
2-Year-Old and 3-Year-Old Italian ALS Patients with Novel ALS2 Mutations: Identification of Key Metabolites in Their Serum and Plasma

 ,
, 
 and
and 
Abstract
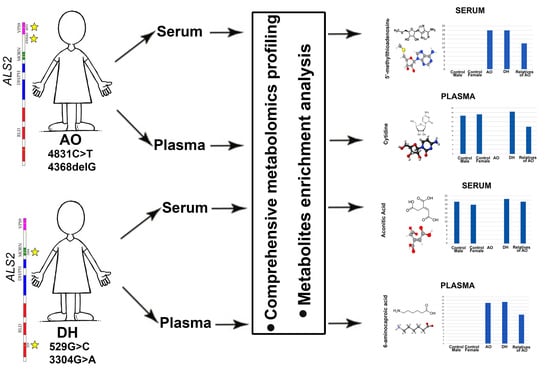
:
1. Introduction
2. Results
2.1. Selection of Patients
2.2. Metabolites Suggest Potential Defects in Key Canonical Pathways
2.3. Metabolites Present in Serum
2.4. Metabolites Present in Plasma
3. Discussion
4. Materials and Methods
4.1. Patient Information
4.2. Genetic Analyses
4.3. Plasma and Serum Sample Collection
4.4. Metabolomics
4.4.1. Metabolite Extraction
4.4.2. Sample Reconstitution after Extraction
4.4.3. Full-Scan Hydrophilic Metabolite Profiling
4.4.4. Metabolite Enrichment Analysis
5. Conclusions
Supplementary Materials
Author Contributions
Funding
Institutional Review Board Statement
Informed Consent Statement
Data Availability Statement
Acknowledgments
Conflicts of Interest
References
- Hadano, S.; Hand, C.K.; Osuga, H.; Yanagisawa, Y.; Otomo, A.; Devon, R.S.; Miyamoto, N.; Showguchi-Miyata, J.; Okada, Y.; Singaraja, R.; et al. A gene encoding a putative GTPase regulator is mutated in familial amyotrophic lateral sclerosis 2. Nat. Genet. 2001, 29, 166–173. [Google Scholar] [CrossRef] [PubMed]
- Yang, Y.; Hentati, A.; Deng, H.X.; Dabbagh, O.; Sasaki, T.; Hirano, M.; Hung, W.Y.; Ouahchi, K.; Yan, J.; Azim, A.C.; et al. The gene encoding alsin, a protein with three guanine-nucleotide exchange factor domains, is mutated in a form of recessive amyotrophic lateral sclerosis. Nat. Genet. 2001, 29, 160–165. [Google Scholar] [CrossRef] [PubMed]
- Sato, K.; Otomo, A.; Ueda, M.T.; Hiratsuka, Y.; Suzuki-Utsunomiya, K.; Sugiyama, J.; Murakoshi, S.; Mitsui, S.; Ono, S.; Nakagawa, S.; et al. Altered oligomeric states in pathogenic ALS2 variants associated with juvenile motor neuron diseases cause loss of ALS2-mediated endosomal function. J. Biol. Chem. 2018, 293, 17135–17153. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Yamanaka, K.; Vande Velde, C.; Eymard-Pierre, E.; Bertini, E.; Boespflug-Tanguy, O.; Cleveland, D.W. Unstable mutants in the peripheral endosomal membrane component ALS2 cause early-onset motor neuron disease. Proc. Natl. Acad. Sci. USA 2003, 100, 16041–16046. [Google Scholar] [CrossRef] [Green Version]
- Eymard-Pierre, E.; Lesca, G.; Dollet, S.; Santorelli, F.M.; di Capua, M.; Bertini, E.; Boespflug-Tanguy, O. Infantile-onset ascending hereditary spastic paralysis is associated with mutations in the alsin gene. Am. J. Hum. Genet. 2002, 71, 518–527. [Google Scholar] [CrossRef] [Green Version]
- Pringle, C.E.; Hudson, A.J.; Munoz, D.G.; Kiernan, J.A.; Brown, W.F.; Ebers, G.C. Primary lateral sclerosis. Clinical features, neuropathology and diagnostic criteria. Brain 1992, 115 Pt 2, 495–520. [Google Scholar] [CrossRef]
- Sprute, R.; Jergas, H.; Olmez, A.; Alawbathani, S.; Karasoy, H.; Dafsari, H.S.; Becker, K.; Daimaguler, H.S.; Nurnberg, P.; Muntoni, F.; et al. Genotype-phenotype correlation in seven motor neuron disease families with novel ALS2 mutations. Am. J. Med. Genet. A 2021, 185, 344–354. [Google Scholar] [CrossRef]
- Hadano, S.; Benn, S.C.; Kakuta, S.; Otomo, A.; Sudo, K.; Kunita, R.; Suzuki-Utsunomiya, K.; Mizumura, H.; Shefner, J.M.; Cox, G.A.; et al. Mice deficient in the Rab5 guanine nucleotide exchange factor ALS2/alsin exhibit age-dependent neurological deficits and altered endosome trafficking. Hum. Mol. Genet. 2006, 15, 233–250. [Google Scholar] [CrossRef] [Green Version]
- Jacquier, A.; Buhler, E.; Schafer, M.K.; Bohl, D.; Blanchard, S.; Beclin, C.; Haase, G. Alsin/Rac1 signaling controls survival and growth of spinal motoneurons. Ann. Neurol. 2006, 60, 105–117. [Google Scholar] [CrossRef]
- Kunita, R.; Otomo, A.; Mizumura, H.; Suzuki-Utsunomiya, K.; Hadano, S.; Ikeda, J.E. The Rab5 activator ALS2/alsin acts as a novel Rac1 effector through Rac1-activated endocytosis. J. Biol. Chem. 2007, 282, 16599–16611. [Google Scholar] [CrossRef] [Green Version]
- Lai, C.; Xie, C.; Shim, H.; Chandran, J.; Howell, B.W.; Cai, H. Regulation of endosomal motility and degradation by amyotrophic lateral sclerosis 2/alsin. Mol. Brain 2009, 2, 23. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Jove, M.; Portero-Otin, M.; Naudi, A.; Ferrer, I.; Pamplona, R. Metabolomics of human brain aging and age-related neurodegenerative diseases. J. Neuropathol. Exp. Neurol. 2014, 73, 640–657. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Kapogiannis, D.; Mattson, M.P. Disrupted energy metabolism and neuronal circuit dysfunction in cognitive impairment and Alzheimer’s disease. Lancet Neurol. 2011, 10, 187–198. [Google Scholar] [CrossRef] [Green Version]
- Muddapu, V.R.; Dharshini, S.A.P.; Chakravarthy, V.S.; Gromiha, M.M. Neurodegenerative Diseases—Is Metabolic Deficiency the Root Cause? Front. Neurosci. 2020, 14, 213. [Google Scholar] [CrossRef] [PubMed]
- Teruya, T.; Chen, Y.J.; Kondoh, H.; Fukuji, Y.; Yanagida, M. Whole-blood metabolomics of dementia patients reveal classes of disease-linked metabolites. Proc. Natl. Acad. Sci. USA 2021, 118, e2022857118. [Google Scholar] [CrossRef] [PubMed]
- Donatti, A.; Canto, A.M.; Godoi, A.B.; da Rosa, D.C.; Lopes-Cendes, I. Circulating Metabolites as Potential Biomarkers for Neurological Disorders-Metabolites in Neurological Disorders. Metabolites 2020, 10, 389. [Google Scholar] [CrossRef]
- Klatt, S.; Doecke, J.D.; Roberts, A.; Boughton, B.A.; Masters, C.L.; Horne, M.; Roberts, B.R. A six-metabolite panel as potential blood-based biomarkers for Parkinson’s disease. NPJ Parkinsons Dis. 2021, 7, 94. [Google Scholar] [CrossRef]
- Oresic, M.; Anderson, G.; Mattila, I.; Manoucheri, M.; Soininen, H.; Hyotylainen, T.; Basignani, C. Targeted Serum Metabolite Profiling Identifies Metabolic Signatures in Patients with Alzheimer’s Disease, Normal Pressure Hydrocephalus and Brain Tumor. Front. Neurosci. 2017, 11, 747. [Google Scholar] [CrossRef]
- Li, J.; Wang, L.; Yu, P.; Shi, L.; Zhang, K.; Sun, Z.S.; Xia, K. Vitamin D-related genes are subjected to significant de novo mutation burdens in autism spectrum disorder. Am. J. Med. Genet. B Neuropsychiatr. Genet. 2017, 174, 568–577. [Google Scholar] [CrossRef]
- Pfohl, S.R.; Kim, R.B.; Coan, G.S.; Mitchell, C.S. Unraveling the Complexity of Amyotrophic Lateral Sclerosis Survival Prediction. Front. Neuroinform. 2018, 12, 36. [Google Scholar] [CrossRef] [Green Version]
- Beghi, E.; Chio, A.; Couratier, P.; Esteban, J.; Hardiman, O.; Logroscino, G.; Millul, A.; Mitchell, D.; Preux, P.M.; Pupillo, E.; et al. The epidemiology and treatment of ALS: Focus on the heterogeneity of the disease and critical appraisal of therapeutic trials. Amyotroph. Lateral Scler. 2011, 12, 1–10. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Gunes, Z.I.; Kan, V.W.Y.; Ye, X.; Liebscher, S. Exciting Complexity: The Role of Motor Circuit Elements in ALS Pathophysiology. Front. Neurosci. 2020, 14, 573. [Google Scholar] [CrossRef] [PubMed]
- Kunita, R.; Otomo, A.; Mizumura, H.; Suzuki, K.; Showguchi-Miyata, J.; Yanagisawa, Y.; Hadano, S.; Ikeda, J.E. Homo-oligomerization of ALS2 through its unique carboxyl-terminal regions is essential for the ALS2-associated Rab5 guanine nucleotide exchange activity and its regulatory function on endosome trafficking. J. Biol. Chem. 2004, 279, 38626–38635. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Otomo, A.; Hadano, S.; Okada, T.; Mizumura, H.; Kunita, R.; Nishijima, H.; Showguchi-Miyata, J.; Yanagisawa, Y.; Kohiki, E.; Suga, E.; et al. ALS2, a novel guanine nucleotide exchange factor for the small GTPase Rab5, is implicated in endosomal dynamics. Hum. Mol. Genet. 2003, 12, 1671–1687. [Google Scholar] [CrossRef] [Green Version]
- Topp, J.D.; Gray, N.W.; Gerard, R.D.; Horazdovsky, B.F. Alsin is a Rab5 and Rac1 guanine nucleotide exchange factor. J. Biol. Chem. 2004, 279, 24612–24623. [Google Scholar] [CrossRef] [Green Version]
- Abiko, H.; Fujiwara, S.; Ohashi, K.; Hiatari, R.; Mashiko, T.; Sakamoto, N.; Sato, M.; Mizuno, K. Rho guanine nucleotide exchange factors involved in cyclic-stretch-induced reorientation of vascular endothelial cells. J. Cell Sci. 2015, 128, 1683–1695. [Google Scholar] [CrossRef] [Green Version]
- Tudor, E.L.; Perkinton, M.S.; Schmidt, A.; Ackerley, S.; Brownlees, J.; Jacobsen, N.J.; Byers, H.L.; Ward, M.; Hall, A.; Leigh, P.N.; et al. ALS2/Alsin regulates Rac-PAK signaling and neurite outgrowth. J. Biol. Chem. 2005, 280, 34735–34740. [Google Scholar] [CrossRef] [Green Version]
- Millecamps, S.; Gentil, B.J.; Gros-Louis, F.; Rouleau, G.; Julien, J.P. Alsin is partially associated with centrosome in human cells. Biochim. Biophys. Acta 2005, 1745, 84–100. [Google Scholar] [CrossRef] [Green Version]
- Pyo, S.H.; Glaser, S.J.; Rehnberg, N.; Hatti-Kaul, R. Clean Production of Levulinic Acid from Fructose and Glucose in Salt Water by Heterogeneous Catalytic Dehydration. ACS Omega 2020, 5, 14275–14282. [Google Scholar] [CrossRef]
- Rand, J.M.; Pisithkul, T.; Clark, R.L.; Thiede, J.M.; Mehrer, C.R.; Agnew, D.E.; Campbell, C.E.; Markley, A.L.; Price, M.N.; Ray, J.; et al. A metabolic pathway for catabolizing levulinic acid in bacteria. Nat. Microbiol. 2017, 2, 1624–1634. [Google Scholar] [CrossRef]
- Signoretto, M.; Taghavi, S.; Ghedini, E.; Menegazzo, F. Catalytic Production of Levulinic Acid (LA) from Actual Biomass. Molecules 2019, 24, 2760. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Abela, L.; Spiegel, R.; Crowther, L.M.; Klein, A.; Steindl, K.; Papuc, S.M.; Joset, P.; Zehavi, Y.; Rauch, A.; Plecko, B.; et al. Plasma metabolomics reveals a diagnostic metabolic fingerprint for mitochondrial aconitase (ACO2) deficiency. PLoS ONE 2017, 12, e0176363. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Montoya, G.; Londono, J.; Cortes, P.; Izquierdo, O. Quantitation of trans-aconitic acid in different stages of the sugar-manufacturing process. J. Agric. Food Chem. 2014, 62, 8314–8318. [Google Scholar] [CrossRef] [PubMed]
- Haschemi, A.; Kosma, P.; Gille, L.; Evans, C.R.; Burant, C.F.; Starkl, P.; Knapp, B.; Haas, R.; Schmid, J.A.; Jandl, C.; et al. The sedoheptulose kinase CARKL directs macrophage polarization through control of glucose metabolism. Cell Metab. 2012, 15, 813–826. [Google Scholar] [CrossRef] [Green Version]
- Nagy, C.; Haschemi, A. Sedoheptulose kinase regulates cellular carbohydrate metabolism by sedoheptulose 7-phosphate supply. Biochem. Soc. Trans. 2013, 41, 674–680. [Google Scholar] [CrossRef] [Green Version]
- Taylor, P.L.; Blakely, K.M.; de Leon, G.P.; Walker, J.R.; McArthur, F.; Evdokimova, E.; Zhang, K.; Valvano, M.A.; Wright, G.D.; Junop, M.S. Structure and function of sedoheptulose-7-phosphate isomerase, a critical enzyme for lipopolysaccharide biosynthesis and a target for antibiotic adjuvants. J. Biol. Chem. 2008, 283, 2835–2845. [Google Scholar] [CrossRef] [Green Version]
- Copeland, C.S.; Neale, S.A.; Salt, T.E. Actions of Xanthurenic acid, a putative endogenous Group II metabotropic glutamate receptor agonist, on sensory transmission in the thalamus. Neuropharmacology 2013, 66, 133–142. [Google Scholar] [CrossRef]
- Fazio, F.; Lionetto, L.; Curto, M.; Iacovelli, L.; Cavallari, M.; Zappulla, C.; Ulivieri, M.; Napoletano, F.; Capi, M.; Corigliano, V.; et al. Xanthurenic Acid Activates mGlu2/3 Metabotropic Glutamate Receptors and is a Potential Trait Marker for Schizophrenia. Sci. Rep. 2015, 5, 17799. [Google Scholar] [CrossRef] [Green Version]
- Thompson, C.M.; Davis, E.; Carrigan, C.N.; Cox, H.D.; Bridges, R.J.; Gerdes, J.M. Inhibitor of the glutamate vesicular transporter (VGLUT). Curr. Med. Chem. 2005, 12, 2041–2056. [Google Scholar] [CrossRef]
- Yoon, S.J.; Lyoo, I.K.; Haws, C.; Kim, T.S.; Cohen, B.M.; Renshaw, P.F. Decreased glutamate/glutamine levels may mediate cytidine’s efficacy in treating bipolar depression: A longitudinal proton magnetic resonance spectroscopy study. Neuropsychopharmacology 2009, 34, 1810–1818. [Google Scholar] [CrossRef] [Green Version]
- Adams, C.J.; Boult, C.H.; Deadman, B.J.; Farr, J.M.; Grainger, M.N.; Manley-Harris, M.; Snow, M.J. Isolation by HPLC and characterisation of the bioactive fraction of New Zealand manuka (Leptospermum scoparium) honey. Carbohydr. Res. 2008, 343, 651–659. [Google Scholar] [CrossRef] [PubMed]
- Mavric, E.; Wittmann, S.; Barth, G.; Henle, T. Identification and quantification of methylglyoxal as the dominant antibacterial constituent of Manuka (Leptospermum scoparium) honeys from New Zealand. Mol. Nutr. Food Res. 2008, 52, 483–489. [Google Scholar] [CrossRef] [PubMed]
- Furuya, A.; Uozaki, M.; Yamasaki, H.; Arakawa, T.; Arita, M.; Koyama, A.H. Antiviral effects of ascorbic and dehydroascorbic acids in vitro. Int. J. Mol. Med. 2008, 22, 541–545. [Google Scholar]
- Manning, K.; Al-Dhalimy, M.; Finegold, M.; Grompe, M. In vivo suppressor mutations correct a murine model of hereditary tyrosinemia type I. Proc. Natl. Acad. Sci. USA 1999, 96, 11928–11933. [Google Scholar] [CrossRef] [Green Version]
- Uozaki, M.; Ikeda, K.; Tsujimoto, K.; Nishide, M.; Yamasaki, H.; Khamsri, B.; Koyama, A.H. Antiviral effects of dehydroascorbic acid. Exp. Ther. Med. 2010, 1, 983–986. [Google Scholar] [CrossRef] [PubMed]
- Magni, G.; Amici, A.; Emanuelli, M.; Orsomando, G.; Raffaelli, N.; Ruggieri, S. Structure and function of nicotinamide mononucleotide adenylyltransferase. Curr. Med. Chem. 2004, 11, 873–885. [Google Scholar] [CrossRef]
- Revollo, J.R.; Korner, A.; Mills, K.F.; Satoh, A.; Wang, T.; Garten, A.; Dasgupta, B.; Sasaki, Y.; Wolberger, C.; Townsend, R.R.; et al. Nampt/PBEF/Visfatin regulates insulin secretion in beta cells as a systemic NAD biosynthetic enzyme. Cell Metab. 2007, 6, 363–375. [Google Scholar] [CrossRef] [Green Version]
- Manoli, I.; Venditti, C.P. Disorders of branched chain amino acid metabolism. Transl. Sci. Rare Dis. 2016, 1, 91–110. [Google Scholar] [CrossRef] [Green Version]
- Davies, O.; Mendes, P.; Smallbone, K.; Malys, N. Characterisation of multiple substrate-specific (d)ITP/(d)XTPase and modelling of deaminated purine nucleotide metabolism. BMB Rep. 2012, 45, 259–264. [Google Scholar] [CrossRef] [Green Version]
- Kim, C.S.; Lee, C.H.; Chung, J.W.; Kim, C.D. Interleukin-1 alpha, interleukin-1 beta and interleukin-8 gene expression in human aural cholesteatomas. Acta Otolaryngol. 1996, 116, 302–306. [Google Scholar] [CrossRef]
- Van Hoeyveld, E.M.; Stevens, E.A. Stabilizing effect of epsilon-aminocaproic acid on allergenic extracts. J. Allergy Clin. Immunol. 1985, 76, 543–550. [Google Scholar] [CrossRef] [Green Version]
- Adibhatla, R.M.; Hatcher, J.F. Cytidine 5′-diphosphocholine (CDP-choline) in stroke and other CNS disorders. Neurochem. Res. 2005, 30, 15–23. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Bekdash, R.A. Choline and the Brain: An Epigenetic Perspective. Adv. Neurobiol. 2016, 12, 381–399. [Google Scholar] [CrossRef] [PubMed]
- Zeisel, S.H.; da Costa, K.A. Choline: An essential nutrient for public health. Nutr. Rev. 2009, 67, 615–623. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Clark, J.B. N-acetyl aspartate: A marker for neuronal loss or mitochondrial dysfunction. Dev. Neurosci. 1998, 20, 271–276. [Google Scholar] [CrossRef]
- Moffett, J.R.; Ross, B.; Arun, P.; Madhavarao, C.N.; Namboodiri, A.M. N-Acetylaspartate in the CNS: From neurodiagnostics to neurobiology. Prog. Neurobiol. 2007, 81, 89–131. [Google Scholar] [CrossRef] [Green Version]
- Pang, Z.; Chong, J.; Zhou, G.; de Lima Morais, D.A.; Chang, L.; Barrette, M.; Gauthier, C.; Jacques, P.E.; Li, S.; Xia, J. MetaboAnalyst 5.0: Narrowing the gap between raw spectra and functional insights. Nucleic Acids Res. 2021, 49, W388–W396. [Google Scholar] [CrossRef]







{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
| Metabolite | Function | References |
|---|---|---|
| Levulinic acid | Important for ATP production when ETC becomes dysfunctional | [29,30,31] |
| Aconitic acid | Intermediate in the citric acid cycle, energy production | [32,33] |
| Sedoheptulose 7-phosphate | Produces ADP and sedoheptulose 7-phosphate, intermediate in the respiratory pathways, generation of energy, reduction of proinflammatory markers | [34,35,36] |
| Xanthurenic acid | Potent vesicular glutamate transporter (VGLUT) inhibitor, involved in neuronal function and firing potentials | [37,38,39] |
| Cytidine | Controls neuronal-glial glutamate cycling, helping decrease glutamate/glutamine ratio | [40] |
| Methylglyoxal | Antiviral, anti-inflammatory, and antioxidant | [41,42] |
| Dehydroascorbic acid | Protective against viruses, involved in metabolic disorder tyrosinemia type I | [43,44,45] |
| Nicotinamide mononucleotide | Key intermediate metabolite in the nicotinate and nicotinamide metabolism pathway, energy production | [46,47] |
| Thiamine | Acts as coenzyme, involved in 2-methyl-3-hydroxybutyryl-coadehydrogenase deficiency pathway | [48] |
| Xanthosine | Intermediate in purine metabolism, involved in purine nucleoside phosphorylase deficiency | [49] |
| 6-aminocaproic acid | Coagulation, shaping and modulation of plasma proteins | [50,51] |
| CDP-choline | Component of cell membranes, generation of acetylcholine, production of homocysteine | [52,53,54] |
| N-Acetylaspartic acid | Fluid balance in the brain, energy production | [55,56] |
Publisher’s Note: MDPI stays neutral with regard to jurisdictional claims in published maps and institutional affiliations. |
© 2022 by the authors. Licensee MDPI, Basel, Switzerland. This article is an open access article distributed under the terms and conditions of the Creative Commons Attribution (CC BY) license (https://creativecommons.org/licenses/by/4.0/).
Share and Cite
Gautam, M.; Carratore, R.D.; Helmold, B.; Tessa, A.; Gozutok, O.; Chandel, N.; Idrisoglu, H.; Bongioanni, P.; Battini, R.; Ozdinler, P.H. 2-Year-Old and 3-Year-Old Italian ALS Patients with Novel ALS2 Mutations: Identification of Key Metabolites in Their Serum and Plasma. Metabolites 2022, 12, 174. https://doi.org/10.3390/metabo12020174
Gautam M, Carratore RD, Helmold B, Tessa A, Gozutok O, Chandel N, Idrisoglu H, Bongioanni P, Battini R, Ozdinler PH. 2-Year-Old and 3-Year-Old Italian ALS Patients with Novel ALS2 Mutations: Identification of Key Metabolites in Their Serum and Plasma. Metabolites. 2022; 12(2):174. https://doi.org/10.3390/metabo12020174
Chicago/Turabian StyleGautam, Mukesh, Renata Del Carratore, Benjamin Helmold, Alessandra Tessa, Oge Gozutok, Navdeep Chandel, Halil Idrisoglu, Paolo Bongioanni, Roberta Battini, and P.Hande Ozdinler. 2022. "2-Year-Old and 3-Year-Old Italian ALS Patients with Novel ALS2 Mutations: Identification of Key Metabolites in Their Serum and Plasma" Metabolites 12, no. 2: 174. https://doi.org/10.3390/metabo12020174
APA StyleGautam, M., Carratore, R. D., Helmold, B., Tessa, A., Gozutok, O., Chandel, N., Idrisoglu, H., Bongioanni, P., Battini, R., & Ozdinler, P. H. (2022). 2-Year-Old and 3-Year-Old Italian ALS Patients with Novel ALS2 Mutations: Identification of Key Metabolites in Their Serum and Plasma. Metabolites, 12(2), 174. https://doi.org/10.3390/metabo12020174