
A Comprehensive Targeted Metabolomics Assay for Crop Plant Sample Analysis



Abstract
:1. Introduction
2. Results and Discussion
2.1. Optimization of Extraction of Metabolites from NIST Standard Reference Material (SRM)
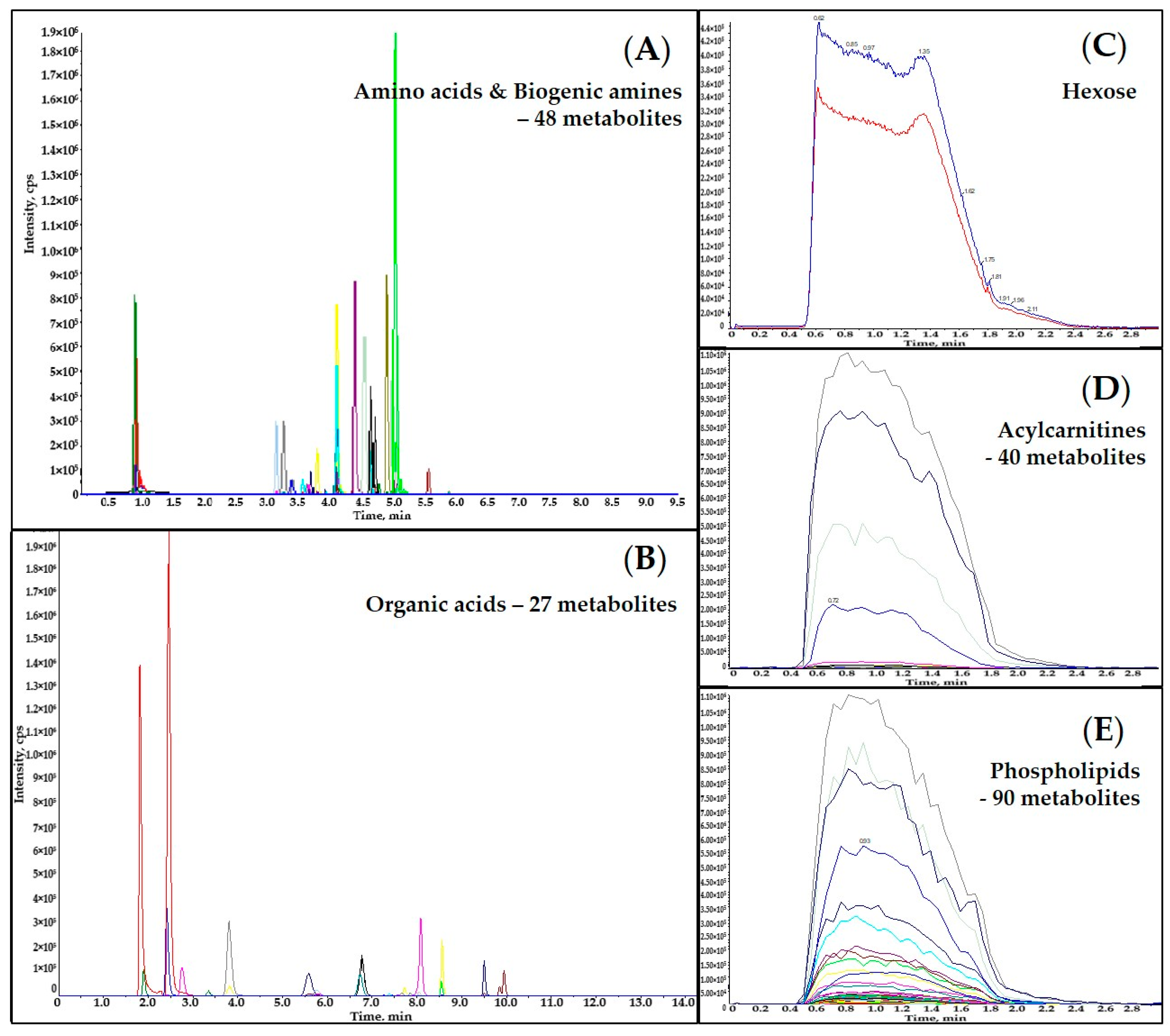
2.2. Liquid Chromatography
2.3. Assay Validation
2.4. Assay Application to Crop Plant Samples
3. Materials and Methods
3.1. Plant Material
3.2. Chemicals, Reagents and Materials
3.3. Stock Solutions, Internal Standard (ISTD) Mixtures, and Calibration Curve Standards
3.4. Plant Extraction
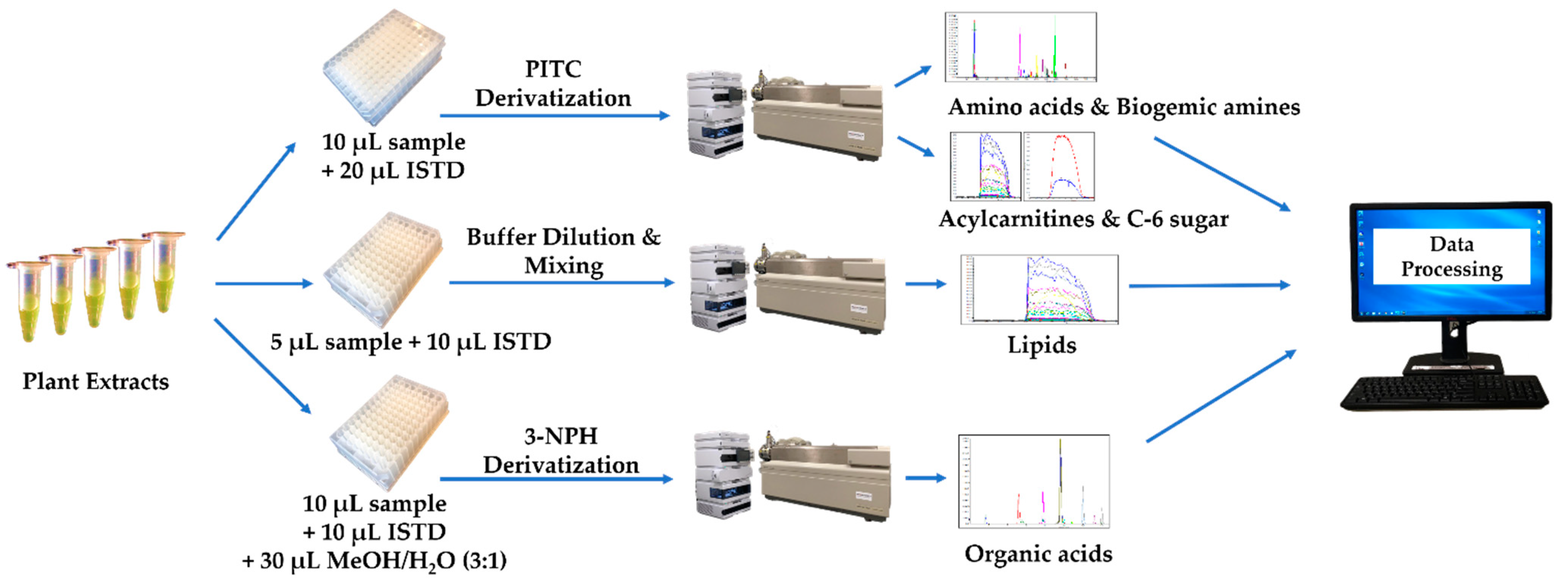
3.5. Plant Extract Analysis
3.6. LC/DFI-MS/MS Analysis
3.7. Method Validation
3.7.1. Calibration Regression
3.7.2. Accuracy and Precision
3.7.3. Recovery
3.7.4. Limits of Detection (LOD) and Quantification (LOQ)
4. Conclusions
Supplementary Materials
Author Contributions
Funding
Institutional Review Board Statement
Informed Consent Statement
Data Availability Statement
Acknowledgments
Conflicts of Interest
References
- Foito, A.; Stewart, D. Metabolomics: A High-throughput Screen for Biochemical and Bioactivity Diversity in Plants and Crops. Curr. Pharm. Des. 2018, 24, 2043–2054. [Google Scholar] [CrossRef] [PubMed]
- Raamsdonk, L.M.; Teusink, B.; Broadhurst, D.; Zhang, N.; Hayes, A.; Walsh, M.C.; Berden, J.A.; Brindle, K.M.; Kell, D.B.; Rowland, J.J.; et al. A functional genomics strategy that uses metabolome data to reveal the phenotype of silent mutations. Nat. Biotechnol. 2001, 19, 45–50. [Google Scholar] [CrossRef] [PubMed]
- Roessner, U.; Luedemann, A.; Brust, D.; Fiehn, O.; Linke, T.; Willmitzer, L.; Fernie, A.R. Metabolic Profiling Allows Comprehensive Phenotyping of Genetically or Environmentally Modified Plant Systems. Plant Cell 2001, 13, 11–29. [Google Scholar] [CrossRef] [Green Version]
- Fiehn, O.; Kopka, J.; Dörmann, P.; Altmann, T.; Trethewey, R.N.; Willmitzer, L. Metabolite profiling for plant functional genomics. Nat. Biotechnol. 2000, 18, 1157–1161. [Google Scholar] [CrossRef]
- Roessner, U.; Willmitzer, L.; Fernie, A.R. High-Resolution Metabolic Phenotyping of Genetically and Environmentally Diverse Potato Tuber Systems. Identification of Phenocopies. Plant Physiol. 2001, 127, 749–764. [Google Scholar] [CrossRef]
- Ott, K.-H.; Araníbar, N.; Singh, B.; Stockton, G.W. Metabonomics classifies pathways affected by bioactive compounds. Artificial neural network classification of NMR spectra of plant extracts. Phytochemistry 2003, 62, 971–985. [Google Scholar] [CrossRef]
- Dias, D.A.; Urban, S.; Roessner, U. A Historical Overview of Natural Products in Drug Discovery. Metabolites 2012, 2, 303–336. [Google Scholar] [CrossRef] [Green Version]
- Mukherjee, P.K.; Harwansh, R.K.; Bahadur, S.; Biswas, S.; Kuchibhatla, L.N.; Tetali, S.D.; Raghavendra, A.S. Metabolomics of medicinal plants—A versatile tool for standardization of herbal products and quality evaluation of Ayurvedic formulations. Curr. Sci. 2016, 111, 1624–1630. [Google Scholar] [CrossRef]
- Graziani, V.; Scognamiglio, M.; Belli, V.; Esposito, A.; D’Abrosca, B.; Chambery, A.; Russo, R.; Panella, M.; Russo, A.; Ciardiello, F.; et al. Metabolomic approach for a rapid identification of natural products with cytotoxic activity against human colorectal cancer cells. Sci. Rep. 2018, 8, 5309. [Google Scholar] [CrossRef] [Green Version]
- Tuyiringire, N.; Tusubira, D.; Munyampundu, J.-P.; Tolo, C.U.; Muvunyi, C.M.; Ogwang, P.E. Application of metabolomics to drug discovery and understanding the mechanisms of action of medicinal plants with anti-tuberculosis activity. Clin. Transl. Med. 2018, 7, 29. [Google Scholar] [CrossRef]
- Selegato, D.M.; Pilon, A.C.; Carnevale Neto, F. Plant Metabolomics Using NMR Spectroscopy. In Methods in Molecular Biology; Humana Press Inc.: Totowa, NJ, USA, 2019; Volume 2037, pp. 345–362. [Google Scholar]
- Krishnan, P.; Kruger, N.J.; Ratcliffe, R.G. Metabolite fingerprinting and profiling in plants using NMR. J. Exp. Bot. 2005, 56, 255–265. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Winning, H.; Roldán-Marín, E.; Dragsted, L.O.; Viereck, N.; Poulsen, M.; Sánchez-Moreno, C.; Cano, M.P.; Engelsen, S.B. An exploratory NMR nutri-metabonomic investigation reveals dimethyl sulfone as a dietary biomarker for onion intake. Analyst 2009, 134, 2344–2351. [Google Scholar] [CrossRef]
- Chen, M.-L.; Huang, Y.-Q.; Liu, J.-Q.; Yuan, B.-F.; Feng, Y.-Q. Highly sensitive profiling assay of acidic plant hormones using a novel mass probe by capillary electrophoresis-time of flight-mass spectrometry. J. Chromatogr. B 2011, 879, 938–944. [Google Scholar] [CrossRef]
- Trenkamp, S.; Eckes, P.; Busch, M.; Fernie, A.R. Temporally resolved GC-MS-based metabolic profiling of herbicide treated plants treated reveals that changes in polar primary metabolites alone can distinguish herbicides of differing mode of action. Metabolomics 2009, 5, 277–291. [Google Scholar] [CrossRef] [Green Version]
- Wang, Y.; Xu, L.; Shen, H.; Wang, J.; Liu, W.; Zhu, X.; Wang, R.; Sun, X.; Liu, L. Metabolomic analysis with GC-MS to reveal potential metabolites and biological pathways involved in Pb & Cd stress response of radish roots. Sci. Rep. 2015, 5, 18296. [Google Scholar] [CrossRef] [Green Version]
- Dai, T.; Chang, X.; Hu, Z.; Liang, L.; Sun, M.; Liu, P.; Liu, X. Untargeted Metabolomics Based on GC-MS and Chemometrics: A New Tool for the Early Diagnosis of Strawberry Anthracnose Caused by Colletotrichum theobromicola. Plant Dis. 2019, 103, 2541–2547. [Google Scholar] [CrossRef] [PubMed]
- Li, X.; Zhang, X.; Ye, L.; Kang, Z.; Jia, D.; Yang, L.; Zhang, B. LC-MS-Based Metabolomic Approach Revealed the Significantly Different Metabolic Profiles of Five Commercial Truffle Species. Front. Microbiol. 2019, 10, 2227. [Google Scholar] [CrossRef]
- Ghosson, H.; Schwarzenberg, A.; Jamois, F.; Yvin, J.-C. Simultaneous untargeted and targeted metabolomics profiling of underivatized primary metabolites in sulfur-deficient barley by ultra-high performance liquid chromatography-quadrupole/time-of-flight mass spectrometry. Plant Methods 2018, 14, 62. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Chen, W.; Gong, L.; Guo, Z.; Wang, W.; Zhang, H.; Liu, X.; Yu, S.; Xiong, L.; Luoa, J. A novel integrated method for large-scale detection, identification, and quantification of widely targeted metabolites: Application in the study of rice metabolomics. Mol. Plant 2013, 6, 1769–1780. [Google Scholar] [CrossRef] [Green Version]
- Sato, S.; Soga, T.; Nishioka, T.; Tomita, M. Simultaneous determination of the main metabolites in rice leaves using capillary electrophoresis mass spectrometry and capillary electrophoresis diode array detection. Plant J. 2004, 40, 151–163. [Google Scholar] [CrossRef]
- Harada, K.; Ohyama, Y.; Tabushi, T.; Kobayashi, A.; Fukusaki, E. Quantitative analysis of anionic metabolites for Catharanthus roseus by capillary electrophoresis using sulfonated capillary coupled with electrospray ionization-tandem mass spectrometry. J. Biosci. Bioeng. 2008, 105, 249–260. [Google Scholar] [CrossRef]
- Zhao, J.; Hu, C.; Zeng, J.; Zhao, Y.; Zhang, J.; Chang, Y.; Li, L.; Zhao, C.; Lu, X.; Xu, G. Study of polar metabolites in tobacco from different geographical origins by using capillary electrophoresis–mass spectrometry. Metabolomics 2014, 10, 805–815. [Google Scholar] [CrossRef]
- Menzel, W.I.; Chen, W.-P.; Hegeman, A.D.; Cohen, J.D. Qualitative and Quantitative Screening of Amino Acids in Plant Tissues in High-Throughput Phenotyping in Plants: Methods and Protocols; Normanly, J., Ed.; Humana Press: Totowa, NJ, USA, 2012; pp. 165–178. ISBN 978-1-61779-995-2. [Google Scholar] [CrossRef]
- Moussa, T.A.A.; Almaghrabi, O.A. Fatty acid constituents of Peganum harmala plant using Gas Chromatography–Mass Spectroscopy. Saudi J. Biol. Sci. 2016, 23, 397–403. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Ratiu, I.A.; Al-Suod, H.; Ligor, M.; Ligor, T.; Krakowska, A.; Górecki, R.; Buszewski, B. Simultaneous Determination of Cyclitols and Sugars Following a Comprehensive Investigation of 40 Plants. Food Anal. Methods 2019, 12, 1466–1478. [Google Scholar] [CrossRef] [Green Version]
- Pan, X.; Wang, X. Profiling of plant hormones by mass spectrometry. J. Chromatogr. B 2009, 877, 2806–2813. [Google Scholar] [CrossRef]
- Bai, Y.; Du, F.; Bai, Y.; Liu, H. Determination strategies of phytohormones: Recent advances. Anal. Methods 2010, 2, 1867–1873. [Google Scholar] [CrossRef]
- Birkemeyer, C.; Kolasa, A.; Kopka, J. Comprehensive chemical derivatization for gas chromatography-mass spectrometry-based multi-targeted profiling of the major phytohormones. J. Chromatogr. A 2003, 993, 89–102. [Google Scholar] [CrossRef]
- Nguyen, T.-D.; Riordan-Short, S.; Dang, T.-T.T.; O’Brien, R.; Noestheden, M. Quantitation of Select Terpenes/Terpenoids and Nicotine Using Gas Chromatography–Mass Spectrometry with High-Temperature Headspace Sampling. ACS Omega 2020, 5, 5565–5573. [Google Scholar] [CrossRef]
- Jordán, M.J.; Tandon, K.; Shaw, P.E.; Goodner, K.L. Aromatic profile of aqueous banana essence and banana fruit by gas chromatography-mass spectrometry (GC-MS) and gas chromatography-olfactometry (GC-O). J. Agric. Food Chem. 2001, 49, 4813–4817. [Google Scholar] [CrossRef]
- Marques, F.G.; de Oliveira Neto, J.R.; da Cunha, L.C.; de Paula, J.R.; Bara, M.T.F. Identification of terpenes and phytosterols in Dipteryx alata (baru) oil seeds obtained through pressing. Rev. Bras. Farmacogn. 2015, 25, 522–525. [Google Scholar] [CrossRef] [Green Version]
- French, K.E.; Harvey, J.; McCullagh, J.S.O. Targeted and Untargeted Metabolic Profiling of Wild Grassland Plants identifies Antibiotic and Anthelmintic Compounds Targeting Pathogen Physiology, Metabolism and Reproduction. Sci. Rep. 2018, 8, 1695. [Google Scholar] [CrossRef] [Green Version]
- De Vos, R.C.H.; Moco, S.; Lommen, A.; Keurentjes, J.J.B.; Bino, R.J.; Hall, R.D. Untargeted large-scale plant metabolomics using liquid chromatography coupled to mass spectrometry. Nat. Protoc. 2007, 2, 778–791. [Google Scholar] [CrossRef]
- Carreno-Quintero, N.; Acharjee, A.; Maliepaard, C.; Bachem, C.W.B.; Mumm, R.; Bouwmeester, H.; Visser, R.G.F.; Keurentjes, J.J.B. Untargeted Metabolic Quantitative Trait Loci Analyses Reveal a Relationship between Primary Metabolism and Potato Tuber Quality. Plant Physiol. 2012, 158, 1306–1318. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Baniasadi, H.; Vlahakis, C.; Hazebroek, J.; Zhong, C.; Asiago, V. Effect of Environment and Genotype on Commercial Maize Hybrids Using LC/MS-Based Metabolomics. J. Agric. Food Chem. 2014, 62, 1412–1422. [Google Scholar] [CrossRef]
- Tohge, T.; Fernie, A.R. Metabolomics-Inspired Insight into Developmental, Environmental and Genetic Aspects of Tomato Fruit Chemical Composition and Quality. Plant Cell Physiol. 2015, 56, 1681–1696. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- David, L.; Kang, J.; Chen, S. Targeted Metabolomics of Plant Hormones and Redox Metabolites in Stomatal Immunity. Methods Mol. Biol. 2020, 2085, 79–92. [Google Scholar] [CrossRef]
- Jander, G.; Norris, S.R.; Joshi, V.; Fraga, M.; Rugg, A.; Yu, S.; Li, L.; Last, R.L. Application of a high-throughput HPLC-MS/MS assay to Arabidopsis mutant screening; evidence that threonine aldolase plays a role in seed nutritional quality. Plant J. 2004, 39, 465–475. [Google Scholar] [CrossRef]
- Liu, Z.; Rochfort, S. A fast liquid chromatography-mass spectrometry (LC-MS) method for quantification of major polar metabolites in plants. J. Chromatogr. B 2013, 912, 8–15. [Google Scholar] [CrossRef] [PubMed]
- Tarazona, P.; Feussner, K.; Feussner, I. An enhanced plant lipidomics method based on multiplexed liquid chromatography-mass spectrometry reveals additional insights into cold- and drought-induced membrane remodeling. Plant J. 2015, 84, 621–633. [Google Scholar] [CrossRef]
- Bajkacz, S.; Baranowska, I.; Buszewski, B.; Kowalski, B.; Ligor, M. Determination of Flavonoids and Phenolic Acids in Plant Materials Using SLE-SPE-UHPLC-MS/MS Method. Food Anal. Methods 2018, 11, 3563–3575. [Google Scholar] [CrossRef] [Green Version]
- Zheng, J.; Zhang, L.; Johnson, M.; Mandal, R.; Wishart, D.S. Comprehensive Targeted Metabolomic Assay for Urine Analysis. Anal. Chem. 2020, 92, 10627–10634. [Google Scholar] [CrossRef]
- Zheng, J.; Mandal, R.; Wishart, D.S. A sensitive, high-throughput LC-MS/MS method for measuring catecholamines in low volume serum. Anal. Chim. Acta 2018, 1037, 159–167. [Google Scholar] [CrossRef] [PubMed]
- Castellano-Escuder, P.; González-Domínguez, R.; Wishart, D.S.; Andrés-Lacueva, C.; Sánchez-Pla, A. FOBI: An ontology to represent food intake data and associate it with metabolomic data. Database 2020, 2020, baaa033. [Google Scholar] [CrossRef] [PubMed]
- Thompson, J.W.; Adams, K.J.; Adamski, J.; Asad, Y.; Borts, D.; Bowden, J.A.; Byram, G.; Dang, V.; Dunn, W.B.; Fernandez, F.; et al. International Ring Trial of a High Resolution Targeted Metabolomics and Lipidomics Platform for Serum and Plasma Analysis. Anal. Chem. 2019, 91, 14407–14416. [Google Scholar] [CrossRef] [PubMed]
- Khaniani, Y.; Lipfert, M.; Bhattacharyya, D.; Pineiro, R.P.; Zheng, J.; Wishart, D.; Khaniani, Y.; Lipfert, M.; Bhattacharyya, D.; Perez Pineiro, R.; et al. A Simple and Convenient Synthesis of Unlabeled and 13C-Labeled 3-(3-Hydroxyphenyl)-3-Hydroxypropionic Acid and Its Quantification in Human Urine Samples. Metabolites 2018, 8, 80. [Google Scholar] [CrossRef] [PubMed] [Green Version]



{kind=link}
{kind=link}
| Metabolite | Concentration Comparison Ratios to 25 mg-Aliquots | ||
|---|---|---|---|
| 12.5 mg-Aliquots | 40 mg-Aliquots | 50 mg-Aliquots | |
| Succinic acid | 0.495 | 1.66 | 1.89 |
| Glutamine | 0.517 | 1.62 | 1.97 |
| Choline | 0.517 | 1.61 | 2.01 |
| Carnitine (C0) | 0.509 | 1.58 | 1.96 |
| Hexose | 0.498 | 1.60 | 1.91 |
| PC aa C38:6 | 0.494 | 1.58 | 2.06 |
| Shikimic acid | 0.516 | 1.29 | 1.33 |
| Glyceric acid | 0.517 | 1.28 | 1.31 |
| Malic acid | 0.509 | 1.27 | 1.44 |
| Alanine | 0.491 | 1.46 | 1.76 |
| Arginine | 0.511 | 1.48 | 1.65 |
| LysoPC a C14:0 | 0.505 | 1.44 | 1.71 |
| Analyte | Correlation Coefficient (R2) | LOD (μM) | LLOQ (μM) | ULOQ (μM) |
|---|---|---|---|---|
| Glycine | 0.9990 | 0.859 | 25.0 | 2000 |
| Proline | 0.9994 | 0.136 | 10.0 | 800 |
| Spermidine | 0.9998 | 0.00954 | 0.250 | 20.0 |
| Choline | 0.9992 | 0.294 | 2.50 | 200 |
| Shikimic acid | 0.9992 | 0.321 | 1.25 | 100 |
| Malic acid | 0.9998 | 0.0574 | 1.25 | 100 |
| Analyte | Intra-Day | Inter-Day | |||
|---|---|---|---|---|---|
| Fortified Concentration (μM) | Accuracy (%) | CV (%) | Accuracy (%) | CV (%) | |
| Glycine | 125 | 105 | 2.52 | 98.0 | 4.23 |
| 500 | 95.7 | 2.43 | 103 | 1.33 | |
| 1500 | 96.3 | 3.90 | 97.5 | 3.25 | |
| Proline | 50 | 112 | 1.63 | 96.7 | 4.36 |
| 200 | 98.6 | 2.69 | 101 | 0.148 | |
| 600 | 96.9 | 2.07 | 101 | 2.44 | |
| Spermidine | 1.25 | 103 | 2.29 | 107 | 8.30 |
| 5.00 | 101 | 2.31 | 98.9 | 3.79 | |
| 15.0 | 98.1 | 3.37 | 95.9 | 5.15 | |
| Choline | 12.5 | 111 | 2.03 | 101 | 2.57 |
| 50.0 | 100 | 2.58 | 101 | 0.794 | |
| 150 | 103 | 2.42 | 101 | 1.95 | |
| Shikimic acid | 2.50 | 95.7 | 1.68 | 97.0 | 2.09 |
| 10.0 | 96.0 | 0.833 | 92.0 | 2.30 | |
| 50.0 | 101 | 2.65 | 98.4 | 0.704 | |
| Malic acid | 2.50 | 97.0 | 3.23 | 99.9 | 1.33 |
| 10.0 | 102 | 2.08 | 103 | 0.900 | |
| 50.0 | 104 | 0.223 | 102 | 2.67 | |
| Analyte | Spiked Concentrations (μM) | Calculated Concentration (μM) | Recovery (%) |
|---|---|---|---|
| Glycine | 125 | 129 | 103 |
| 500 | 530 | 106 | |
| 1500 | 1646 | 110 | |
| Proline | 50 | 53.3 | 107 |
| 200 | 224 | 112 | |
| 600 | 616 | 103 | |
| Spermidine | 1.25 | 1.39 | 111 |
| 5.00 | 5.58 | 112 | |
| 15.0 | 17.1 | 114 | |
| Choline | 12.5 | 12.5 | 100 |
| 50.0 | 56.8 | 114 | |
| 150 | 158 | 105 | |
| Shikimic acid | 2.50 | 2.58 | 103 |
| 10.0 | 10.3 | 103 | |
| 50.0 | 46.0 | 91.9 | |
| Malic acid | 2.50 | 2.83 | 113 |
| 10.0 | 10.1 | 101 | |
| 50.0 | 53.9 | 108 |
| Analyte | Accuracy (%) | CV (%) | Recovery (%) | LOD (μM) |
|---|---|---|---|---|
| LysoPC a C18:0 | 105 | 5.17 | 104 | 0.274 |
| PC aa C36:0 | 104 | 6.22 | 101 | 0.0898 |
| C0 | 92.6 | 4.28 | 98.5 | 0.222 |
| Hexose | 96.3 | 2.91 | 103 | 18.5 |
| Metabolite Class | Number of Metabolites Detected and Quantified | |||
|---|---|---|---|---|
| NIST® SRM® 1575a Pine Needles | Canola Root Samples | Commercial Cannabis Buds | Spruce and Pine Needles | |
| Amino acids and derivatives | 24 | 27 | 27 | 26 |
| Biogenic amines and derivatives | 8 | 12 | 13 | 14 |
| Organic acids (phytohormones included) | 15 | 21 | 24 | 18 |
| Acylcarnitines | 1 | 6 | 1 | 1 |
| Phospholipids | 44 | 43 | 12 | 46 |
| Hexose | 1 | 1 | 1 | 1 |
| Total | 93 | 110 | 78 | 106 |
Publisher’s Note: MDPI stays neutral with regard to jurisdictional claims in published maps and institutional affiliations. |
© 2021 by the authors. Licensee MDPI, Basel, Switzerland. This article is an open access article distributed under the terms and conditions of the Creative Commons Attribution (CC BY) license (https://creativecommons.org/licenses/by/4.0/).
Share and Cite
Zheng, J.; Johnson, M.; Mandal, R.; Wishart, D.S. A Comprehensive Targeted Metabolomics Assay for Crop Plant Sample Analysis. Metabolites 2021, 11, 303. https://doi.org/10.3390/metabo11050303
Zheng J, Johnson M, Mandal R, Wishart DS. A Comprehensive Targeted Metabolomics Assay for Crop Plant Sample Analysis. Metabolites. 2021; 11(5):303. https://doi.org/10.3390/metabo11050303
Chicago/Turabian StyleZheng, Jiamin, Mathew Johnson, Rupasri Mandal, and David S. Wishart. 2021. "A Comprehensive Targeted Metabolomics Assay for Crop Plant Sample Analysis" Metabolites 11, no. 5: 303. https://doi.org/10.3390/metabo11050303
APA StyleZheng, J., Johnson, M., Mandal, R., & Wishart, D. S. (2021). A Comprehensive Targeted Metabolomics Assay for Crop Plant Sample Analysis. Metabolites, 11(5), 303. https://doi.org/10.3390/metabo11050303