
A Modular and Expandable Ecosystem for Metabolomics Data Annotation in R

 ,
,  , , ,
, , ,  ,
,  , ,
, ,  , , and
, , and 
Abstract
:1. Introduction
2. Results
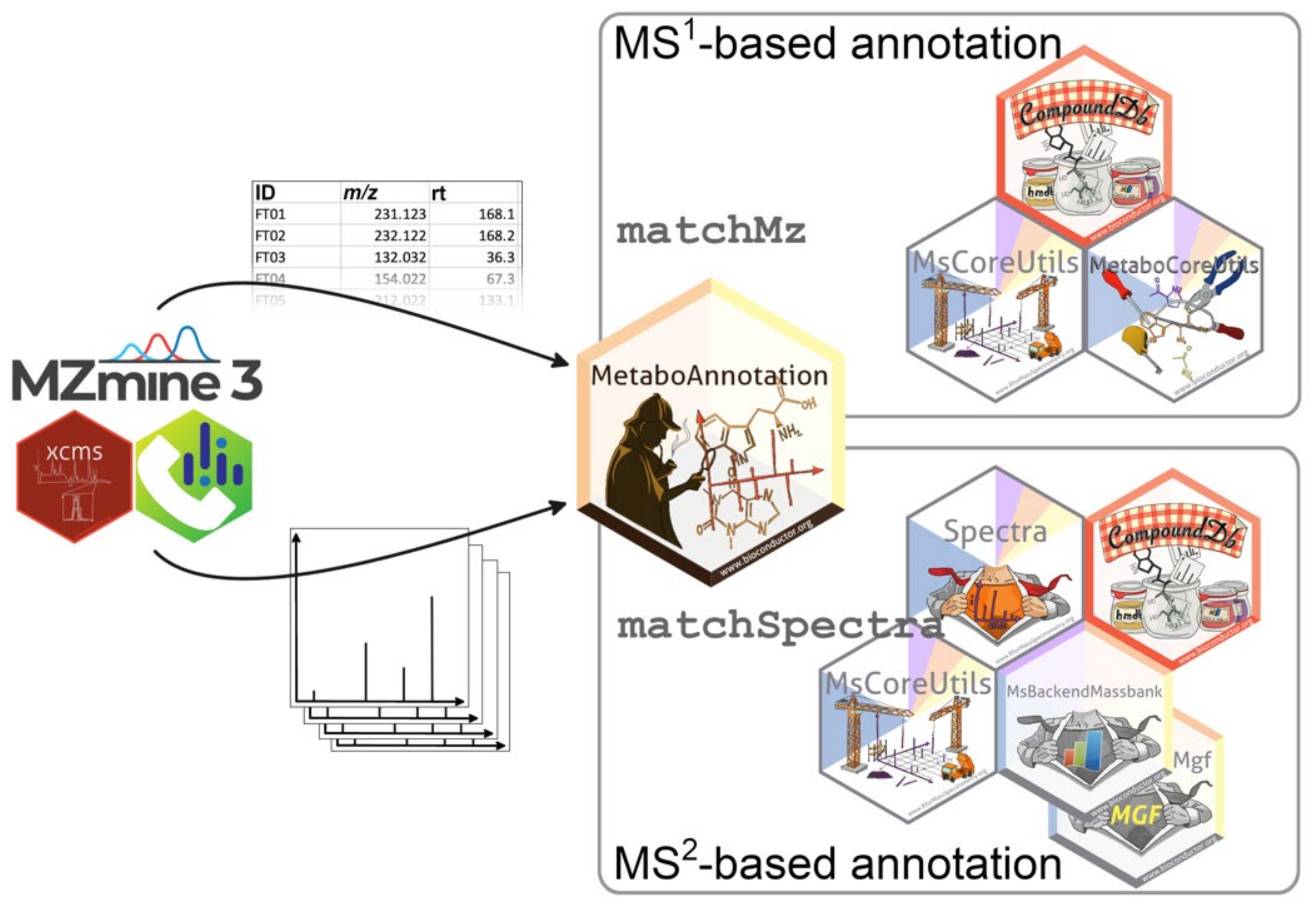
2.1. Package Ecosystem
2.1.1. Overview and Architecture
2.1.2. Development and Maintenance Processes
2.2. Utility Functions
2.3. Creating, Managing and Using Reference Databases
2.4. Functions for Working with Retention and Migration Times
2.5. MS1 Annotation
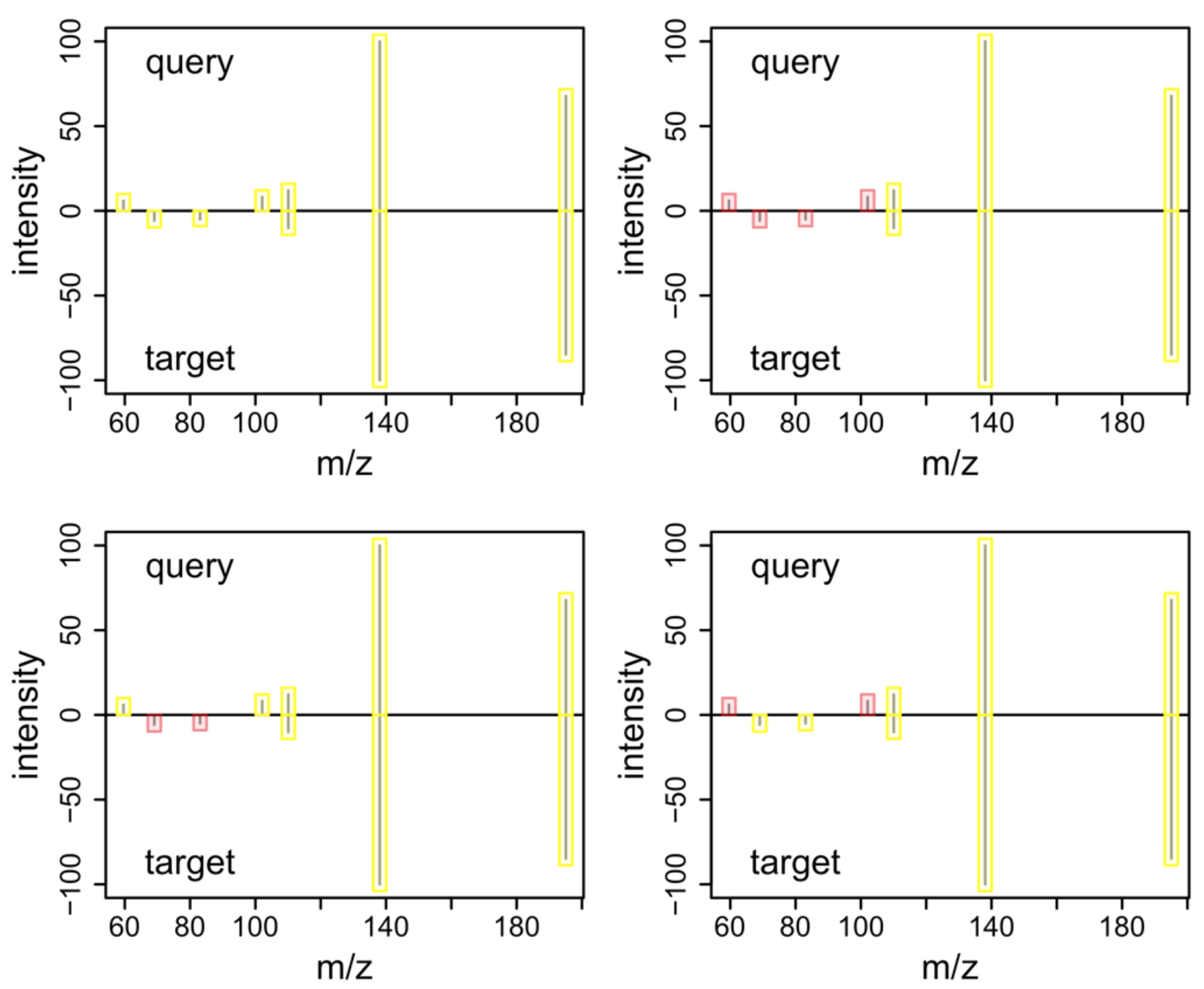
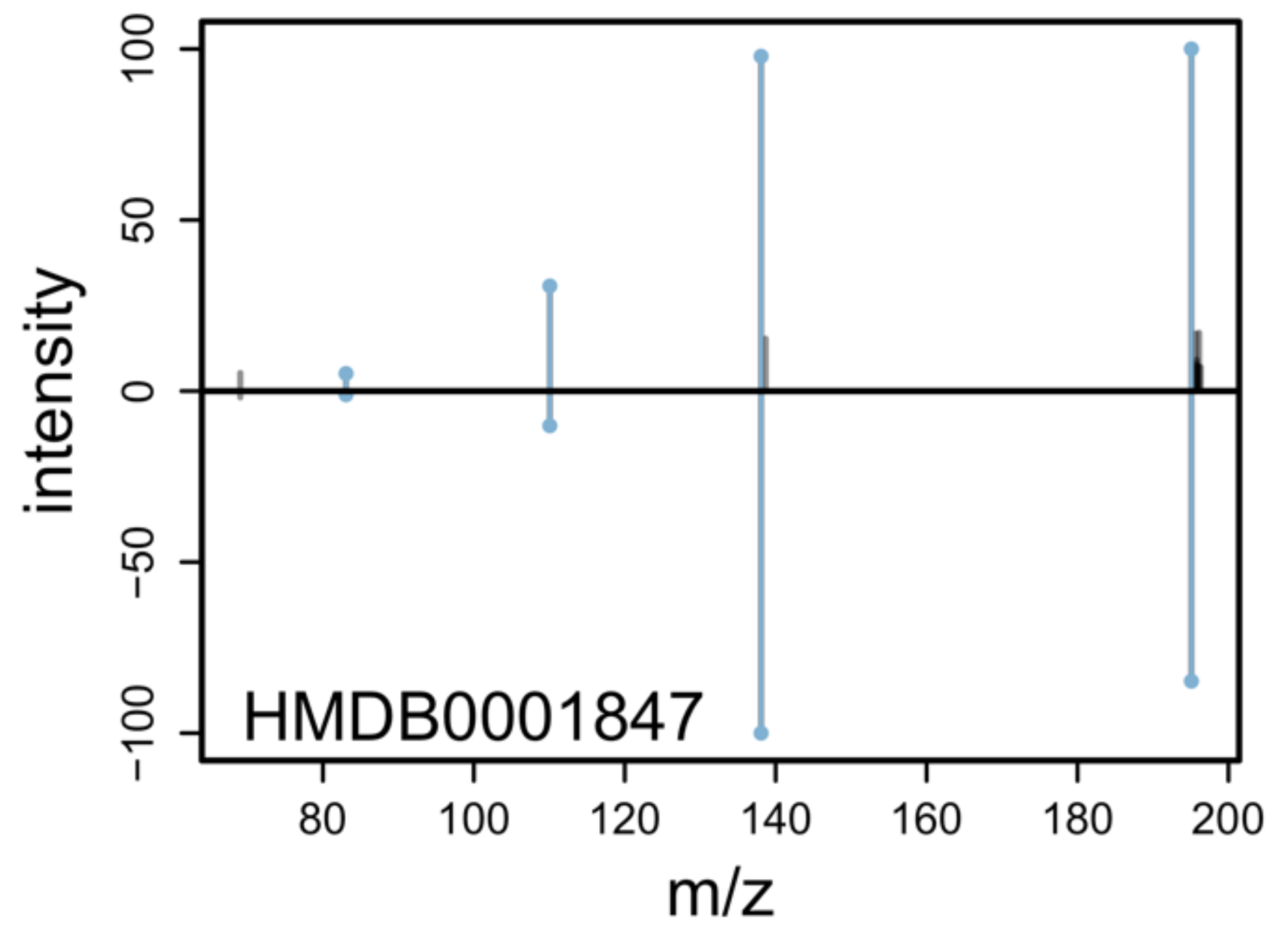
2.6. MS2 Annotation
2.7. Examples and Use Cases
3. Discussion
4. Materials and Methods
4.1. Acquisition of the Example Dataset for MS¹- and MS²-Based Annotation
4.2. Retention Indexing Example
5. Conclusions
Supplementary Materials
Author Contributions
Funding
Institutional Review Board Statement
Informed Consent Statement
Data Availability Statement
Acknowledgments
Conflicts of Interest
References
- Sumner, L.W.; Amberg, A.; Barrett, D.; Beale, M.H.; Beger, R.; Daykin, C.A.; Fan, T.W.-M.; Fiehn, O.; Goodacre, R.; Griffin, J.L.; et al. Proposed minimum reporting standards for chemical analysis. Metabolomics 2007, 3, 211–221. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Schymanski, E.L.; Jeon, J.; Gulde, R.; Fenner, K.; Ruff, M.; Singer, H.P.; Hollender, J. Identifying small molecules via high resolution mass spectrometry: Communicating confidence. Environ. Sci. Technol. 2014, 48, 2097–2098. [Google Scholar] [CrossRef] [PubMed]
- Misra, B.B. New software tools, databases, and resources in metabolomics: Updates from 2020. Metabolomics 2021, 17, 49. [Google Scholar] [CrossRef] [PubMed]
- Dührkop, K.; Shen, H.; Meusel, M.; Rousu, J.; Böcker, S. Searching molecular structure databases with tandem mass spectra using CSI:FingerID. Proc. Natl. Acad. Sci. USA 2015, 112, 12580–12585. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Hoffmann, M.A.; Nothias, L.-F.; Ludwig, M.; Fleischauer, M.; Gentry, E.C.; Witting, M.; Dorrestein, P.C.; Dührkop, K.; Böcker, S. High-confidence structural annotation of metabolites absent from spectral libraries. Nat. Biotechnol. 2021, 1–11. [Google Scholar] [CrossRef] [PubMed]
- Stanstrup, J.; Broeckling, C.D.; Helmus, R.; Hoffmann, N.; Mathé, E.; Naake, T.; Nicolotti, L.; Peters, K.; Rainer, J.; Salek, R.M.; et al. The metaRbolomics toolbox in bioconductor and beyond. Metabolites 2019, 9, 200. [Google Scholar] [CrossRef] [Green Version]
- Chong, J.; Yamamoto, M.; Xia, J. MetaboAnalystR 2.0: From raw spectra to biological insights. Metabolites 2019, 9, 57. [Google Scholar] [CrossRef] [Green Version]
- Helmus, R.; ter Laak, T.L.; van Wezel, A.P.; de Voogt, P.; Schymanski, E.L. patRoon: Open-source software platform for environmental mass spectrometry based non-target screening. J. Cheminform. 2021, 13, 1–25. [Google Scholar] [CrossRef]
- Shen, X.; Wu, S.; Liang, L.; Chen, S.; Contrepois, K.; Zhu, Z.-J.; Snyder, M. metID: An R package for automatable compound annotation for LC−MS-based data. Bioinformatics 2021, 38, 568–569. [Google Scholar] [CrossRef]
- Li, S.; Park, Y.; Duraisingham, S.; Strobel, F.H.; Khan, N.; Soltow, Q.A.; Jones, D.P.; Pulendran, B. Predicting network activity from high throughput metabolomics. PLOS Comput. Biol. 2013, 9, e1003123. [Google Scholar] [CrossRef] [Green Version]
- Lawson, T.N.; Weber, R.J.M.; Jones, M.R.; Chetwynd, A.J.; Rodríguez-Blanco, G.; Di Guida, R.; Viant, M.R.; Dunn, W.B. msPurity: Automated evaluation of precursor ion purity for mass spectrometry-based fragmentation in metabolomics. Anal. Chem. 2017, 89, 2432–2439. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Hill, E.A. On a system of indexing chemical literature: Adopted by the classification division of the U.S. Patent Office. J. Am. Chem. Soc. 1900, 22, 478–494. [Google Scholar] [CrossRef] [Green Version]
- Stein, S.E.; Scott, D.R. Optimization and testing of mass spectral library search algorithms for compound identification. J. Am. Soc. Mass Spectrom. 1994, 5, 859–866. [Google Scholar] [CrossRef] [Green Version]
- Toprak, U.H.; Gillet, L.; Maiolica, A.; Navarro, P.; Leitner, A.; Aebersold, R. Conserved peptide fragmentation as a benchmarking tool for mass spectrometers and a discriminating feature for targeted proteomics. Mol. Cell. Proteom. 2014, 13, 2056–2071. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Hastings, J.; Owen, G.; Dekker, A.; Ennis, M.; Kale, N.; Muthukrishnan, V.; Turner, S.; Swainston, N.; Mendes, P.; Steinbeck, C. ChEBI in 2016: Improved services and an expanding collection of metabolites. Nucleic Acids Res. 2015, 44, D1214–D1219. [Google Scholar] [CrossRef] [PubMed]
- Wishart, D.S.; Guo, A.; Oler, E.; Wang, F.; Anjum, A.; Peters, H.; Dizon, R.; Sayeeda, Z.; Tian, S.; Lee, B.L.; et al. HMDB 5.0: The Human Metabolome Database for 2022. Nucleic Acids Res. 2021, 50, D622–D631. [Google Scholar] [CrossRef]
- Horai, H.; Arita, M.; Kanaya, S.; Nihei, Y.; Ikeda, T.; Suwa, K.; Ojima, Y.; Tanaka, K.; Tanaka, S.; Aoshima, K.; et al. MassBank: A public repository for sharing mass spectral data for life sciences. J. Mass Spectrom. 2010, 45, 703–714. [Google Scholar] [CrossRef]
- Kim, S.; Chen, J.; Cheng, T.; Gindulyte, A.; He, J.; He, S.; Li, Q.; Shoemaker, B.A.; Thiessen, P.A.; Yu, B.; et al. PubChem in 2021: New data content and improved web interfaces. Nucleic Acids Res. 2021, 49, D1388–D1395. [Google Scholar] [CrossRef]
- Strehmel, N.; Hummel, J.; Erban, A.; Strassburg, K.; Kopka, J. Retention index thresholds for compound matching in GC–MS metabolite profiling. J. Chromatogr. B 2008, 871, 182–190. [Google Scholar] [CrossRef]
- Stoffel, R.; Quilliam, M.A.; Hardt, N.; Fridstrom, A.; Witting, M. N-Alkylpyridinium sulfonates for retention time indexing in reversed-phase-liquid chromatography-mass spectrometry-based metabolomics. Anal. Bioanal. Chem. 2021, 1–12. [Google Scholar] [CrossRef]
- Aalizadeh, R.; Alygizakis, N.A.; Schymanski, E.L.; Krauss, M.; Schulze, T.; Ibáñez, M.; McEachran, A.D.; Chao, A.; Williams, A.J.; Gago-Ferrero, P.; et al. Development and application of liquid chromatographic retention time indices in HRMS-based suspect and nontarget screening. Anal. Chem. 2021, 93, 11601–11611. [Google Scholar] [CrossRef] [PubMed]
- Schmitt-Kopplin, P.; Garmash, A.V.; Kudryavtsev, A.V.; Menzinger, F.; Perminova, I.V.; Hertkorn, N.; Freitag, D.; Petrosyan, V.S.; Kettrup, A. Quantitative and qualitative precision improvements by effective mobility-scale data transformation in capillary electrophoresis analysis. Electrophoresis 2001, 22, 77–87. [Google Scholar] [CrossRef]
- González-Ruiz, V.; Gagnebin, Y.; Drouin, N.; Codesido, S.; Rudaz, S.; Schappler, J. ROMANCE: A new software tool to improve data robustness and feature identification in CE-MS metabolomics. Electrophoresis 2018, 39, 1222–1232. [Google Scholar] [CrossRef] [PubMed]
- Ikuta, N.; Yamada, Y.; Yoshiyama, T.; Hirokawa, T. New method for standardization of electropherograms obtained in capillary zone electrophoresis. J. Chromatogr. A 2000, 894, 11–17. [Google Scholar] [CrossRef]
- Wägele, B.; Witting, M.; Schmitt-Kopplin, P.; Suhre, K. MassTRIX reloaded: Combined analysis and visualization of transcriptome and metabolome data. PLoS ONE 2012, 7, e39860. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Hernández-De-Diego, R.; Tarazona, S.; Martínez-Mira, C.; Balzano-Nogueira, L.; Furió-Tarí, P.; Pappas, G.J.; Conesa, A. PaintOmics 3: A web resource for the pathway analysis and visualization of multi-omics data. Nucleic Acids Res. 2018, 46, W503–W509. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Drost, H.-G. Philentropy: Information theory and distance quantification with R. J. Open Source Softw. 2018, 3, 765. [Google Scholar] [CrossRef]
- Burke, M.C.; Mirokhin, Y.A.; Tchekhovskoi, D.V.; Markey, S.P.; Thompson, J.H.; Larkin, C.; Stein, S.E. The hybrid search: A mass spectral library search method for discovery of modifications in proteomics. J. Proteome Res. 2017, 16, 1924–1935. [Google Scholar] [CrossRef]
- Cooper, B.T.; Yan, X.; Simón-Manso, Y.; Tchekhovskoi, D.V.; Mirokhin, Y.A.; Stein, S.E. Hybrid search: A method for identifying metabolites absent from tandem mass spectrometry libraries. Anal. Chem. 2019, 91, 13924–13932. [Google Scholar] [CrossRef]
- Watrous, J.; Roach, P.; Alexandrov, T.; Heath, B.S.; Yang, J.Y.; Kersten, R.D.; van der Voort, M.; Pogliano, K.; Gross, H.; Raaijmakers, J.M.; et al. Mass spectral molecular networking of living microbial colonies. Proc. Natl. Acad. Sci. USA 2012, 109, E1743–E1752. [Google Scholar] [CrossRef] [Green Version]
- Xing, S.; Hu, Y.; Yin, Z.; Liu, M.; Tang, X.; Fang, M.; Huan, T. Retrieving and utilizing hypothetical neutral losses from tandem mass spectra for spectral similarity analysis and unknown metabolite annotation. Anal. Chem. 2020, 92, 14476–14483. [Google Scholar] [CrossRef] [PubMed]
- Wishart, D.S.; Feunang, Y.D.; Marcu, A.; Guo, A.C.; Liang, K.; Vázquez-Fresno, R.; Sajed, T.; Johnson, D.; Li, C.; Karu, N.; et al. HMDB 4.0: The human metabolome database for 2018. Nucleic Acids Res. 2018, 46, D608–D617. [Google Scholar] [CrossRef] [PubMed]
- Nothias, L.-F.; Petras, D.; Schmid, R.; Dührkop, K.; Rainer, J.; Sarvepalli, A.; Protsyuk, I.; Ernst, M.; Tsugawa, H.; Fleischauer, M.; et al. Feature-based molecular networking in the GNPS analysis environment. Nat. Methods 2020, 17, 905–908. [Google Scholar] [CrossRef] [PubMed]




{kind=link}
{kind=link}
{kind=link}
| Function | Description | Package |
|---|---|---|
| countElements | Counts elements in chemical formulas. | MetaboCoreUtils |
| pasteElements | Converts element counts to chemical formulas. | MetaboCoreUtils |
| subtractElements | Removes elements from chemical formulas. | MetaboCoreUtils |
| addElements | Adds elements to chemical formulas. | MetaboCoreUtils |
| standardizeFormula | Standardizes formulas according to the Hill notation [12]. | MetaboCoreUtils |
| calculateMass | Calculates exact masses from chemical formulas. | MetaboCoreUtils |
| mass2mz, mz2mass | Converts between masses and m/z values. | MetaboCoreUtils |
| isotopologues | Groups potential isotopologue peaks in MS1 data. | MetaboCoreUtils |
| closest | Matches numeric values accepting differences. | MsCoreUtils |
| ndotproduct | Normalized dot product [13]. | MsCoreUtils |
| neuclidian | Normalized Euclidian distance [13]. | MsCoreUtils |
| navdist | Normalized absolute values distance [13]. | MsCoreUtils |
| nspectraangle | Normalized spectra angle [14]. | MsCoreUtils |
| Function | Parameter Object | Description |
|---|---|---|
| matchMz | MzParam | Performs m/z matching between query and target. |
| matchMz | MzRtParam | Matches m/z values and retention times from query and target. |
| matchMz | Mass2MzParam | Performs m/z matching after converting target masses to m/z values. |
| matchMz | Mass2MzRtParam | Matches m/z values and retention times between query and target after conversion of target masses to m/z values. |
| Function | Parameter Object | Description |
|---|---|---|
| joinPeaks | - | Maps peaks between two spectra accepting differences between the peaks’ m/z values that can be defined by ppm and tolerance. |
| joinPeaksGnps | - | Hybrid search approach [28,29,30,31]: also peaks for which the difference in m/z values matches the difference of the precursor m/z of the two spectra are considered matching. |
| compareSpectra | - | Calculates pairwise similarity scores between two spectra objects. |
| matchSpectra | CompareSpectraParam | Identifies spectra with a similarity score above a user-defined threshold. |
| matchSpectra | MatchForwardReverseParam | Identifies spectra with a similarity score above a user-defined threshold and calculates in addition the reverse score. |
Publisher’s Note: MDPI stays neutral with regard to jurisdictional claims in published maps and institutional affiliations. |
© 2022 by the authors. Licensee MDPI, Basel, Switzerland. This article is an open access article distributed under the terms and conditions of the Creative Commons Attribution (CC BY) license (https://creativecommons.org/licenses/by/4.0/).
Share and Cite
Rainer, J.; Vicini, A.; Salzer, L.; Stanstrup, J.; Badia, J.M.; Neumann, S.; Stravs, M.A.; Verri Hernandes, V.; Gatto, L.; Gibb, S.; et al. A Modular and Expandable Ecosystem for Metabolomics Data Annotation in R. Metabolites 2022, 12, 173. https://doi.org/10.3390/metabo12020173
Rainer J, Vicini A, Salzer L, Stanstrup J, Badia JM, Neumann S, Stravs MA, Verri Hernandes V, Gatto L, Gibb S, et al. A Modular and Expandable Ecosystem for Metabolomics Data Annotation in R. Metabolites. 2022; 12(2):173. https://doi.org/10.3390/metabo12020173
Chicago/Turabian StyleRainer, Johannes, Andrea Vicini, Liesa Salzer, Jan Stanstrup, Josep M. Badia, Steffen Neumann, Michael A. Stravs, Vinicius Verri Hernandes, Laurent Gatto, Sebastian Gibb, and et al. 2022. "A Modular and Expandable Ecosystem for Metabolomics Data Annotation in R" Metabolites 12, no. 2: 173. https://doi.org/10.3390/metabo12020173
APA StyleRainer, J., Vicini, A., Salzer, L., Stanstrup, J., Badia, J. M., Neumann, S., Stravs, M. A., Verri Hernandes, V., Gatto, L., Gibb, S., & Witting, M. (2022). A Modular and Expandable Ecosystem for Metabolomics Data Annotation in R. Metabolites, 12(2), 173. https://doi.org/10.3390/metabo12020173