 Fatty Acids. J. Clin. Med. 2016, 5, 19. [Google Scholar] [CrossRef] [Green Version]
- Connor, A.J.; Watts, J.L. Chapter 7—Omega-3 and Omega-6 Fatty Acid Metabolism: Modeling Growth and Disease Using Caenorhabditis elegans. In Omega Fatty Acids in Brain and Neurological Health, 2nd ed.; Watson, R.R., Preedy, V.R., Eds.; Academic Press: Cambridge, MA, USA, 2019; pp. 107–116. [Google Scholar] [CrossRef]
- Shi, X.; Li, J.; Zou, X.; Greggain, J.; Rodkaer, S.V.; Faergeman, N.J.; Liang, B.; Watts, J.L. Regulation of lipid droplet size and phospholipid composition by stearoyl-CoA desaturase. J. Lipid Res. 2013, 54, 2504–2514. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Shi, X.; Tarazona, P.; Brock, T.J.; Browse, J.; Feussner, I.; Watts, J.L. A Caenorhabditis elegans model for ether lipid biosynthesis and function. J. Lipid Res. 2016, 57, 265–275. [Google Scholar] [CrossRef] [Green Version]
- Witting, M.; Schmitt-Kopplin, P. The Caenorhabditis elegans lipidome: A primer for lipid analysis in Caenorhabditis elegans. Arch Biochem. Biophys. 2016, 589, 27–37. [Google Scholar] [CrossRef]
- Perez, C.L.; Van Gilst, M.R. A 13C isotope labeling strategy reveals the influence of insulin signaling on lipogenesis in C. elegans. Cell Metab. 2008, 8, 266–274. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Dancy, B.C.; Chen, S.W.; Drechsler, R.; Gafken, P.R.; Olsen, C.P. 13C- and 15N-Labeling Strategies Combined with Mass Spectrometry Comprehensively Quantify Phospholipid Dynamics in C. elegans. PLoS ONE 2015, 10, e0141850. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Montigny, C.; Lyons, J.; Champeil, P.; Nissen, P.; Lenoir, G. On the molecular mechanism of flippase- and scramblase-mediated phospholipid transport. Biochim. Biophys. Acta 2016, 1861, 767–783. [Google Scholar] [CrossRef] [PubMed]
- Gumienny, T.L.; Lambie, E.; Hartwieg, E.; Horvitz, H.R.; Hengartner, M.O. Genetic control of programmed cell death in the Caenorhabditis elegans hermaphrodite germline. Development 1999, 126, 1011–1022. [Google Scholar] [PubMed]
- Lant, B.; Derry, W.B. Analysis of apoptosis in Caenorhabditis elegans. Cold Spring Harb. Protoc. 2014, 2014. [Google Scholar] [CrossRef] [Green Version]
- Sulston, J.; Schierenberg, E.; White, J.; Thomson, J. The embryonic Cell Lineage of the Nematode Caenorhabditis elegans. Dev. Biol. 1983, 100, 64–119. [Google Scholar] [CrossRef]
- Sulston, J.; Horvitz, H. Postembryonic cell lineages of the nematode Caenorhabditis elegans. Dev. Biol. 1977, 56, 110–156. [Google Scholar] [CrossRef]
- Gumienny, T.L.; Brugnera, E.; Tosello-Trampont, A.-C.; Kinchen, J.M.; Haney, L.B.; Nishiwaki, K.; Walk, S.F.; Nemergut, M.E.; Macara, I.G.; Francis, R.; et al. CED-12/ELMO, a Novel Member of the CrkII/Dock180/Rac Pathway, Is Required for Phagocytosis and Cell Migration. Cell 2001, 107, 27–41. [Google Scholar] [CrossRef] [Green Version]
- Stergiou, L.; Hengartner, M.O. Death and more: DNA damage response pathways in the nematode C. elegans. Cell Death Differ. 2004, 11, 21–28. [Google Scholar] [CrossRef] [Green Version]
- Lettre, G.; Hengartner, M.O. Developmental apoptosis in C. elegans: A complex CEDnario. Nat. Rev. Mol. Cell Biol. 2006, 7, 97–108. [Google Scholar] [CrossRef]
- Hofmann, E.R.; Milstein, S.; Boulton, S.J.; Ye, M.; Hofmann, J.J.; Stergiou, L.; Gartner, A.; Vidal, M.; Hengartner, M.O. Caenorhabditis elegans HUS-1 Is a DNA Damage Checkpoint Protein Required for Genome Stability and EGL-1-Mediated Apoptosis. Curr. Biol. 2002, 12, 1908–1918. [Google Scholar] [CrossRef] [Green Version]
- Elmore, S. Apoptosis: A review of programmed cell death. Toxicol. Pathol. 2007, 35, 495–516. [Google Scholar] [CrossRef] [PubMed]
- Horvitz, H.R. Genetic Control of Programmed Cell Death in the Nematode Caenorhabditis elegans. Cancer Res. 1999, 59, 1701s–1706s. [Google Scholar] [PubMed]
- Bailly, A.; Gartner, A. Germ cell apoptosis and DNA damage responses. Adv. Exp. Med. Biol. 2013, 757, 249–276. [Google Scholar] [CrossRef] [Green Version]
- Bretscher, M.S. Asymmetrical lipid bilayer structure for biological membranes. Nat. New Biol. 1972, 236, 11–12. [Google Scholar] [CrossRef]
- Kamp, J.A.F.O.d. Lipid Asymmetry in Membranes. Annu. Rev. Biochem. 1979, 48, 47–71. [Google Scholar] [CrossRef]
- Fadok, V.A.; Voelker, D.R.; Campbell, P.A.; Cohen, J.J.; Bratton, D.L.; Henson, P.M. Exposure of phosphatidylserine on the surface of apoptotic lymphocytes triggers specific recognition and removal by macrophages. J. Immunol. 1992, 148, 2207–2216. [Google Scholar] [PubMed]
- Martin, S.J.; Reutelingsperger, C.P.; McGahon, A.J.; Rader, J.A.; van Schie, R.C.; LaFace, D.M.; Green, D.R. Early redistribution of plasma membrane phosphatidylserine is a general feature of apoptosis regardless of the initiating stimulus: Inhibition by overexpression of Bcl-2 and Abl. J. Exp. Med. 1995, 182, 1545–1556. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Savill, J.; Fadok, V. Corpse clearance defines the meaning of cell death. Nature 2000, 407, 784–788. [Google Scholar] [CrossRef]
- Segawa, K.; Nagata, S. An Apoptotic ‘Eat Me’ Signal: Phosphatidylserine Exposure. Trends Cell Biol. 2015, 25, 639–650. [Google Scholar] [CrossRef]
- Shlomovitz, I.; Speir, M.; Gerlic, M. Flipping the dogma—phosphatidylserine in non-apoptotic cell death. Cell Commun. Signal. 2019, 17, 139. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Robertson, A.M.G.; Thomson, J.N. Morphology of programmed cell death in the ventral nerve cord of Caenorhabditis elegans larvae. J. Embryol. Exp. Morphol. 1982, 67, 89–100. [Google Scholar]
- Zhou, Z.; Hartwieg, E.; Horvitz, H.R. CED-1 Is a Transmembrane Receptor that Mediates Cell Corpse Engulfment in C. elegans. Cell 2001, 104, 43–56. [Google Scholar] [CrossRef] [Green Version]
- Wang, X.; Wang, J.; Gengyo-Ando, K.; Gu, L.; Sun, C.-L.; Yang, C.; Shi, Y.; Kobayashi, T.; Shi, Y.; Mitani, S.; et al. C. elegans mitochondrial factor WAH-1 promotes phosphatidylserine externalization in apoptotic cells through phospholipid scramblase SCRM-1. Nat. Cell Biol. 2007, 9, 541–549. [Google Scholar] [CrossRef] [PubMed]
- Fadeel, B.; Xue, D. The ins and outs of phospholipid asymmetry in the plasma membrane: Roles in health and disease. Crit. Rev. Biochem. Mol. Biol. 2009, 44, 264–277. [Google Scholar] [CrossRef]
- Wang, X.; Yang, C. Programmed cell death and clearance of cell corpses in Caenorhabditis elegans. Cell. Mol. Life Sci. 2016, 73, 2221–2236. [Google Scholar] [CrossRef]
- Kobayashi, T.; Menon, A.K. Transbilayer lipid asymmetry. Curr. Biol. 2018, 28, R386–R391. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Venegas, V.; Zhou, Z. Two alternative mechanisms that regulate the presentation of apoptotic cell engulfment signal in Caenorhabditis elegans. Mol. Biol. Cell 2007, 18, 3180–3192. [Google Scholar] [CrossRef] [Green Version]
- Preta, G.; Fadeel, B. AIF and Scythe (Bat3) Regulate Phosphatidylserine Exposure and Macrophage Clearance of Cells Undergoing Fas (APO-1)-Mediated Apoptosis. PLoS ONE 2012, 7, e47328. [Google Scholar] [CrossRef] [Green Version]
- Chen, Y.-Z.; Mapes, J.; Lee, E.-S.; Robert Skeen-Gaar, R.; Xue, D. Caspase-mediated activation of Caenorhabditis elegans CED-8 promotes apoptosis and phosphatidylserine externalization. Nat. Commun. 2013, 4, 2726. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Stanfield, G.M.; Horvitz, H.R. The ced-8 gene controls the timing of programmed cell deaths in C. elegans. Mol. Cell 2000, 5, 423–433. [Google Scholar] [CrossRef]
- Suzuki, J.; Imanishi, E.; Nagata, S. Xkr8 phospholipid scrambling complex in apoptotic phosphatidylserine exposure. Proc. Natl. Acad. Sci. USA 2016, 113, 9509–9514. [Google Scholar] [CrossRef] [Green Version]
- Züllig, S.; Neukomm, L.J.; Jovanovic, M.; Charette, S.J.; Lyssenko, N.N.; Halleck, M.S.; Reutelingsperger, C.P.; Schlegel, R.A.; Hengartner, M.O. Aminophospholipid Translocase TAT-1 Promotes Phosphatidylserine Exposure during C. elegans Apoptosis. Curr. Biol. 2007, 17, 994–999. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Darland-Ransom, M.; Wang, X.; Sun, C.-L.; Mapes, J.; Gengyo-Ando, K.; Mitani, S.; Xue, D. Role of C. elegans TAT-1 Protein in Maintaining Plasma Membrane Phosphatidylserine Asymmetry. Science 2008, 320, 528–531. [Google Scholar] [CrossRef] [Green Version]
- Segawa, K.; Kurata, S.; Yanagihashi, Y.; Brummelkamp, T.R.; Matsuda, F.; Nagata, S. Caspase-mediated cleavage of phospholipid flippase for apoptotic phosphatidylserine exposure. Science 2014, 344, 1164–1168. [Google Scholar] [CrossRef] [Green Version]
- Ruaud, A.-F.; Nilsson, L.; Richard, F.; Larsen, M.K.; Bessereau, J.-L.; Tuck, S. The C. elegans P4-ATPase TAT-1 Regulates Lysosome Biogenesis and Endocytosis. Traffic 2009, 10, 88–100. [Google Scholar] [CrossRef]
- Chen, B.; Jiang, Y.; Zeng, S.; Yan, J.; Li, X.; Zhang, Y.; Zou, W.; Wang, X. Endocytic sorting and recycling require membrane phosphatidylserine asymmetry maintained by TAT-1/CHAT-1. PLoS Genet. 2010, 6, e1001235. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Chen, Y.Z.; Klöditz, K.; Lee, E.S.; Nguyen, D.P.; Yuan, Q.; Johnson, J.; Lee-Yow, Y.; Hall, A.; Mitani, S.; Xia, N.S.; et al. Structure and function analysis of the C. elegans aminophospholipid translocase TAT-1. J. Cell Sci. 2019, 132. [Google Scholar] [CrossRef] [Green Version]
- Li, Z.; Venegas, V.; Nagaoka, Y.; Morino, E.; Raghavan, P.; Audhya, A.; Nakanishi, Y.; Zhou, Z. Necrotic cells actively attract phagocytes through the collaborative action of two distinct PS-exposure mechanisms. PLoS Genet. 2015, 11, e1005285. [Google Scholar]
- Yang, H.; Chen, Y.-Z.; Zhang, Y.; Wang, X.; Zhao, X.; Godfroy, J.I.; Liang, Q.; Zhang, M.; Zhang, T.; Yuan, Q.; et al. A lysine-rich motif in the phosphatidylserine receptor PSR-1 mediates recognition and removal of apoptotic cells. Nat. Commun. 2015, 6, 5717. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Reddien, P.W.; Horvitz, H.R. The engulfment process of programmed cell death in Caenorhabditis elegans. Annu. Rev. Cell Dev. Biol. 2004, 20, 193–221. [Google Scholar] [CrossRef] [Green Version]
- Yu, X.; Odera, S.; Chuang, C.-H.; Lu, N.; Zhou, Z. C. elegans Dynamin Mediates the Signaling of Phagocytic Receptor CED-1 for the Engulfment and Degradation of Apoptotic Cells. Dev. Cell 2006, 10, 743–757. [Google Scholar] [CrossRef] [Green Version]
- Grimsley, C.; Ravichandran, K.S. Cues for apoptotic cell engulfment: Eat-me, don’t eat-me and come-get-me signals. Trends Cell Biol. 2003, 13, 648–656. [Google Scholar] [CrossRef]
- Wu, Y.-C.; Horvitz, H.R. C. elegans phagocytosis and cell-migration protein CED-5 is similar to human DOCK180. Nature 1998, 392, 501–504. [Google Scholar] [CrossRef] [PubMed]
- Wang, X.; Wu, Y.-C.; Fadok, V.A.; Lee, M.-C.; Gengyo-Ando, K.; Cheng, L.-C.; Ledwich, D.; Hsu, P.-K.; Chen, J.-Y.; Chou, B.-K.; et al. Cell Corpse Engulfment Mediated by C. elegans Phosphatidylserine Receptor Through CED-5 and CED-12. Science 2003, 302, 1563–1566. [Google Scholar] [CrossRef]
- Neumann, B.; Coakley, S.; Giordano-Santini, R.; Linton, C.; Lee, E.S.; Nakagawa, A.; Xue, D.; Hilliard, M.A. EFF-1-mediated regenerative axonal fusion requires components of the apoptotic pathway. Nature 2015, 517, 219–222. [Google Scholar] [CrossRef] [PubMed]
- Lu, N.; Zhou, Z. Membrane Trafficking and Phagosome Maturation During the Clearance of Apoptotic Cells. In International Review of Cell and Molecular Biology; Jeon, K.W., Ed.; Academic Press: Cambridge, MA, USA, 2012; Chapter 8; Volume 293, pp. 269–309. [Google Scholar]
- Vicinanza, M.; D’Angelo, G.; Di Campli, A.; De Matteis, M.A. Function and dysfunction of the PI system in membrane trafficking. EMBO J. 2008, 27, 2457–2470. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Margaria, J.P.; Ratto, E.; Gozzelino, L.; Li, H.; Hirsch, E. Class II PI3Ks at the Intersection between Signal Transduction and Membrane Trafficking. Biomolecules 2019, 9, 104. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Flannagan, R.S.; Jaumouillé, V.; Grinstein, S. The cell biology of phagocytosis. Annu. Rev. Pathol. 2012, 7, 61–98. [Google Scholar] [CrossRef]
- Lu, N.; Shen, Q.; Mahoney, T.R.; Neukomm, L.J.; Wang, Y.; Zhou, Z. Two PI 3-kinases and one PI 3-phosphatase together establish the cyclic waves of phagosomal PtdIns(3)P critical for the degradation of apoptotic cells. PLoS Biol. 2012, 10, e1001245. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Cheng, S.; Wang, K.; Zou, W.; Miao, R.; Huang, Y.; Wang, H.; Wang, X. PtdIns(4,5)P2 and PtdIns3P coordinate to regulate phagosomal sealing for apoptotic cell clearance. J. Cell Biol. 2015, 210, 485–502. [Google Scholar] [CrossRef] [Green Version]
- Kinchen, J.M.; Doukoumetzidis, K.; Almendinger, J.; Stergiou, L.; Tosello-Trampont, A.; Sifri, C.D.; Hengartner, M.O.; Ravichandran, K.S. A pathway for phagosome maturation during engulfment of apoptotic cells. Nat. Cell Biol. 2008, 10, 556–566. [Google Scholar] [CrossRef] [Green Version]
- Abdu, Y.; Maniscalco, C.; Heddleston, J.M.; Chew, T.-L.; Nance, J. Developmentally programmed germ cell remodelling by endodermal cell cannibalism. Nat. Cell Biol. 2016, 18, 1302–1310. [Google Scholar] [CrossRef] [PubMed]
- Bohdanowicz, M.; Balkin, D.M.; De Camilli, P.; Grinstein, S. Recruitment of OCRL and Inpp5B to phagosomes by Rab5 and APPL1 depletes phosphoinositides and attenuates Akt signaling. Mol. Biol. Cell 2012, 23, 176–187. [Google Scholar] [CrossRef]
- Niedergang, F.; Grinstein, S. How to build a phagosome: New concepts for an old process. Curr. Opin. Cell Biol. 2018, 50, 57–63. [Google Scholar] [CrossRef] [PubMed]
- Baba, Y.; Higa, J.K.; Shimada, B.K.; Horiuchi, K.M.; Suhara, T.; Kobayashi, M.; Woo, J.D.; Aoyagi, H.; Marh, K.S.; Kitaoka, H.; et al. Protective effects of the mechanistic target of rapamycin against excess iron and ferroptosis in cardiomyocytes. Am. J. Physiol. Heart Circ. Physiol. 2018, 314, H659–H668. [Google Scholar] [CrossRef] [Green Version]
- Friedmann Angeli, J.P.; Schneider, M.; Proneth, B.; Tyurina, Y.Y.; Tyurin, V.A.; Hammond, V.J.; Herbach, N.; Aichler, M.; Walch, A.; Eggenhofer, E.; et al. Inactivation of the ferroptosis regulator Gpx4 triggers acute renal failure in mice. Nat. Cell Biol. 2014, 16, 1180–1191. [Google Scholar] [CrossRef] [Green Version]
- Liu, P.; Feng, Y.; Li, H.; Chen, X.; Wang, G.; Xu, S.; Li, Y.; Zhao, L. Ferrostatin-1 alleviates lipopolysaccharide-induced acute lung injury via inhibiting ferroptosis. Cell. Mol. Biol. Lett. 2020, 25, 10. [Google Scholar] [CrossRef] [Green Version]
- Wang, W.; Green, M.; Choi, J.E.; Gijón, M.; Kennedy, P.D.; Johnson, J.K.; Liao, P.; Lang, X.; Kryczek, I.; Sell, A.; et al. CD8(+) T cells regulate tumour ferroptosis during cancer immunotherapy. Nature 2019, 569, 270–274. [Google Scholar] [CrossRef] [PubMed]
- Shintoku, R.; Takigawa, Y.; Yamada, K.; Kubota, C.; Yoshimoto, Y.; Takeuchi, T.; Koshiishi, I.; Torii, S. Lipoxygenase-mediated generation of lipid peroxides enhances ferroptosis induced by erastin and RSL3. Cancer Sci. 2017, 108, 2187–2194. [Google Scholar] [CrossRef]
- Yang, W.S.; SriRamaratnam, R.; Welsch, M.E.; Shimada, K.; Skouta, R.; Viswanathan, V.S.; Cheah, J.H.; Clemons, P.A.; Shamji, A.F.; Clish, C.B.; et al. Regulation of ferroptotic cancer cell death by GPX4. Cell 2014, 156, 317–331. [Google Scholar] [CrossRef] [Green Version]
- Hinman, A.; Holst, C.R.; Latham, J.C.; Bruegger, J.J.; Ulas, G.; McCusker, K.P.; Amagata, A.; Davis, D.; Hoff, K.G.; Kahn-Kirby, A.H.; et al. Vitamin E hydroquinone is an endogenous regulator of ferroptosis via redox control of 15-lipoxygenase. PLoS ONE 2018, 13, e0201369. [Google Scholar] [CrossRef]
- Ingold, I.; Berndt, C.; Schmitt, S.; Doll, S.; Poschmann, G.; Buday, K.; Roveri, A.; Peng, X.; Porto Freitas, F.; Seibt, T.; et al. Selenium Utilization by GPX4 Is Required to Prevent Hydroperoxide-Induced Ferroptosis. Cell 2018, 172, 409–422.e21. [Google Scholar] [CrossRef] [Green Version]
- Forcina, G.C.; Dixon, S.J. GPX4 at the Crossroads of Lipid Homeostasis and Ferroptosis. Proteomics 2019, 19, 1800311. [Google Scholar] [CrossRef] [PubMed]
- Halliwell, B.; Gutteridge, J.M. Oxygen toxicity, oxygen radicals, transition metals and disease. Biochem. J. 1984, 219, 1–14. [Google Scholar] [CrossRef]
- Feng, H.; Stockwell, B.R. Unsolved mysteries: How does lipid peroxidation cause ferroptosis? PLoS Biol. 2018, 16, e2006203. [Google Scholar] [CrossRef] [PubMed]
- Doll, S.; Proneth, B.; Tyurina, Y.Y.; Panzilius, E.; Kobayashi, S.; Ingold, I.; Irmler, M.; Beckers, J.; Aichler, M.; Walch, A.; et al. ACSL4 dictates ferroptosis sensitivity by shaping cellular lipid composition. Nat. Chem. Biol. 2017, 13, 91–98. [Google Scholar] [CrossRef] [PubMed]
- Kagan, V.E.; Mao, G.; Qu, F.; Angeli, J.P.; Doll, S.; Croix, C.S.; Dar, H.H.; Liu, B.; Tyurin, V.A.; Ritov, V.B.; et al. Oxidized arachidonic and adrenic PEs navigate cells to ferroptosis. Nat. Chem. Biol. 2017, 13, 81–90. [Google Scholar] [CrossRef] [PubMed]
- Yang, W.S.; Kim, K.J.; Gaschler, M.M.; Patel, M.; Shchepinov, M.S.; Stockwell, B.R. Peroxidation of polyunsaturated fatty acids by lipoxygenases drives ferroptosis. Proc. Natl. Acad. Sci. USA 2016, 113, E4966. [Google Scholar] [CrossRef] [Green Version]
- Sakamoto, T.; Maebayashi, K.; Nakagawa, Y.; Imai, H. Deletion of the four phospholipid hydroperoxide glutathione peroxidase genes accelerates aging in Caenorhabditis elegans. Genes Cells 2014, 19, 778–792. [Google Scholar] [CrossRef] [PubMed]
- Drummen, G.P.C.; van Liebergen, L.C.M.; Op den Kamp, J.A.F.; Post, J.A. C11-BODIPY581/591, an oxidation-sensitive fluorescent lipid peroxidation probe: (micro)spectroscopic characterization and validation of methodology. Free Radic. Biol. Med. 2002, 33, 473–490. [Google Scholar] [CrossRef]
- Magtanong, L.; Ko, P.J.; To, M.; Cao, J.Y.; Forcina, G.C.; Tarangelo, A.; Ward, C.C.; Cho, K.; Patti, G.J.; Nomura, D.K.; et al. Exogenous Monounsaturated Fatty Acids Promote a Ferroptosis-Resistant Cell State. Cell Chem. Biol. 2019, 26, 420–432.e9. [Google Scholar] [CrossRef]
- Gillingham, L.G.; Harris-Janz, S.; Jones, P.J. Dietary monounsaturated fatty acids are protective against metabolic syndrome and cardiovascular disease risk factors. Lipids 2011, 46, 209–228. [Google Scholar] [CrossRef] [PubMed]
- de Oliveira, P.A.; Kovacs, C.; Moreira, P.; Magnoni, D.; Saleh, M.H.; Faintuch, J. Unsaturated Fatty Acids Improve Atherosclerosis Markers in Obese and Overweight Non-diabetic Elderly Patients. Obes. Surg. 2017, 27, 2663–2671. [Google Scholar] [CrossRef]
- Angeli, J.P.F.; Shah, R.; Pratt, D.A.; Conrad, M. Ferroptosis Inhibition: Mechanisms and Opportunities. Trends Pharmacol. Sci. 2017, 38, 489–498. [Google Scholar] [CrossRef]
- Han, C.; Liu, Y.; Dai, R.; Ismail, N.; Su, W.; Li, B. Ferroptosis and Its Potential Role in Human Diseases. Front. Pharmacol. 2020, 11. [Google Scholar] [CrossRef] [PubMed]
- Fenton, H.J.H. LXXIII.—Oxidation of tartaric acid in presence of iron. J. Chem. Soc. Trans. 1894, 65, 899–910. [Google Scholar] [CrossRef] [Green Version]
- Zou, Y.; Li, H.; Graham, E.T.; Deik, A.A.; Eaton, J.K.; Wang, W.; Sandoval-Gomez, G.; Clish, C.B.; Doench, J.G.; Schreiber, S.L. Cytochrome P450 oxidoreductase contributes to phospholipid peroxidation in ferroptosis. Nat. Chem. Biol. 2020, 16, 302–309. [Google Scholar] [CrossRef]
- Yan, B.; Ai, Y.; Sun, Q.; Ma, Y.; Cao, Y.; Wang, J.; Zhang, Z.; Wang, X. Membrane Damage during Ferroptosis Is Caused by Oxidation of Phospholipids Catalyzed by the Oxidoreductases POR and CYB5R1. Mol. Cell 2020. [Google Scholar] [CrossRef]
- Hou, W.; Xie, Y.; Song, X.; Sun, X.; Lotze, M.T.; Zeh, H.J.; Kang, R.; Tang, D. Autophagy promotes ferroptosis by degradation of ferritin. Autophagy 2016, 12, 1425–1428. [Google Scholar] [CrossRef]
- Li, Q.; Han, X.; Lan, X.; Gao, Y.; Wan, J.; Durham, F.; Cheng, T.; Yang, J.; Wang, Z.; Jiang, C.; et al. Inhibition of neuronal ferroptosis protects hemorrhagic brain. Jci Insight 2017, 2. [Google Scholar] [CrossRef] [Green Version]
- Chen, X.; Yu, C.; Kang, R.; Tang, D. Iron Metabolism in Ferroptosis. Front. Cell Dev. Biol. 2020, 8. [Google Scholar] [CrossRef]
- Gao, M.; Monian, P.; Quadri, N.; Ramasamy, R.; Jiang, X. Glutaminolysis and Transferrin Regulate Ferroptosis. Mol. Cell 2015, 59, 298–308. [Google Scholar] [CrossRef] [Green Version]
- Torii, S.; Shintoku, R.; Kubota, C.; Yaegashi, M.; Torii, R.; Sasaki, M.; Suzuki, T.; Mori, M.; Yoshimoto, Y.; Takeuchi, T.; et al. An essential role for functional lysosomes in ferroptosis of cancer cells. Biochem. J. 2016, 473, 769–777. [Google Scholar] [CrossRef] [Green Version]
- Becerril-Ortega, J.; Bordji, K.; Fréret, T.; Rush, T.; Buisson, A. Iron overload accelerates neuronal amyloid-β production and cognitive impairment in transgenic mice model of Alzheimer’s disease. Neurobiol. Aging 2014, 35, 2288–2301. [Google Scholar] [CrossRef]
- Lei, P.; Ayton, S.; Finkelstein, D.I.; Spoerri, L.; Ciccotosto, G.D.; Wright, D.K.; Wong, B.X.; Adlard, P.A.; Cherny, R.A.; Lam, L.Q.; et al. Tau deficiency induces parkinsonism with dementia by impairing APP-mediated iron export. Nat. Med. 2012, 18, 291–295. [Google Scholar] [CrossRef] [PubMed]
- James, S.A.; Roberts, B.R.; Hare, D.J.; de Jonge, M.D.; Birchall, I.E.; Jenkins, N.L.; Cherny, R.A.; Bush, A.I.; McColl, G. Direct in vivo imaging of ferrous iron dyshomeostasis in ageing Caenorhabditis elegans. Chem. Sci. 2015, 6, 2952–2962. [Google Scholar] [CrossRef] [Green Version]
- Jiménez-Rojo, N.; Riezman, H. On the road to unraveling the molecular functions of ether lipids. FEBS Lett. 2019, 593, 2378–2389. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Rangholia, N.; Leisner, T.M.; Holly, S.P. Bioactive Ether Lipids: Primordial Modulators of Cellular Signaling. Metabolites 2021, 11, 41. [Google Scholar] [CrossRef]
- de Vet, E.C.; Ijlst, L.; Oostheim, W.; Dekker, C.; Moser, H.W.; van Den Bosch, H.; Wanders, R.J. Ether lipid biosynthesis: Alkyl-dihydroxyacetonephosphate synthase protein deficiency leads to reduced dihydroxyacetonephosphate acyltransferase activities. J. Lipid Res. 1999, 40, 1998–2003. [Google Scholar] [CrossRef]
- James, P.F.; Lake, A.C.; Hajra, A.K.; Larkins, L.K.; Robinson, M.; Buchanan, F.G.; Zoeller, R.A. An animal cell mutant with a deficiency in acyl/alkyl-dihydroxyacetone-phosphate reductase activity. Effects on the biosynthesis of ether-linked and diacyl glycerolipids. J. Biol. Chem. 1997, 272, 23540–23546. [Google Scholar] [CrossRef] [Green Version]
- Dean, J.M.; Lodhi, I.J. Structural and functional roles of ether lipids. Protein Cell 2018, 9, 196–206. [Google Scholar] [CrossRef] [PubMed]
- Wood, P.L.; Locke, V.A.; Herling, P.; Passaro, A.; Vigna, G.B.; Volpato, S.; Valacchi, G.; Cervellati, C.; Zuliani, G. Targeted lipidomics distinguishes patient subgroups in mild cognitive impairment (MCI) and late onset Alzheimer’s disease (LOAD). BBA Clin. 2016, 5, 25–28. [Google Scholar] [CrossRef] [Green Version]
- Fabelo, N.; Martín, V.; Santpere, G.; Marín, R.; Torrent, L.; Ferrer, I.; Díaz, M. Severe Alterations in Lipid Composition of Frontal Cortex Lipid Rafts from Parkinson’s Disease and Incidental Parkinson’s Disease. Mol. Med. 2011, 17, 1107–1118. [Google Scholar] [CrossRef]
- Braverman, N.; Steel, G.; Obie, C.; Moser, A.; Moser, H.; Gould, S.J.; Valle, D. Human PEX7 encodes the peroxisomal PTS2 receptor and is responsible for rhizomelic chondrodysplasia punctata. Nat. Genet. 1997, 15, 369–376. [Google Scholar] [CrossRef] [PubMed]
- Stadelmann-Ingrand, S.; Favreliere, S.; Fauconneau, B.; Mauco, G.; Tallineau, C. Plasmalogen degradation by oxidative stress: Production and disappearance of specific fatty aldehydes and fatty α-hydroxyaldehydes. Free Radic. Biol. Med. 2001, 31, 1263–1271. [Google Scholar] [CrossRef]
- Ebenezer, D.L.; Fu, P.; Ramchandran, R.; Ha, A.W.; Putherickal, V.; Sudhadevi, T.; Harijith, A.; Schumacher, F.; Kleuser, B.; Natarajan, V. S1P and plasmalogen derived fatty aldehydes in cellular signaling and functions. Biochim. Biophys. Acta (BBA) Mol. Cell Biol. Lipids 2020, 1865, 158681. [Google Scholar] [CrossRef]
- Zou, Y.; Henry, W.S.; Ricq, E.L.; Graham, E.T.; Phadnis, V.V.; Maretich, P.; Paradkar, S.; Boehnke, N.; Deik, A.A.; Reinhardt, F.; et al. Plasticity of ether lipids promotes ferroptosis susceptibility and evasion. Nature 2020, 585, 603–608. [Google Scholar] [CrossRef] [PubMed]
- Aldrovandi, M.; Conrad, M. Ferroptosis: The Good, the Bad and the Ugly. Cell Res. 2020, 30, 1061–1062. [Google Scholar] [CrossRef] [PubMed]
- Saitoh, M.; Itoh, M.; Takashima, S.; Mizuguchi, M.; Iwamori, M. Phosphatidyl ethanolamine with increased polyunsaturated fatty acids in compensation for plasmalogen defect in the Zellweger syndrome brain. Neurosci. Lett. 2009, 449, 164–167. [Google Scholar] [CrossRef] [PubMed]
- Gross, R.W. High plasmalogen and arachidonic acid content of canine myocardial sarcolemma: A fast atom bombardment mass spectroscopic and gas chromatography-mass spectroscopic characterization. Biochemistry 1984, 23, 158–165. [Google Scholar] [CrossRef]





{kind=link}
{kind=link}
{kind=link}
{kind=link}
| Apoptosis | ||
| Feature | Mammals | C. elegans |
| Phosphotidylserine presentation as “eat me signal” | Inactivation of ATP11C and CDC50A activity promotes PS exposure and cell death [103]. | Inactivation of TAT-1 leads to PS exposure and cell death [98,101,102]. |
| Activation of PLSCR1 via translocation of AIF from mitochondria [97]. | Translocation of WAH-1 from CED-3 cleavage for SCRM-1 activation [92,96]. | |
| Activation of XRC8 via Caspase 3 leads to PS exposure [100]. | Activation of CED-8 via CED-3 leads to PS exposure [98,99]. | |
| Corpse engulfment via phosphotidylserine | LRP1 associates with ABCA1 to activate GULP and further activates Dyn2 for actin reorganization and elongation of cell around apoptotic corpse [111]. | CED-1 associates with CED-7 further activating CED-6 for DYN-1 associated pseudopodal elongation around apoptotic corpse [91,110]. |
| CRKII complexes with DOCK180 and ELMO to activate RAC for cytoskeletal rearrangement and elongation of pseudopodal arms around apoptotic cell [111]. | CED-2 complexes with CED-5 and CED-12 to activate downstream CED-10 for elongation of pseudopodal arms for engulfment of apoptotic corpse [76,108,112,113]. | |
| Phosphotidylinositol in corpse consumption | Phagosome sealing involves many factors, including the complex of PIK3C2A, PIK3C3, and MTM1 that recruit SNX9 and DYN1; this further recruits OCRL to seal phagosome by increasing PIP2, the phospholipid type that accumulates in sealed phagosomes [116,117,123]. | Phagosome sealing involves complexing of PIKI-1, VPS-34, and MTM-1 that recruits LST-4 and DYN-1; this further recruits OCRL-1 to increase PIP2 that accumulates for sealing [119,120]. |
| Ferroptosis | ||
| Feature | Mammals | C. elegans |
| Requirement of oxidized ω-6 polyunsaturated fatty acids | Oxidized PE-AA, PE-AdA and DGLA act as executioners of ferroptosis in cancer cells [43,136,137,138]. | Oxidized DGLA, and to some extent AA, induce ferroptosis of germ cells [43,55,59,60]. |
| Monounsaturated fatty acids in protection from ferroptosis | Exogenous OA is incorporated into the plasma membrane via ACSL3 to promote a ferroptosis-resistant membrane [141]. | Worms fed OA are rescued from dietary DGLA-induced ferroptosis of germ cells [43]. |
| Requirement of iron in ferroptosis | Cancer cells are more resistant to ferroptosis via downregulation or transferrin receptors reducing iron intake involved in the and are rescued from ferroptosis when treated with iron chelation reducing substrate for the fenton reaction [20,151,152]. Iron also acts as a cofactor for lipid oxidizing enzymes to generate ferroptosis initiators [138,147,148]. | Worms fed DGLA in conjunction with iron chelators are rescued from germline ferroptosis reducing fenton reaction substrates and lipid oxidizing enzyme cofactors [43,60]. Iron chelation in older worms increased lifespan of worms reducing frailty-induced ferroptosis [44]. |
| Ether lipids in ferroptosis | Ether depletion through small molecule inhibition of AGPS led to cancer cell ferroptosis when treated with either DGLA or RSL3 [43]. In contrast, ether lipid synthesis inhibition that reduced PUFA levels led to rescue from ferroptosis [167]. | Genetic depletion of ether lipids in ads-1 strain worms are extremely sensitive to DGLA-induced ferroptosis, suggesting a protective role in ferroptosis [43]. |
Publisher’s Note: MDPI stays neutral with regard to jurisdictional claims in published maps and institutional affiliations. |
© 2021 by the authors. Licensee MDPI, Basel, Switzerland. This article is an open access article distributed under the terms and conditions of the Creative Commons Attribution (CC BY) license (http://creativecommons.org/licenses/by/4.0/).
Share and Cite
Perez, M.A.; Watts, J.L. Worms, Fat, and Death: Caenorhabditis elegans Lipid Metabolites Regulate Cell Death. Metabolites 2021, 11, 125. https://doi.org/10.3390/metabo11020125
Perez MA, Watts JL. Worms, Fat, and Death: Caenorhabditis elegans Lipid Metabolites Regulate Cell Death. Metabolites. 2021; 11(2):125. https://doi.org/10.3390/metabo11020125
Chicago/Turabian StylePerez, Marcos A., and Jennifer L. Watts. 2021. "Worms, Fat, and Death: Caenorhabditis elegans Lipid Metabolites Regulate Cell Death" Metabolites 11, no. 2: 125. https://doi.org/10.3390/metabo11020125
APA StylePerez, M. A., & Watts, J. L. (2021). Worms, Fat, and Death: Caenorhabditis elegans Lipid Metabolites Regulate Cell Death. Metabolites, 11(2), 125. https://doi.org/10.3390/metabo11020125