
Microbiome, Mycobiome and Related Metabolites Alterations in Patients with Metabolic Syndrome—A Pilot Study


 ,
,  ,
,  ,
,  and
and
Abstract
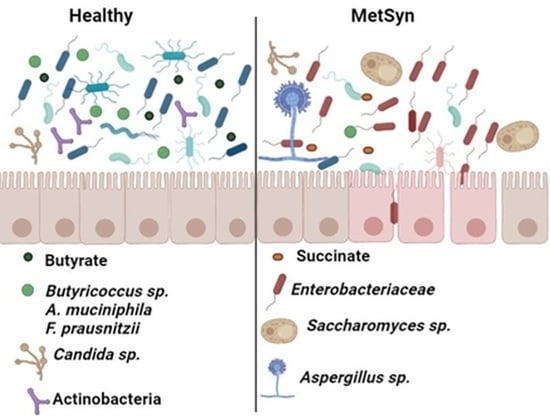
:
1. Introduction
2. Results
3. Discussion
4. Materials and Methods
4.1. Study Group
4.2. Microbiota and Mycobiota Analysis
4.3. Metabolite Analysis
4.4. Statistical Analysis
5. Conclusions
Supplementary Materials
Author Contributions
Funding
Institutional Review Board Statement
Informed Consent Statement
Data Availability Statement
Acknowledgments
Conflicts of Interest
References
- Saklayen, M.G. The global epidemic of the metabolic syndrome. Curr. Hypertens. Rep. 2018, 20, 12. [Google Scholar] [CrossRef] [Green Version]
- Carrizales-Sánchez, A.K.; García-Cayuela, T.; Hernández-Brenes, C.; Senés-Guerrero, C. Gut microbiota associations with metabolic syndrome and relevance of its study in pediatric subjects. Gut Microbes 2021, 13, 1960135. [Google Scholar] [CrossRef]
- Popa, S.; Moţa, M.; Popa, A.; Moţa, E.; Serafinceanu, C.; Guja, C.; Catrinoiu, D.; Hâncu, N.; Lichiardopol, R.; Bala, C.; et al. Prevalence of overweight/obesity, abdominal obesity and metabolic syndrome and atypical cardiometabolic phenotypes in the adult Romanian population: PREDATORR study. J. Endocrinol. Investig. 2016, 39, 1045–1053. [Google Scholar] [CrossRef] [PubMed]
- Lazar, V.; Ditu, L.-M.; Pircalabioru, G.G.; Gheorghe, I.; Curutiu, C.; Holban, A.M.; Picu, A.; Petcu, L.; Chifiriuc, M.C. Aspects of Gut Microbiota and Immune System Interactions in Infectious Diseases, Immunopathology, and Cancer. Front. Immunol. 2018, 9, 1830. [Google Scholar] [CrossRef] [Green Version]
- Lazar, V.; Ditu, L.-M.; Pircalabioru, G.G.; Picu, A.; Petcu, L.; Cucu, N.; Chifiriuc, M.C. Gut Microbiota, Host Organism, and Diet Trialogue in Diabetes and Obesity. Front. Nutr. 2019, 6, 21. [Google Scholar] [CrossRef] [Green Version]
- Chelariu, M.; Grosu, M.; Gheorghe, I.; Gradisteanu, G.; Picu, A.; Petcu, L.; Popa, M.; Chifiriuc, M.C.; Ditu, L.M.; Lazar, V. Host metabolic syndrome can disrupt the intestinal microbiota and promote the acquisition of resistance and virulence genes in Enterobacteriaceae stains. Rom. Biotechnol. Lett. 2017, 22, 12643–12650. [Google Scholar]
- Mazidi, M.; Rezaie, P.; Kengne, A.P.; Mobarhan, M.G.; Ferns, G.A. Gut microbiome and metabolic syndrome. Diabetes Metab. Syndr. Clin. Res. Rev. 2016, 10, S150–S157. [Google Scholar] [CrossRef]
- Clarke, S.; Murphy, E.F.; O’Sullivan, O.; Lucey, A.; Humphreys, M.; Hogan, A.; Hayes, P.; O’Reilly, M.; Jeffery, I.; Wood-Martin, R.; et al. Exercise and associated dietary extremes impact on gut microbial diversity. Gut 2014, 63, 1913–1920. [Google Scholar] [CrossRef] [Green Version]
- Gildner, T.E. Links between metabolic syndrome and the microbiome. Evol. Med. Public Health 2020, 2020, 45–46. [Google Scholar] [CrossRef]
- Mocanu, V.; Zhang, Z.; Deehan, E.C.; Kao, D.H.; Hotte, N.; Karmali, S.; Birch, D.W.; Samarasinghe, K.K.; Walter, J.; Madsen, K.L. Fecal microbial transplantation and fiber supplementation in patients with severe obesity and metabolic syndrome: A randomized double-blind, placebo-controlled phase 2 trial. Nat. Med. 2021, 27, 1272–1279. [Google Scholar] [CrossRef]
- Malnick, S.; Fisher, D.; Somin, M.; Neuman, M. Treating the Metabolic Syndrome by Fecal Transplantation—Current Status. Biology 2021, 10, 447. [Google Scholar] [CrossRef]
- Porras, A.M.; Shi, Q.; Zhou, H.; Callahan, R.; Montenegro-Bethancourt, G.; Solomons, N.; Brito, I.L. Geographic differences in gut microbiota composition impact susceptibility to enteric infection. Cell Rep. 2021, 36, 109457. [Google Scholar] [CrossRef]
- Jiang, T.T.; Shao, T.Y.; Ang, W.X.G.; Kinder, J.M.; Turner, L.H.; Pham, G. Commensal fungi recapitulate the protective benefits of intestinal bacteria. Cell Host Microbe 2017, 22, 809–816. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Nash, A.K.; Auchtung, T.A.; Wong, M.C.; Smith, D.P.; Gesell, J.R.; Ross, M.C. The gut mycobiome of the Human Microbiome Project healthy cohort. Microbiome 2017, 5, 153. [Google Scholar] [CrossRef]
- Eckburg, P.B.; Bik, E.M.; Bernstein, C.N.; Purdom, E.; Dethlefsen, L.; Sargent, M.; Gill, S.R.; Nelson, K.E.; Relman, D.A. Diversity of the human intestinal microbial flora. Science 2005, 308, 1635–1638. [Google Scholar] [CrossRef] [Green Version]
- Ley, R.E.; Bäckhed, F.; Turnbaugh, P.; Lozupone, C.A.; Knight, R.D.; Gordon, J.I. Obesity alters gut microbial ecology. Proc. Natl. Acad. Sci. USA 2005, 102, 11070–11075. [Google Scholar] [CrossRef] [Green Version]
- Binda, C.; Lopetuso, L.R.; Rizzatti, G.; Gibiino, G.; Cennamo, V.; Gasbarrini, A. Actinobacteria: A relevant minority for the maintenance of gut homeostasis. Dig. Liver Dis. 2018, 50, 421–428. [Google Scholar] [CrossRef]
- Hayashi, H.; Sakamoto, M.; Benno, Y. Phylogenetic analysis of the human gut microbiota using 16S rDNA clone libraries and strictly anaerobic culture-based methods. Microbiol. Immunol. 2002, 46, 535–548. [Google Scholar] [CrossRef]
- Baldelli, V.; Scaldaferri, F.; Putignani, L.; Del Chierico, F. The Role of Enterobacteriaceae in Gut Microbiota Dysbiosis in Inflammatory Bowel Diseases. Microorganisms 2021, 9, 697. [Google Scholar] [CrossRef]
- Gradisteanu, G.P.; Stoica, R.A.; Petcu, L.; Picu, A.; Suceveanu, A.P.; Salmen, T.; Stefan, D.S.; Serafinceanu, C.; Chifiriuc, M.C.; Stoian, A.P. Microbiota signatures in type-2 diabetic patients with chronic kidney disease—A Pilot Study. J. Mind Med. Sci. 2019, 6, 130–136. [Google Scholar] [CrossRef]
- Rausch, P.; Steck, N.; Suwandi, A.; Seidel, J.A.; Künzel, S.; Bhullar, K.; Basic, M.; Bleich, A.; Johnsen, J.M.; Vallance, B.; et al. Expression of the Blood-Group-Related Gene B4galnt2 Alters Susceptibility to Salmonella Infection. PLoS Pathog. 2015, 11, e1005008. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Forbes, J.D.; Van Domselaar, G.; Bernstein, C.N. The Gut Microbiota in Immune-Mediated Inflammatory Diseases. Front. Microbiol. 2016, 7, 1081. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Cuív, P.Ó.; Klaassens, E.S.; Durkin, A.S.; Harkins, D.M.; Foster, L.; McCorrison, J.; Torralba, M.; Nelson, K.E.; Morrison, M. Draft Genome Sequence of Turicibacter sanguinis PC909, Isolated from Human Feces. J. Bacteriol. 2011, 193, 1288–1289. [Google Scholar] [CrossRef] [Green Version]
- Pircalabioru, G.G.; Aviello, G.; Kubica, M.; Zhdanov, A.; Paclet, M.-H.; Brennan, L.; Hertzberger, R.; Papkovsky, D.; Bourke, B.; Knaus, U.G. Defensive Mutualism Rescues NADPH Oxidase Inactivation in Gut Infection. Cell Host Microbe 2016, 19, 651–663. [Google Scholar] [CrossRef] [PubMed]
- Heeney, D.D.; Gareau, M.G.; Marco, M.L. Intestinal Lactobacillus in health and disease, a driver or just along for the ride? Curr. Opin. Biotechnol. 2018, 49, 140–147. [Google Scholar] [CrossRef] [Green Version]
- Leylabadlo, H.E.; Ghotaslou, R.; Feizabadi, M.M.; Farajnia, S.; Moaddab, S.Y.; Ganbarov, K.; Khodadadi, E.; Tanomand, A.; Sheykhsaran, E.; Yousefi, B.; et al. The critical role of Faecalibacterium prausnitzii in human health: An overview. Microb. Pathog. 2020, 149, 104344. [Google Scholar] [CrossRef]
- Zhou, K. Strategies to promote abundance of Akkermansia muciniphila, an emerging probiotics in the gut, evidence from dietary intervention studies. J. Funct. Foods 2017, 33, 194–201. [Google Scholar] [CrossRef]
- Qin, J.; Li, Y.; Cai, Z.; Li, S.; Zhu, J.; Zhang, F.; Liang, S.; Zhang, W.; Guan, Y.; Shen, D.; et al. A metagenome-wide association study of gut microbiota in type 2 diabetes. Nature 2012, 490, 55–60. [Google Scholar] [CrossRef]
- Tilg, H.; Moschen, A.R. Microbiota and diabetes: An evolving relationship. Gut 2014, 63, 1513–1521. [Google Scholar] [CrossRef]
- Albenberg, L.; Esipova, T.V.; Judge, C.P.; Bittinger, K.; Chen, J.; Laughlin, A.; Grunberg, S.; Baldassano, R.N.; Lewis, J.D.; Li, H.; et al. Correlation Between Intraluminal Oxygen Gradient and Radial Partitioning of Intestinal Microbiota. Gastroenterology 2014, 147, 1055–1063.e8. [Google Scholar] [CrossRef] [Green Version]
- Ribaldone, D.G.; Pellicano, R.; Actis, G.C. Inflammation: A highly conserved, Janus-like phenomenon—A gastroenterologist’ perspective. Klin. Wochenschr. 2018, 96, 861–871. [Google Scholar] [CrossRef]
- Rigottier-Gois, L. Dysbiosis in inflammatory bowel diseases: The oxygen hypothesis. ISME J. 2013, 7, 1256–1261. [Google Scholar] [CrossRef] [PubMed]
- Colombo, A.V.; Sadler, R.K.; Llovera, G.; Singh, V.; Roth, S.; Heindl, S.; Monasor, L.S.; Verhoeven, A.; Peters, F.; Parhizkar, S.; et al. Microbiota-derived short chain fatty acids modulate microglia and promote Aβ plaque deposition. eLife 2021, 10, e59826. [Google Scholar] [CrossRef] [PubMed]
- Luo, M.; Zhuang, X.; Tian, Z.; Xiong, L. Alterations in short-chain fatty acids and serotonin in irritable bowel syndrome: A systematic review and meta-analysis. BMC Gastroenterol. 2021, 21, 14. [Google Scholar] [CrossRef]
- Canani, R.B.; Di Costanzo, M.; Leone, L.; Pedata, M.; Meli, R.; Calignano, A. Potential beneficial effects of butyrate in intestinal and extraintestinal diseases. World J. Gastroenterol. 2011, 17, 1519–1528. [Google Scholar] [CrossRef] [PubMed]
- Forslund, K.; Hildebrand, F.; Nielsen, T.; Falony, G.; Le Chatelier, E.; Sunagawa, S.; Prifti, E.; Vieira-Silva, S.; Gudmundsdottir, V.; Krogh Pedersen, H.; et al. Disentangling type 2 diabetes and metformin treatment signatures in the human gut microbiota. Nature 2015, 528, 262–266. [Google Scholar] [CrossRef]
- Shin, N.R.; Lee, J.C.; Lee, H.Y.; Kim, M.S.; Whon, T.W.; Lee, M.S.; Bae, J.W. An increase in the Akkermansia spp. population induced by metformin treatment improves glucose homeostasis in diet-induced obese mice. Gut 2014, 63, 727–735. [Google Scholar] [CrossRef] [Green Version]
- Burton, J.; Johnson, M.; Johnson, J.; Hsia, D.; Greenway, F.L.; Heiman, M.L. Addition of a Gastrointestinal Microbiome Modulator to Metformin Improves Metformin Tolerance and Fasting Glucose Levels. J. Diabetes Sci. Technol. 2015, 9, 808–814. [Google Scholar] [CrossRef] [Green Version]
- Vital, M.; Karch, A.; Pieper, D.H. Colonic Butyrate-Producing Communities in Humans: An Overview Using Omics Data. mSystems. 2017, 6, e00130. [Google Scholar] [CrossRef] [Green Version]
- Delzenne, N.M.; Williams, C.M. Prebiotics and lipid metabolism. Curr. Opin. Lipidol. 2002, 13, 61–67. [Google Scholar] [CrossRef] [Green Version]
- Jan, G.; Belzacq, A.-S.; Haouzi, D.; Rouault, A.; Métivier, D.; Kroemer, G.; Brenner, C. Propionibacteria induce apoptosis of colorectal carcinoma cells via short-chain fatty acids acting on mitochondria. Cell Death Differ. 2002, 9, 179–188. [Google Scholar] [CrossRef] [PubMed]
- Louis, P.; Flint, H.J. Formation of propionate and butyrate by the human colonic microbiota. Environ. Microbiol. 2017, 19, 29–41. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Abedi, E.; Hashemi, S.M.B. Lactic acid production—Producing microorganisms and substrates sources-state of art. Heliyon 2020, 6, e04974. [Google Scholar] [CrossRef] [PubMed]
- Connors, J.; Dawe, N.; Van Limbergen, J. The Role of Succinate in the Regulation of Intestinal Inflammation. Nutrients 2018, 11, 25. [Google Scholar] [CrossRef] [Green Version]
- Zhao, L.; Yang, W.; Chen, Y.; Huang, F.; Lu, L.; Lin, C.; Huang, T.; Ning, Z.; Zhai, L.; Zhong, L.L.; et al. A Clostridia-rich microbiota enhances bile acid excretion in diarrhea-predominant irritable bowel syndrome. J. Clin. Investig. 2020, 130, 438–450. [Google Scholar] [CrossRef] [Green Version]
- Imae, M.; Asano, T.; Murakami, S. Potential role of taurine in the prevention of diabetes and metabolic syndrome. Amino Acids 2014, 46, 81–88. [Google Scholar] [CrossRef]
- Wójcik, O.P.; Koenig, K.L.; Zeleniuch-Jacquotte, A.; Pearte, C.; Costa, M.; Chen, Y. Serum taurine and risk of coronary heart disease: A prospective, nested case–control study. Eur. J. Nutr. 2013, 52, 169–178. [Google Scholar] [CrossRef]
- Beheshti-Maal, A.; Shahrokh, S.; Ansari, S.; Mirsamadi, E.S.; Yadegar, A.; Mirjalali, H.; Zali, M.R. Gut mycobiome: The probable determinative role of fungi in IBD patients. Mycoses 2021, 64, 468–476. [Google Scholar] [CrossRef]
- Tampakakis, E.; Peleg, A.Y.; Mylonakis, E. Interaction of Candida albicans with an Intestinal Pathogen, Salmonella enterica Serovar Typhimurium. Eukaryot. Cell 2009, 8, 732–737. [Google Scholar] [CrossRef] [Green Version]
- van Tilburg Bernardes, E.; Pettersen, V.K.; Gutierrez, M.W.; Laforest-Lapointe, I.; Jendzjowsky, N.G.; Cavin, J.B.; Vicentini, F.A.; Keenan, C.M.; Ramay, H.R.; Samara, J.; et al. Intestinal fungi are causally implicated in microbiome assembly and immune development in mice. Nat. Commun. 2020, 11, 2577. [Google Scholar] [CrossRef]
- Wheeler, M.L.; Limon, J.J.; Bar, A.S.; Leal, C.A.; Gargus, M.; Tang, J.; Brown, J.; Funari, V.A.; Wang, H.L.; Crother, T.; et al. Immunological Consequences of Intestinal Fungal Dysbiosis. Cell Host Microbe 2016, 19, 865–873. [Google Scholar] [CrossRef] [Green Version]
- Corb Aron, R.A.; Abid, A.; Vesa, C.M.; Nechifor, A.C.; Behl, T.; Ghitea, T.C.; Munteanu, M.A.; Fratila, O.; Andronie-Cioara, F.L.; Toma, M.M.; et al. Recognizing the Benefits of Pre-/Probiotics in Metabolic Syndrome and Type 2 Diabetes Mellitus Considering the Influence of Akkermansia muciniphila as a Key Gut Bacterium. Microorganisms 2021, 9, 618. [Google Scholar] [CrossRef]
- Asnicar, F.; Berry, S.E.; Valdes, A.M.; Nguyen, L.H.; Piccinno, G.; Drew, D.A.; Leeming, E.; Gibson, R.; Le Roy, C.; Al Khatib, H.; et al. Microbiome connections with host metabolism and habitual diet from 1,098 deeply phenotyped individuals. Nat. Med. 2021, 27, 321–332. [Google Scholar] [CrossRef]
- Alberti, K.G.M.M.; Zimmet, P.; Shaw, J. Metabolic syndrome—A new world-wide definition. A Consensus Statement from the International Diabetes Federation. Diabet. Med. 2006, 23, 469–480. [Google Scholar] [CrossRef]
- Rinttila, T.; Kassinen, A.; Malinen, E.; Krogius, L.; Palva, A. Development of an extensive set of 16S rDNA-targeted primers for quantification of pathogenic and indigenous bacteria in faecal samples by real-time PCR. J. Appl. Microbiol. 2004, 97, 1166–1177. [Google Scholar] [CrossRef]
- McAuley, J.; Linden, S.K.; Png, C.W.; King, R.M.; Pennington, H.L.; Gendler, S.J.; Florin, T.H.; Hill, G.; Korolik, V.; McGuckin, M.A. MUC1 cell surface mucin is a critical element of the mucosal barrier to infection. J. Clin. Investig. 2007, 117, 2313–2324. [Google Scholar] [CrossRef]
- Matsuki, T.; Watanabe, K.; Fujimoto, J.; Takada, T.; Tanaka, R. Use of 16S rRNA Gene-Targeted Group-Specific Primers for Real-Time PCR Analysis of Predominant Bacteria in Human Feces. Appl. Environ. Microbiol. 2004, 70, 7220–7228. [Google Scholar] [CrossRef] [Green Version]





{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
| Healthy (n = 30) | MetSyn (n = 30) | p Value | |
|---|---|---|---|
| Sex | 22 females, 8 males | 25 females, 5 males | - |
| Age | 56 ± 11.25 | 62 ± 11.39 | 0.0446 |
| BMI | 25.3 ± 1.255 | 32 ± 5.39 | p < 0.0001 |
| HbAc | 5.4 ± 0.385 | 6.6 ± 1.472 | p < 0.0001 |
| TG | 88 ± 20.15 | 124 ± 54.5 | 0.0012 |
| HDL | 66 ±5.75 | 49 ± 8.43 | p < 0.0001 |
| LDL | 99 ±19.95 | 115 ± 35.64 | 0.0361 |
| Total cholesterol | 178 ± 29.56 | 199 ± 49.92 | 0.0522 |
| Type 2 diabetes | - | 22/30 (73.33%) | - |
| Hypertension (%) | 3/30 (10%) | 25/30 (83.33%) | - |
| Insulin (%) | - | 17/30 (56.66%) | - |
| Metformin (%) | - | 13/30 (43.33%) | - |
| Statins (%) | 0 | 23/30 (76.66%) | - |
| Taxonomic Target | Sequence |
|---|---|
| Actinobacteria | TGTAGCGGTGGAATGCGC |
| AATTAAGCCACATGCTCCGCT | |
| Tenericutes | ATGTGTAGCGGTAAAATGCGTAA |
| CATACTTGCGTACGTACTACT | |
| Verrucomicrobia | TCAGGTCAGTATGGCCCTTAT |
| CAGTTTTCAGGATTTCCTCCGCC | |
| Bacteroides | CCTACGATGGATAGGGGTT |
| CACGCTACTTGGCTGGTTCAG | |
| Betaproteobacteria | AACGCGAAAAACCTTACCTACC |
| TGCCCTTTCGTAGCAACTAGTG | |
| Butyricicoccus sp. | ACCTGAAGAATAAGCTCC |
| GATAACGCTTGCTCCCTACGT | |
| Gamma proteobacteria | GCTAACGCATTAAGTACCCCG |
| GCCATGCAGCACCTGTCT | |
| Akkermansia muciniphila | GCGTAGGCTGTTTCGTAAGTCGTGTGTGAAAG |
| GAGTGTTCCCGATATCTACGCATTTCA | |
| Eubacteria | ACTCCTACGGGAGGCAGCAGT |
| ATTACCGCGGCTGCTGGC | |
| Lactobacillus | ACGAGTAGGGAAATCTTCCA |
| CACCGCTACACATGGAG | |
| BPP | GGTGTCGGCTTAAGTGCCAT |
| CGGACGTAAGGGCCGTGC | |
| Clostridium leptum | GCACAAGCAGTGGAGT |
| CTTCCTCCGTTTTGTCAA | |
| Clostridium cocoides | GACGCCGCGTGAAGGA |
| AGCCCCAGCCTTTCACATC | |
| Ruminococcus sp. | ACTGAGAGGTTGAACGGCCA |
| CCTTTACACCCAGTAATTCCGGA | |
| Turicibacter sp. | CAGACGGGGACAACGATTGGA |
| TACGCATCGTCGCCTTGGGTA | |
| Firmicutes | GGAGCATGTGGTTTAATTCGAAGCA |
| AGCTGACGACAACCATGCAC | |
| Bacteroidetes | GGAACATGTGGTTTAATTCGATGAT |
| AGCTGACGACAACCATGCAG | |
| F. prausnitzii | CCCTTCAGTGCCGCAGT |
| GTCGCAGGATGTCAAGAC | |
| ARNr 18S | ATTGGAGGGCAAGTCTGGTG |
| CCGATCCCTAGTCGGCATAG | |
| Saccharomyces sp. | AGGAGTGCGGTTCTTTG |
| TACTTACCGAGGCAAGCTACA | |
| Candida sp. | TTTATCAACTTGTCACACCAGA |
| ATCCCGCCTTACCACTACCG | |
| Aspergillus sp. | GTGGAGTGATTTGTCTGCTTAATTG |
| TCTAAGGGCATCACAGACCTGTT |
Publisher’s Note: MDPI stays neutral with regard to jurisdictional claims in published maps and institutional affiliations. |
© 2022 by the authors. Licensee MDPI, Basel, Switzerland. This article is an open access article distributed under the terms and conditions of the Creative Commons Attribution (CC BY) license (https://creativecommons.org/licenses/by/4.0/).
Share and Cite
Gradisteanu Pircalabioru, G.; Ilie, I.; Oprea, L.; Picu, A.; Petcu, L.M.; Burlibasa, L.; Chifiriuc, M.-C.; Musat, M. Microbiome, Mycobiome and Related Metabolites Alterations in Patients with Metabolic Syndrome—A Pilot Study. Metabolites 2022, 12, 218. https://doi.org/10.3390/metabo12030218
Gradisteanu Pircalabioru G, Ilie I, Oprea L, Picu A, Petcu LM, Burlibasa L, Chifiriuc M-C, Musat M. Microbiome, Mycobiome and Related Metabolites Alterations in Patients with Metabolic Syndrome—A Pilot Study. Metabolites. 2022; 12(3):218. https://doi.org/10.3390/metabo12030218
Chicago/Turabian StyleGradisteanu Pircalabioru, Gratiela, Iuliana Ilie, Luciana Oprea, Ariana Picu, Laura Madalina Petcu, Liliana Burlibasa, Mariana-Carmen Chifiriuc, and Madalina Musat. 2022. "Microbiome, Mycobiome and Related Metabolites Alterations in Patients with Metabolic Syndrome—A Pilot Study" Metabolites 12, no. 3: 218. https://doi.org/10.3390/metabo12030218
APA StyleGradisteanu Pircalabioru, G., Ilie, I., Oprea, L., Picu, A., Petcu, L. M., Burlibasa, L., Chifiriuc, M. -C., & Musat, M. (2022). Microbiome, Mycobiome and Related Metabolites Alterations in Patients with Metabolic Syndrome—A Pilot Study. Metabolites, 12(3), 218. https://doi.org/10.3390/metabo12030218