


Early Life Polychlorinated Biphenyl 126 Exposure Disrupts Gut Microbiota and Metabolic Homeostasis in Mice Fed with High-Fat Diet in Adulthood
 , , and
, , and
Abstract
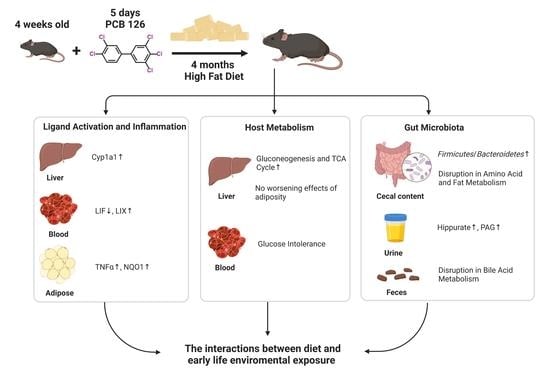
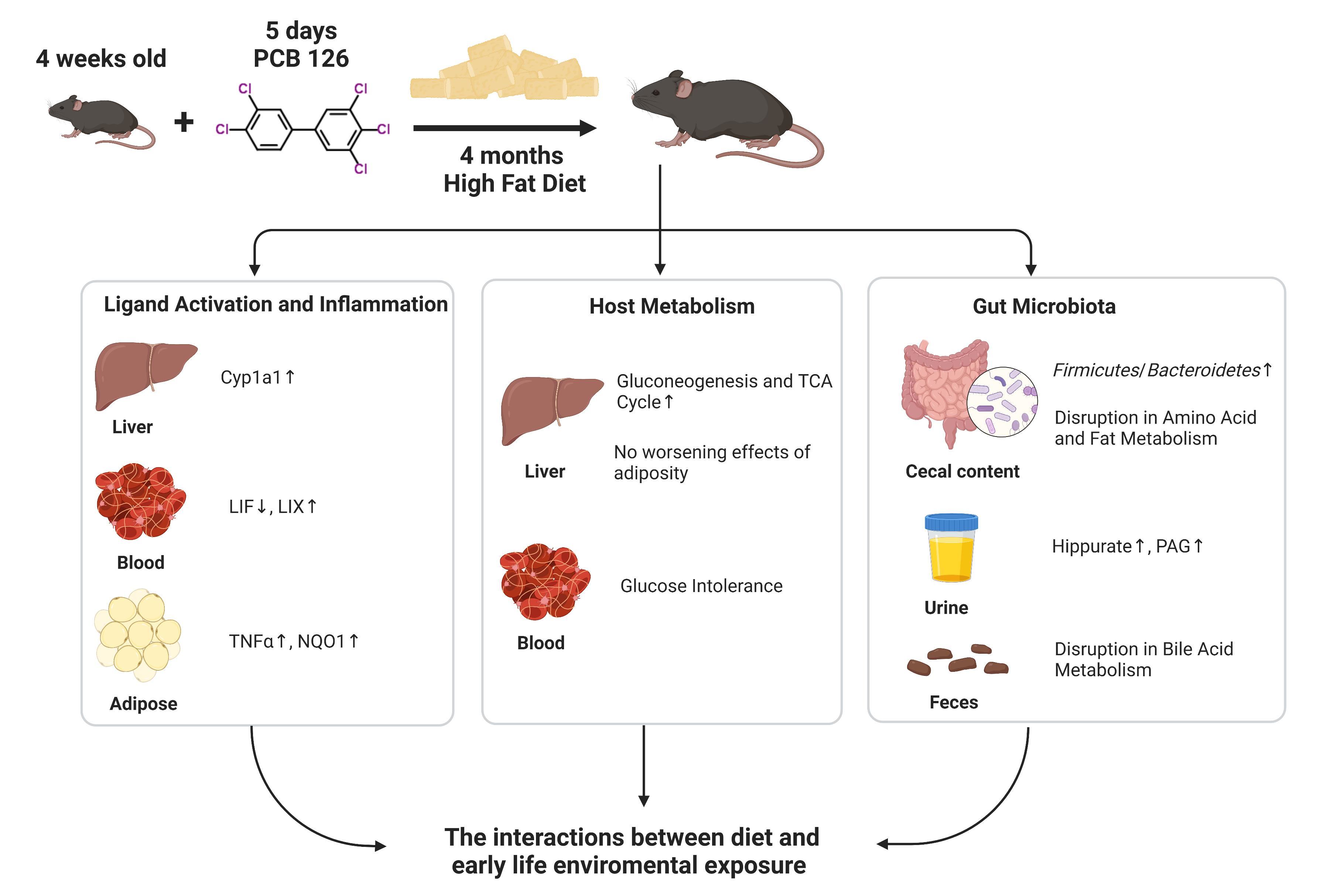
:
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
1. Introduction
2. Materials and Methods
2.1. Chemicals
2.2. Animals and Diets
2.3. Histopathology
2.4. Blood Clinical Biochemistry and Cytokine Analysis
2.5. Glucose Tolerance Test
2.6. Liver Triglyceride and Glutathione Quantification
2.7. Tissue RNA Isolation and qPCR
2.8. 1H NMR Based Metabolomics Experiments
2.9. Bile Acid Quantitation by UPLC-MS/MS
2.10. Metagenomic Analysis
2.11. Statistics
3. Results
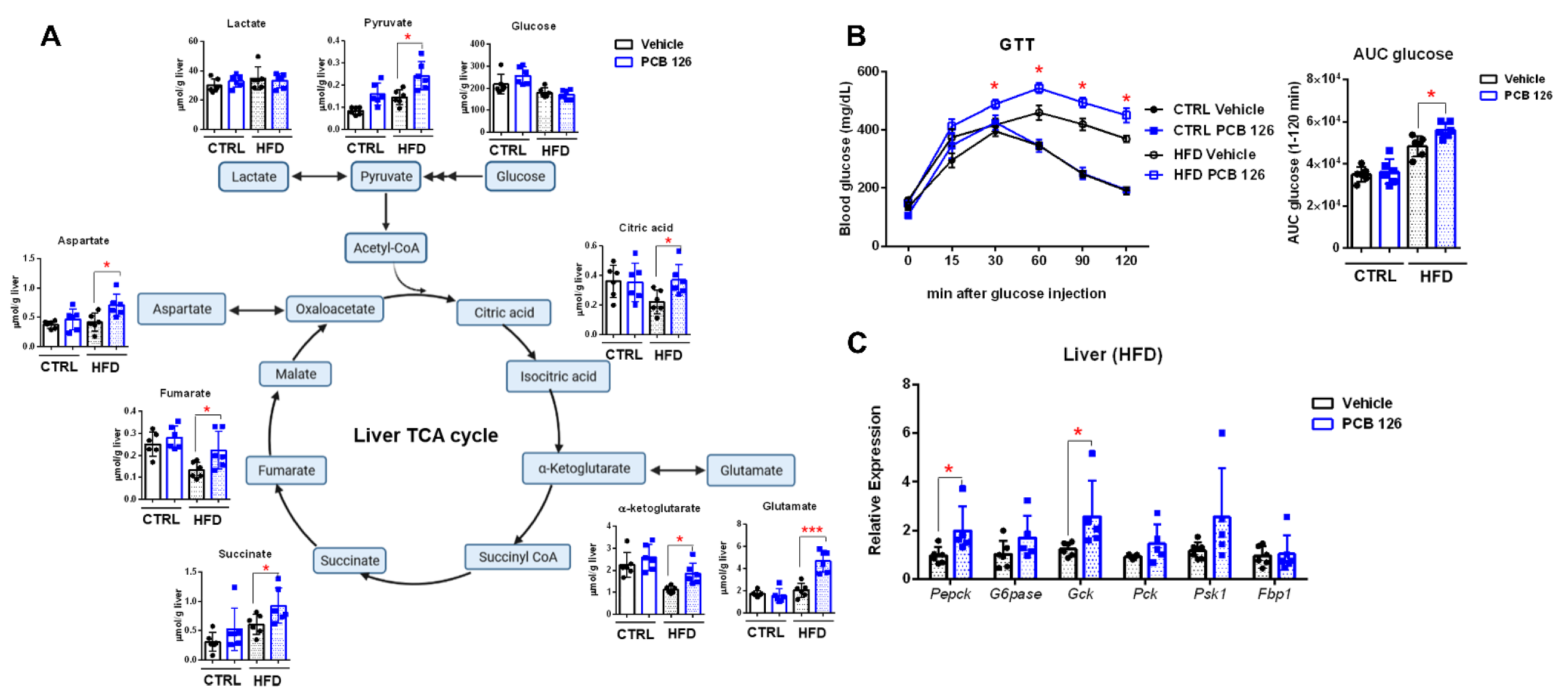
3.1. Early Life PCB 126 Exposure Persistently Affects AHR Signaling and Liver Nucleotide Metabolism in Both CTRL and HFD-Fed Mice in Adulthood
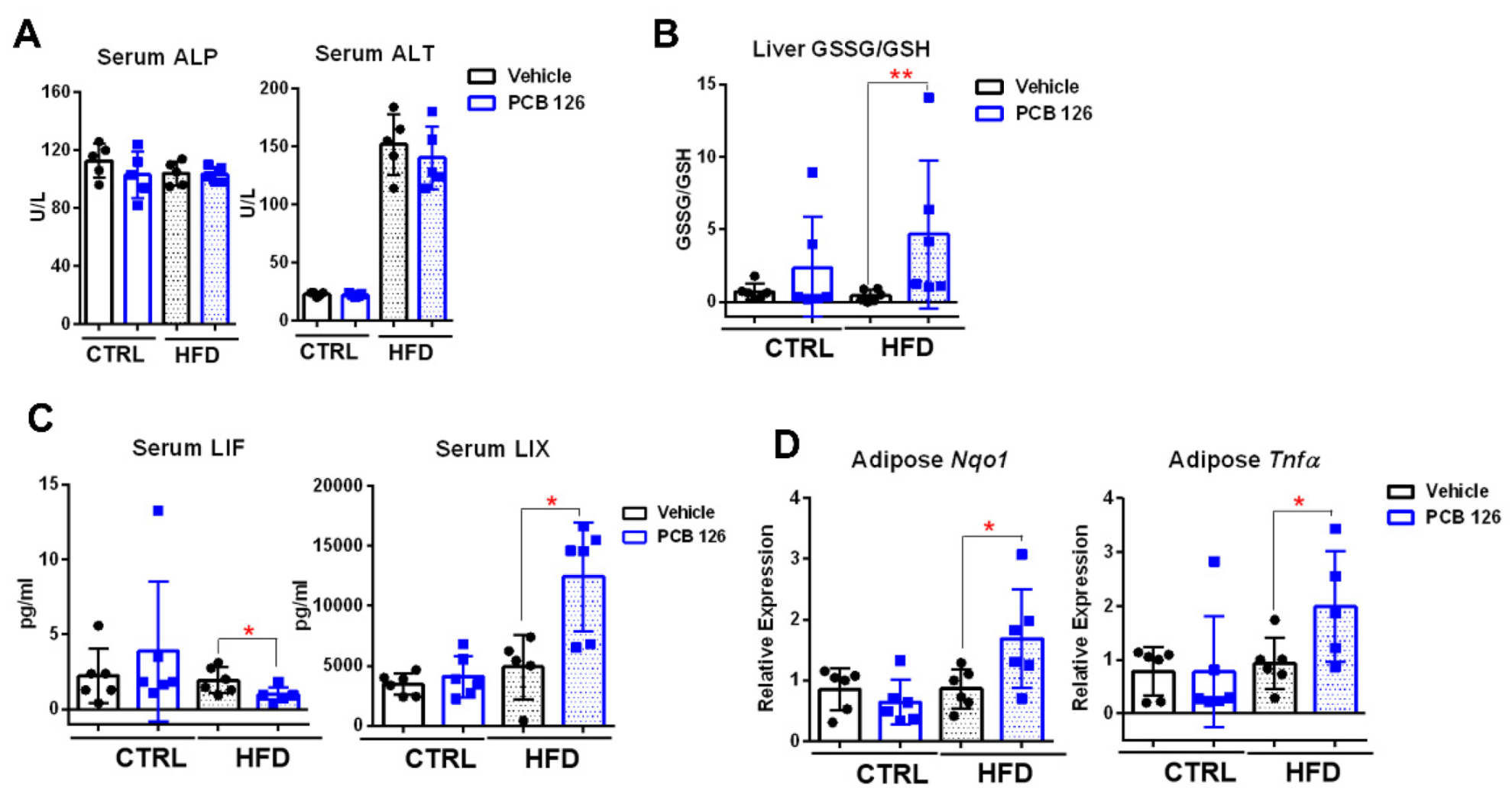
3.2. Early Life PCB 126 Exposure Causes Inflammatory Response in HFD-Fed Mice in Adulthood
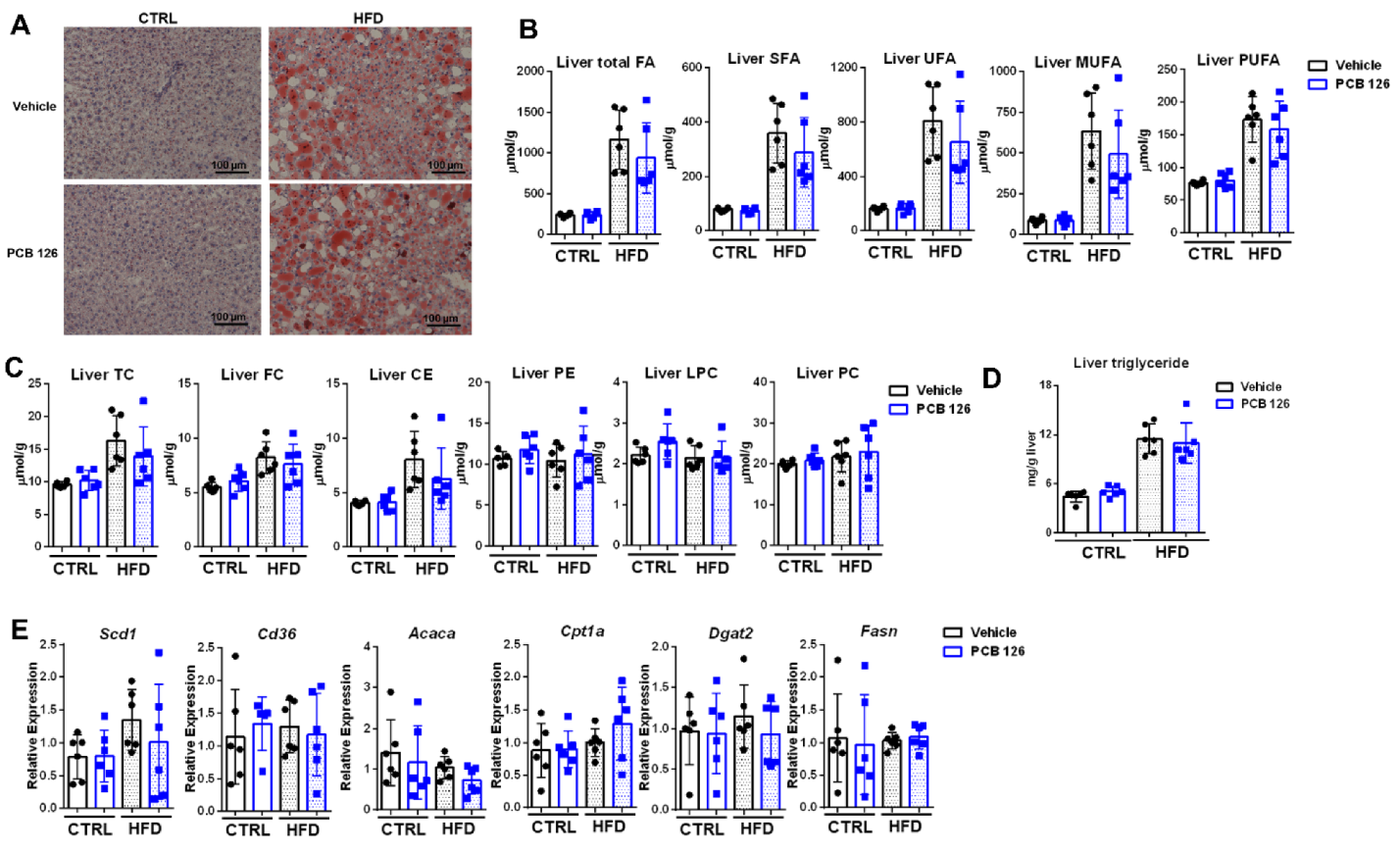
3.3. Early Life PCB 126 Exposure Does Not Worsen the Effects of HFD Related to Adiposity in Adulthood
3.4. Early Life PCB 126 Exposure Aggravates Impaired Glucose Homeostasis in HFD-Fed Mice in Adulthood
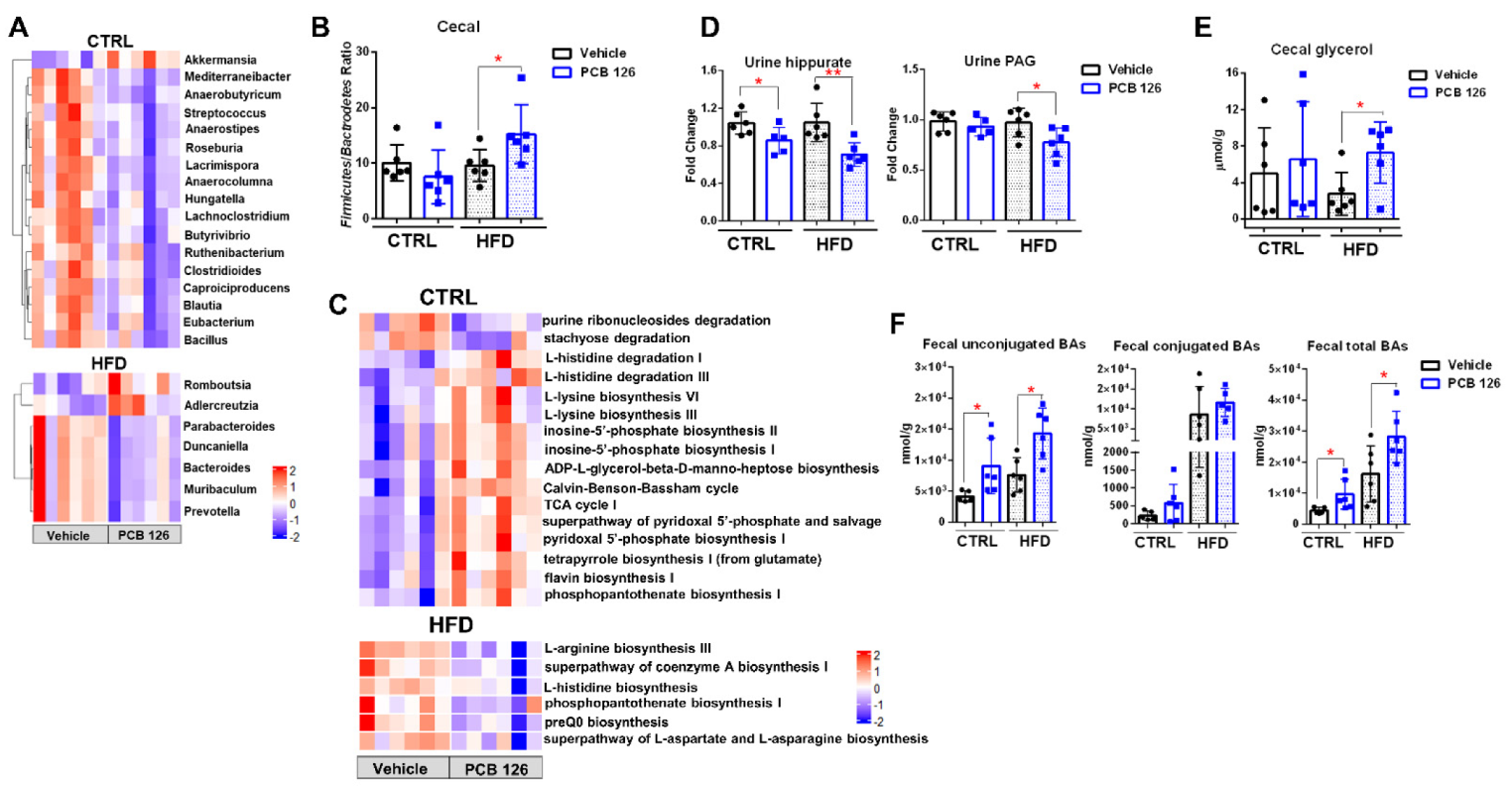
3.5. Early Life PCB 126 Exposure Results in Diet-Dependent Changes in Bacteria Community, Gene Levels, and Metabolism in Adulthood
4. Discussion
5. Conclusions
Supplementary Materials
Author Contributions
Funding
Institutional Review Board Statement
Informed Consent Statement
Data Availability Statement
Conflicts of Interest
References
- Guo, W.J.; Pan, B.H.; Sakkiah, S.; Yavas, G.; Ge, W.G.; Zou, W.; Tong, W.D.; Hong, H.X. Persistent organic pollutants in food: Contamination sources, health effects and detection methods. Int. J. Environ. Res. 2019, 16, 4361. [Google Scholar] [CrossRef] [PubMed]
- Lee, Y.M.; Kim, K.S.; Jacobs, D.R.; Lee, D.H. Persistent organic pollutants in adipose tissue should be considered in obesity research. Obes. Rev. 2017, 18, 129–139. [Google Scholar] [CrossRef] [PubMed]
- Ruzzin, J. Public health concern behind the exposure to persistent organic pollutants and the risk of metabolic diseases. BMC Public Health 2012, 12, 298. [Google Scholar] [CrossRef] [PubMed]
- Koual, M.; Cano-Sancho, G.; Bats, A.S.; Tomkiewicz, C.; Kaddouch-Amar, Y.; Douay-Hauser, N.; Ngo, C.; Bonsang, H.; Delomenie, M.; Lecuru, F.; et al. Associations between persistent organic pollutants and risk of breast cancer metastasis. Environ. Int. 2019, 132, 105028. [Google Scholar] [CrossRef]
- Ren, A.G.; Qiu, X.H.; Jin, L.; Ma, J.; Li, Z.W.; Zhang, L.; Zhu, H.P.; Finnell, R.H.; Zhu, T. Association of selected persistent organic pollutants in the placenta with the risk of neural tube defects. Proc. Natl. Acad. Sci. USA 2011, 108, 12770–12775. [Google Scholar] [CrossRef]
- Crinnion, W.J. Polychlorinated biphenyls: Persistent pollutants with immunological, neurological, and endocrinological consequences. Altern. Med. Rev. 2011, 16, 5–13. [Google Scholar]
- Safe, S.; Bandiera, S.; Sawyer, T.; Robertson, L.; Safe, L.; Parkinson, A.; Thomas, P.E.; Ryan, D.E.; Reik, L.M.; Levin, W.; et al. PCBs—Structure-function-relationships and mechanism of action. Environ. Health Perspect. 1985, 60, 47–56. [Google Scholar]
- ATSDR. Toxicological Profile for Polychlorinated Biphenyls (PCBs); U.S. Department of Health and Human Services, Public Health Service: Atlanta, GA, USA, 2000.
- Jin, J.; Wahlang, B.; Shi, H.X.; Hardesty, J.E.; Falkner, K.C.; Head, K.Z.; Srivastava, S.; Merchant, M.L.; Rai, S.N.; Cave, M.C.; et al. Dioxin-like and non-dioxin-like PCBs differentially regulate the hepatic proteome and modify diet-induced nonalcoholic fatty liver disease severity. Med. Chem. Res. 2020, 29, 1247–1263. [Google Scholar] [CrossRef]
- Loiola, R.A.; dos Anjos, F.M.; Shimada, A.L.; Cruz, W.S.; Drewes, C.C.; Rodrigues, S.F.; Cardozo, K.H.M.; Carvalho, V.M.; Pinto, E.; Farsky, S.H. Long-term in vivo polychlorinated biphenyl 126 exposure induces oxidative stress and alters proteomic profile on islets of Langerhans. Sci. Rep. 2016, 6, 27882. [Google Scholar] [CrossRef]
- Eti, N.A.; Flor, S.; Iqbal, K.; Scott, R.L.; Klenov, V.E.; Gibson-Corley, K.N.; Soares, M.J.; Ludewig, G.; Robertson, L.W. PCB126 induced toxic actions on liver energy metabolism is mediated by AhR in rats. Toxicology 2022, 466, 153054. [Google Scholar] [CrossRef]
- Lai, I.; Chai, Y.T.; Simmons, D.; Luthe, G.; Coleman, M.C.; Spitz, D.; Haschek, W.M.; Ludewig, G.; Robertson, L.W. Acute toxicity of 3,3′,4,4′,5-pentachlorobiphenyl (PCB 126) in male Sprague-Dawley rats: Effects on hepatic oxidative stress, glutathione and metals status. Environ. Int. 2010, 36, 918–923. [Google Scholar] [CrossRef]
- Zhang, Y.R.; Cui, R.N.; Shi, G.H.; Dai, Y.; Dong, J.H.; Wu, Q.; Zhang, H.X.; Dai, J.Y. Dioxin-like polychlorinated biphenyl 126 (PCB126) disrupts gut microbiota-host metabolic dysfunction in mice via aryl hydrocarbon receptor activation. Ecotoxicol. Environ. Saf. 2022, 236, 113448. [Google Scholar] [CrossRef]
- Thayer, K.A.; Heindel, J.J.; Bucher, J.R.; Gallo, M.A. Role of Environmental Chemicals in Diabetes and Obesity: A National Toxicology Program Workshop Review. Environ. Health Perspect. 2012, 120, 779–789. [Google Scholar] [CrossRef]
- Tian, Y.; Rimal, B.; Gui, W.; Koo, I.; Yokoyama, S.; Perdew, G.H.; Patterson, A.D. Early Life Short-Term Exposure to Polychlorinated Biphenyl 126 in Mice Leads to Metabolic Dysfunction and Microbiota Changes in Adulthood. Int. J. Mol. Sci. 2022, 23, 8220. [Google Scholar] [CrossRef]
- Uemura, H.; Arisawa, K.; Hiyoshi, M.; Kitayama, A.; Takami, H.; Sawachika, F.; Dakeshita, S.; Nii, K.; Satoh, H.; Sumiyoshi, Y.; et al. Prevalence of Metabolic Syndrome Associated with Body Burden Levels of Dioxin and Related Compounds among Japan’s General Population. Environ. Health Perspect. 2009, 117, 568–573. [Google Scholar] [CrossRef]
- Everett, C.J.; Frithsen, I.L.; Diaz, V.A.; Koopman, R.J.; Simpson, W.M.; Mainous, A.G. Association of a polychlorinated dibenzo-p-dioxin, a polychlorinated biphenyl, and DDT with diabetes in the 1999–2002 National Health and Nutrition Examination Survey. Environ. Res. 2007, 103, 413–418. [Google Scholar] [CrossRef]
- Silverstone, A.E.; Rosenbaum, P.F.; Weinstock, R.S.; Bartell, S.M.; Foushee, H.R.; Shelton, C.; Pavuk, M.; Anniston Environm Hlth Res, C. Polychlorinated biphenyl (PCB) exposure and diabetes: Results from the Anniston Community Health Survey. Environ. Health Perspect. 2012, 120, 727–732. [Google Scholar] [CrossRef]
- Wahlang, B.; Falkner, K.C.; Gregory, B.; Ansert, D.; Young, D.; Conklin, D.J.; Bhatnagar, A.; McClain, C.J.; Cave, M. Polychlorinated biphenyl 153 is a diet-dependent obesogen that worsens nonalcoholic fatty liver disease in male C57BL6/J mice. J. Nutr. Biochem. 2013, 24, 1587–1595. [Google Scholar] [CrossRef]
- Klinge, C.M.; Piell, K.M.; Petri, B.J.; He, L.; Zhang, X.; Pan, J.; Rai, S.N.; Andreeva, K.; Rouchka, E.C.; Wahlang, B.; et al. Combined exposure to polychlorinated biphenyls and high-fat diet modifies the global epitranscriptomic landscape in mouse liver. Environ. Epigenet. 2021, 7, dvab008. [Google Scholar]
- Petriello, M.C.; Hoffman, J.B.; Vsevolozhskaya, O.; Morris, A.J.; Hennig, B. Dioxin-like PCB 126 increases intestinal inflammation and disrupts gut microbiota and metabolic homeostasis. Environ Pollut. 2018, 242, 1022–1032. [Google Scholar] [CrossRef]
- Virtue, S.; Vidal-Puig, A. GTTs and ITTs in mice: Simple tests, complex answers. Nat. Metab. 2021, 3, 883–886. [Google Scholar] [CrossRef] [PubMed]
- Tian, Y.; Nichols, R.G.; Cai, J.W.; Patterson, A.D.; Cantorna, M.T. Vitamin A deficiency in mice alters host and gut microbial metabolism leading to altered energy homeostasis. J. Nutr. Biochem. 2018, 54, 28–34. [Google Scholar] [CrossRef] [PubMed]
- Amiel, A.; Tremblay-Franco, M.; Gautier, R.; Ducheix, S.; Montagner, A.; Polizzi, A.; Debrauwer, L.; Guillou, H.; Bertrand-Michel, J.; Canlet, C. Proton NMR enables the absolute quantification of aqueous metabolites and lipid classes in unique mouse liver samples. Metabolites 2020, 10, 9. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Tian, Y.; Cai, J.; Allman, E.L.; Smith, P.B.; Patterson, A.D. Quantitative Analysis of Bile Acid with UHPLC-MS/MS. Methods Mol. Biol. 2021, 2194, 291–300. [Google Scholar] [PubMed]
- Bolger, A.M.; Lohse, M.; Usadel, B. Trimmomatic: A flexible trimmer for Illumina sequence data. Bioinformatics 2014, 30, 2114–2120. [Google Scholar] [CrossRef]
- Franzosa, E.A.; McIver, L.J.; Rahnavard, G.; Thompson, L.R.; Schirmer, M.; Weingart, G.; Lipson, K.S.; Knight, R.; Caporaso, J.G.; Segata, N.; et al. Species-level functional profiling of metagenomes and metatranscriptomes. Nat. Methods 2018, 15, 962–968. [Google Scholar] [CrossRef]
- Wood, D.E.; Lu, J.; Langmead, B. Improved metagenomic analysis with Kraken 2. Genome Biol. 2019, 20, 257. [Google Scholar] [CrossRef]
- Lu, J.; Breitwieser, F.P.; Thielen, P.; Salzberg, S.L. Bracken: Estimating species abundance in metagenomics data. Peerj Comput. Sci. 2017, 3, e104. [Google Scholar] [CrossRef]
- Beghini, F.; McIver, L.J.; Blanco-Miguez, A.; Dubois, L.; Asnicar, F.; Maharjan, S.; Mailyan, A.; Manghi, P.; Scholz, M.; Thomas, A.M.; et al. Integrating taxonomic, functional, and strain-level profiling of diverse microbial communities with bioBakery 3. eLife 2021, 10, e65088. [Google Scholar] [CrossRef]
- Buchfink, B.; Reuter, K.; Drost, H.G. Sensitive protein alignments at tree-of-life scale using DIAMOND. Nat. Methods 2021, 18, 366–368. [Google Scholar] [CrossRef]
- Milbrath, M.O.; Wenger, Y.; Chang, C.W.; Emond, C.; Garabrant, D.; Gillespie, B.W.; Jolliet, O. Apparent half-lives of dioxins, furans, and polychlorinated biphenyls as a function of age, body fat, smoking status, and breast-feeding. Environ. Health Perspect. 2009, 117, 417–425. [Google Scholar] [CrossRef] [PubMed]
- Hectors, T.L.M.; Vanparys, C.; van der Ven, K.; Martens, G.A.; Jorens, P.G.; Van Gaal, L.F.; Covaci, A.; De Coen, W.; Blust, R. Environmental pollutants and type 2 diabetes: A review of mechanisms that can disrupt beta cell function. Diabetologia 2011, 54, 1273–1290. [Google Scholar] [CrossRef] [PubMed]
- Jakobsdottir, G.; Xu, J.; Molin, G.; Ahrne, S.; Nyman, M. High-Fat Diet Reduces the Formation of Butyrate, but Increases Succinate, Inflammation, Liver Fat and Cholesterol in Rats, while Dietary Fibre Counteracts These Effects. PLoS ONE 2013, 8, e80476. [Google Scholar] [CrossRef] [Green Version]
- Cordain, L.; Eaton, S.B.; Sebastian, A.; Mann, N.; Lindeberg, S.; Watkins, B.A.; O’Keefe, J.H.; Brand-Miller, J. Origins and evolution of the Western diet: Health implications for the 21st century. Am. J. Clin. Nutr. 2005, 81, 341–354. [Google Scholar] [CrossRef] [PubMed]
- Zhang, W.S.; Sargis, R.M.; Volden, P.A.; Carmean, C.M.; Sun, X.J.; Brady, M.J. PCB 126 and other dioxin-Like PCBs specifically suppress hepatic PEPCK expression via the aryl hydrocarbon receptor. PLoS ONE 2012, 7, e37103. [Google Scholar] [CrossRef]
- Reyes-Caballero, H.; Rao, X.Q.; Sun, Q.S.; Warmoes, M.O.; Penghui, L.; Sussan, T.E.; Park, B.; Fan, T.W.M.; Maiseyeu, A.; Rajagopalan, S.; et al. Air pollution-derived particulate matter dysregulates hepatic Krebs cycle, glucose and lipid metabolism in mice. Sci. Rep. 2019, 9, 17423. [Google Scholar] [CrossRef]
- Ariav, Y.; Ch’ng, J.H.; Christofk, H.R.; Ron-Harel, N.; Erez, A. Targeting nucleotide metabolism as the nexus of viral infections, cancer, and the immune response. Sci. Adv. 2021, 7, eabg6165. [Google Scholar] [CrossRef]
- Lustig, R.H.; Collier, D.; Kassotis, C.; Roepke, T.A.; Kim, M.J.; Blanc, E.; Barouki, R.; Bansal, A.; Cave, M.C.; Chatterjee, S.; et al. Obesity I: Overview and molecular and biochemical mechanisms. Biochem. Pharmacol. 2022, 199, 115012. [Google Scholar] [CrossRef]
- Fioravante, M.; Bombassaro, B.; Ramalho, A.F.; Dragano, N.R.; Morari, J.; Solon, C.; Tobar, N.; Ramos, C.D.; Velloso, L.A. Inhibition of hypothalamic leukemia inhibitory factor exacerbates diet-induced obesity phenotype. J. Neuroinflammation 2017, 14, 178. [Google Scholar] [CrossRef]
- Fenton, J.I.; Nunez, N.P.; Yakar, S.; Perkins, S.N.; Hord, N.G.; Hursting, S.D. Diet-induced adiposity alters the serum profile of inflammation in C57BL/6N mice as measured by antibody array. Diabetes Obes. Metab. 2009, 11, 343–354. [Google Scholar] [CrossRef]
- Zitka, O.; Skalickova, S.; Gumulec, J.; Masarik, M.; Adam, V.; Hubalek, J.; Trnkova, L.; Kruseova, J.; Eckschlager, T.; Kizek, R. Redox status expressed as GSH:GSSG ratio as a marker for oxidative stress in paediatric tumour patients. Oncol. Lett. 2012, 4, 1247–1253. [Google Scholar] [CrossRef]
- Mesnage, R.; Biserni, M.; Balu, S.; Frainay, C.; Poupin, N.; Jourdan, F.; Wozniak, E.; Xenakis, T.; Mein, C.A.; Antoniou, M.N. Integrated transcriptomics and metabolomics reveal signatures of lipid metabolism dysregulation in HepaRG liver cells exposed to PCB 126. Arch. Toxicol. 2018, 92, 2533–2547. [Google Scholar] [CrossRef] [PubMed]
- Gadupudi, G.S.; Klaren, W.D.; Olivier, A.K.; Klingelhutz, A.J.; Robertson, L.W. PCB126-induced disruption in gluconeogenesis and fatty acid oxidation precedes fatty liver in male rats. Toxicol. Sci. 2016, 149, 98–110. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Liu, Q.; Zhang, L.; Allman, E.L.; Hubbard, T.D.; Murray, I.A.; Hao, F.; Tian, Y.; Gui, W.; Nichols, R.G.; Smith, P.B.; et al. The aryl hydrocarbon receptor activates ceramide biosynthesis in mice contributing to hepatic lipogenesis. Toxicology 2021, 458, 152831. [Google Scholar] [CrossRef] [PubMed]
- Ahola, A.J.; Lassenius, M.I.; Forsblom, C.; Harjutsalo, V.; Lehto, M.; Groop, P.H. Dietary patterns reflecting healthy food choices are associated with lower serum LPS activity. Sci. Rep. 2017, 7, 6511. [Google Scholar] [CrossRef] [PubMed]
- Metidji, A.; Omenetti, S.; Crotta, S.; Li, Y.; Nye, E.; Ross, E.; Li, V.; Maradana, M.R.; Schiering, C.; Stockinger, B. The Environmental Sensor AHR Protects from Inflammatory Damage by Maintaining Intestinal Stem Cell Homeostasis and Barrier Integrity. Immunity 2018, 49, 353–362. [Google Scholar] [CrossRef]
- Daniel, H.; Gholami, A.M.; Berry, D.; Desmarchelier, C.; Hahne, H.; Loh, G.; Mondot, S.; Lepage, P.; Rothballer, M.; Walker, A.; et al. High-fat diet alters gut microbiota physiology in mice. ISME J. 2014, 8, 295–308. [Google Scholar] [CrossRef]
- Wu, G.D.; Chen, J.; Hoffmann, C.; Bittinger, K.; Chen, Y.Y.; Keilbaugh, S.A.; Bewtra, M.; Knights, D.; Walters, W.A.; Knight, R.; et al. Linking long-term dietary patterns with gut microbial enterotypes. Science 2011, 334, 105–108. [Google Scholar] [CrossRef] [PubMed]
- Guthrie, L.; Wolfson, S.; Kelly, L. The human gut chemical landscape predicts microbe-mediated biotransformation of foods and drugs. eLife 2019, 8, e42866. [Google Scholar] [CrossRef]
- Stojanov, S.; Berlec, A.; Strukelj, B. The influence of probiotics on the Firmicutes/Bacteroidetes ratio in the treatment of obesity and inflammatory bowel disease. Microorganisms 2020, 8, 1715. [Google Scholar] [CrossRef]
- Klasson Wehler, E.; Jönsson, J.; Bergman, A.; Brandt, I.; Darnerud, P.O. 3,3′,4,4′-tetrachlorobiphenyl and 3,3′,4,4′,5-pentachlorobiphenyl-tissue-localization and metabolic fate in the mouse. Chemosphere 1989, 19, 809–812. [Google Scholar] [CrossRef]
- Pallister, T.; Jackson, M.A.; Martin, T.C.; Zierer, J.; Jennings, A.; Mohney, R.P.; MacGregor, A.; Steves, C.J.; Cassidy, A.; Spector, T.D.; et al. Hippurate as a metabolomic marker of gut microbiome diversity: Modulation by diet and relationship to metabolic syndrome. Sci. Rep. 2017, 7, 13670. [Google Scholar] [CrossRef]
- Metges, C.C. Contribution of microbial amino acids to amino acid homeostasis of the host. J. Nutr. 2000, 130, 1857S–1864S. [Google Scholar] [CrossRef] [Green Version]
- Neis, E.; Dejong, C.H.C.; Rensen, S.S. The role of microbial amino acid metabolism in host metabolism. Nutrients 2015, 7, 2930–2946. [Google Scholar] [CrossRef] [PubMed]
- Leonardi, R.; Jackowski, S. Biosynthesis of pantothenic acid and coenzyme A. Ecosal Plus 2007, 2, 1–28. [Google Scholar] [CrossRef] [PubMed]
- De Weirdt, R.; Possemiers, S.; Vermeulen, G.; Moerdijk-Poortvliet, T.C.W.; Boschker, H.T.S.; Verstraete, W.; Van de Wiele, T. Human faecal microbiota display variable patterns of glycerol metabolism. FEMS Microbiol. Ecol. 2010, 74, 601–611. [Google Scholar] [CrossRef]
- Marchesi, J.R.; Holmes, E.; Khan, F.; Kochhar, S.; Scanlan, P.; Shanahan, F.; Wilson, I.D.; Wang, Y.L. Rapid and noninvasive metabonomic characterization of inflammatory bowel disease. J. Proteome Res. 2007, 6, 546–551. [Google Scholar] [CrossRef] [PubMed]





Publisher’s Note: MDPI stays neutral with regard to jurisdictional claims in published maps and institutional affiliations. |
© 2022 by the authors. Licensee MDPI, Basel, Switzerland. This article is an open access article distributed under the terms and conditions of the Creative Commons Attribution (CC BY) license (https://creativecommons.org/licenses/by/4.0/).
Share and Cite
Tian, Y.; Rimal, B.; Gui, W.; Koo, I.; Smith, P.B.; Yokoyama, S.; Patterson, A.D. Early Life Polychlorinated Biphenyl 126 Exposure Disrupts Gut Microbiota and Metabolic Homeostasis in Mice Fed with High-Fat Diet in Adulthood. Metabolites 2022, 12, 894. https://doi.org/10.3390/metabo12100894
Tian Y, Rimal B, Gui W, Koo I, Smith PB, Yokoyama S, Patterson AD. Early Life Polychlorinated Biphenyl 126 Exposure Disrupts Gut Microbiota and Metabolic Homeostasis in Mice Fed with High-Fat Diet in Adulthood. Metabolites. 2022; 12(10):894. https://doi.org/10.3390/metabo12100894
Chicago/Turabian StyleTian, Yuan, Bipin Rimal, Wei Gui, Imhoi Koo, Philip B. Smith, Shigetoshi Yokoyama, and Andrew D. Patterson. 2022. "Early Life Polychlorinated Biphenyl 126 Exposure Disrupts Gut Microbiota and Metabolic Homeostasis in Mice Fed with High-Fat Diet in Adulthood" Metabolites 12, no. 10: 894. https://doi.org/10.3390/metabo12100894
APA StyleTian, Y., Rimal, B., Gui, W., Koo, I., Smith, P. B., Yokoyama, S., & Patterson, A. D. (2022). Early Life Polychlorinated Biphenyl 126 Exposure Disrupts Gut Microbiota and Metabolic Homeostasis in Mice Fed with High-Fat Diet in Adulthood. Metabolites, 12(10), 894. https://doi.org/10.3390/metabo12100894