
MetaboListem and TABoLiSTM: Two Deep Learning Algorithms for Metabolite Named Entity Recognition


Abstract
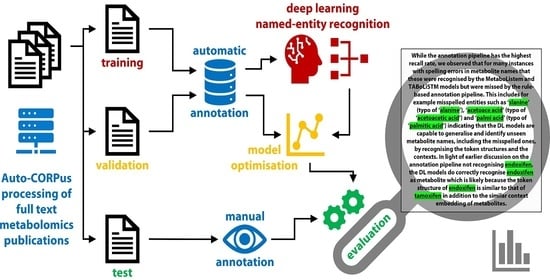
:
1. Introduction
2. Results
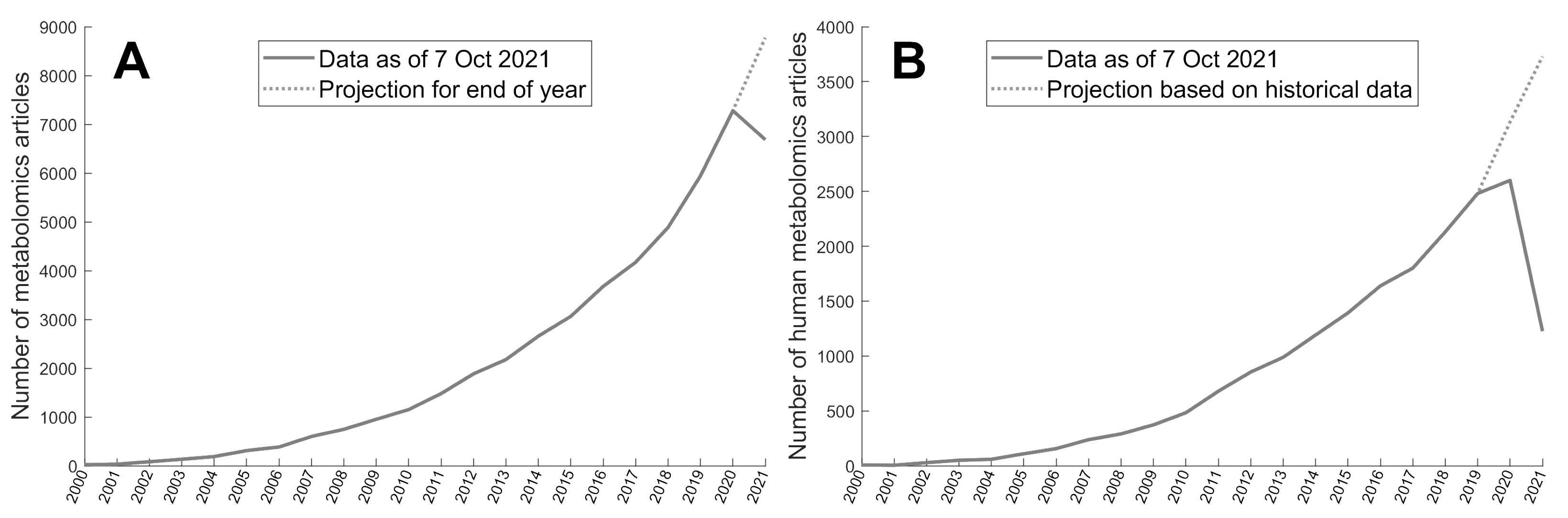
2.1. Metabolomics Corpus
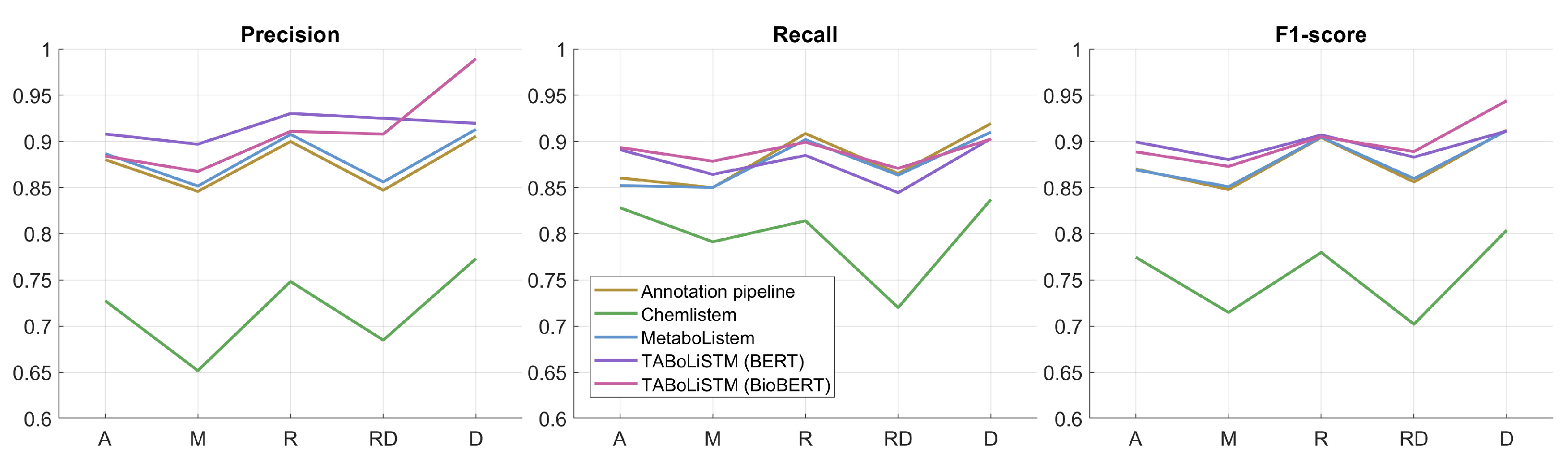
2.2. Model Performance
2.3. Automated Literature Review of the Metabolomics Corpus
3. Discussion
3.1. Annotation Pipeline
3.2. Metabolomics Corpus
3.3. Deep Learning Models
3.4. Biological Interpretation
4. Materials and Methods
4.1. Dataset
4.2. Rule-Based Annotation Pipeline
4.2.1. Pre-Processing
4.2.2. Metabolite Identification
4.2.3. Post-Processing
4.3. Training, Validation and Test Set Generation and Evaluation
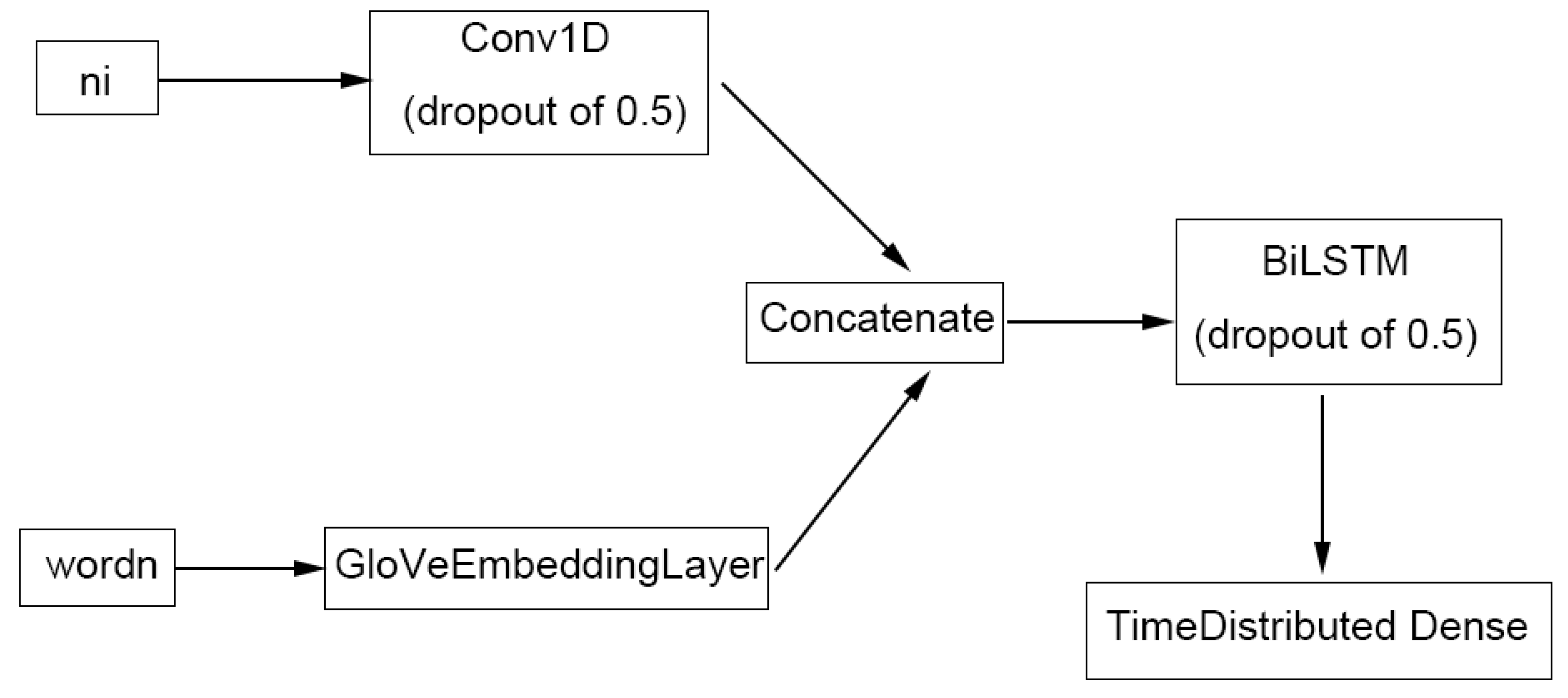
4.4. Metabolite NER Using LSTM
4.4.1. Pre-Processing
4.4.2. ChemListem and MetaboListem
4.4.3. TABoLiSTM (Transformer-Affixed Bidirectional LSTM)
4.4.4. Post-Processing
4.5. Metabolite Co-Occurence Network
5. Conclusions
Author Contributions
Funding
Institutional Review Board Statement
Informed Consent Statement
Data Availability Statement
Acknowledgments
Conflicts of Interest
Abbreviations
| Auto-CORPus | Automated and Consistent Outputs from Research Publications |
| BERT | Bidirectional Encoder Representations from Transformers |
| BiLSTM | Bidirectional Long Short-Term Memory |
| BioBERT | BERT for Biomedical Text Mining |
| CRF | Conditional Random Fields |
| DL | Deep Learning |
| GloVe | Global Vectors for word representation |
| HMDB | Human Metabolome DataBase |
| HTML | HyperText Markup Language |
| IAO | Information Artefact Ontology |
| JSON | JavaScript Object Notation |
| NER | Named Entity Recognition |
| NLP | Natural Language Processing |
| OA | Open Access |
| PMC | PubMed Central |
| PMCID | PMC Identifier |
| TABoLiSTM | Transformer-Affixed Bidirectional LSTM |
Appendix A. Metabolomics Studies PubMed Search

Appendix B. Network Representing the Co-Occurrence of Metabolites in Smoking Articles

Appendix C. Regular Expression Table

{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
| Regular Expression | Example Metabolite |
|---|---|
| (\ (?\ )?-?\w+(oxy|yl|ol|ine|ene|ose|ane|ate|ester |isomer|ium|ino|ide|one|oxo|en|estra))+ (-\w+)s?\ (?\ )?(ic)?(?=[-,\.\!\?; ]) | 3-Methoxy-4-hydroxy-phenyl-acetate |
| ([a-z]|-|[0-9])*([0-9]+([a-z]|alpha|beta|sigma| omega|tau|epsilon|zeta|[-])* [ \), \+ \- \] \}])+(([a-z]|[0-9])+-)+ | 5alpha-Androstan-3alpha,17beta-diol disulfate |
| (([a-z]?[0-9]+(:)[0-9]+)(/[a-z]?[0-9]+(:)[0-9]+)*) | d18:1/16:0 |
| \d+[a-z]?(,\d+[a-z]?)+\)?- | (1S,10R,11S,15S)-5-hydroxy-4-methoxy -15-methyltetracycloheptadeca -2(7),3,5-trien-14-one |
| ([1-9]|\()((alpha|beta|sigma|omega|tau|epsilon| zeta|[-]|Z|E|L|R|S|-|\+),?)+\)- | (11E)-Octadecenoic acid |
| (oline|dino|etin|olin|ecine|idine|noid|oate|gmine| [a-z]dine|none|igine|zole|pine|nime|anide|acene| prost|sone|idone|hane|cine|cin|tril|erol| [a-wy-z]amine|zime|[a-z]toxin(e?)|otin(e?)|ione| oside|zine|olone|xone|[a-z]dene|lene|lasin(e?)| atin(e?)|ffin(e?)|[a-np-z]yline|xin(e?)|zin(e?)| bene|stine|ythin(e?)|astin(e?)|xitin(e?))(s?)$ | Ribofuranosylcreatine |
Appendix D. PMC Search Terms for Metabolomics
References
- Wishart, D.S. Metabolomics for Investigating Physiological and Pathophysiological Processes. Physiol. Rev. 2019, 99, 1819–1875. [Google Scholar] [CrossRef] [PubMed]
- Miggiels, P.; Wouters, B.; van Westen, G.J.; Dubbelman, A.C.; Hankemeier, T. Novel technologies for metabolomics: More for less. TrAC Trends Anal. Chem. 2019, 120, 115323. [Google Scholar] [CrossRef]
- Bornmann, L.; Mutz, R. Growth rates of modern science: A bibliometric analysis based on the number of publications and cited references: Growth Rates of Modern Science: A Bibliometric Analysis Based on the Number of Publications and Cited References. J. Assoc. Inf. Sci. Technol. 2015, 66, 2215–2222. [Google Scholar] [CrossRef] [Green Version]
- Jackson, R.G.; Patel, R.; Jayatilleke, N.; Kolliakou, A.; Ball, M.; Gorrell, G.; Roberts, A.; Dobson, R.J.; Stewart, R. Natural language processing to extract symptoms of severe mental illness from clinical text: The Clinical Record Interactive Search Comprehensive Data Extraction (CRIS-CODE) project. BMJ Open 2017, 7, e012012. [Google Scholar] [CrossRef] [Green Version]
- Sheikhalishahi, S.; Miotto, R.; Dudley, J.T.; Lavelli, A.; Rinaldi, F.; Osmani, V. Natural Language Processing of Clinical Notes on Chronic Diseases: Systematic Review. JMIR Med. Inform. 2019, 7, e12239. [Google Scholar] [CrossRef] [Green Version]
- Milosevic, N.; Gregson, C.; Hernandez, R.; Nenadic, G. A framework for information extraction from tables in biomedical literature. Int. J. Doc. Anal. Recognit. (IJDAR) 2019, 22, 55–78. [Google Scholar] [CrossRef] [Green Version]
- Kuleshov, V.; Ding, J.; Vo, C.; Hancock, B.; Ratner, A.; Li, Y.; Ré, C.; Batzoglou, S.; Snyder, M. A machine-compiled database of genome-wide association studies. Nat. Commun. 2019, 10, 3341. [Google Scholar] [CrossRef] [Green Version]
- Nobata, C.; Dobson, P.D.; Iqbal, S.A.; Mendes, P.; Tsujii, J.; Kell, D.B.; Ananiadou, S. Mining metabolites: Extracting the yeast metabolome from the literature. Metabolomics 2010, 7, 94–101. [Google Scholar] [CrossRef] [Green Version]
- Kongburan, W.; Padungweang, P.; Krathu, W.; Chan, J.H. Metabolite Named Entity Recognition: A Hybrid Approach. In International Conference on Neural Information Processing: Neural Information Processing; Hirose, A., Ozawa, S., Doya, K., Ikeda, K., Lee, M., Liu, D., Eds.; Springer International Publishing: Berlin/Heidelberg, Germany, 2016; pp. 451–460. [Google Scholar]
- Majumder, E.L.W.; Billings, E.M.; Benton, H.P.; Martin, R.L.; Palermo, A.; Guijas, C.; Rinschen, M.M.; Domingo-Almenara, X.; Montenegro-Burke, J.R.; Tagtow, B.A.; et al. Cognitive analysis of metabolomics data for systems biology. Nat. Protoc. 2021, 16, 1376–1418. [Google Scholar] [CrossRef]
- Kodra, D.; Pousinis, P.; Vorkas, P.A.; Kademoglou, K.; Liapikos, T.; Pechlivanis, A.; Virgiliou, C.; Wilson, I.D.; Gika, H.; Theodoridis, G. Is Current Practice Adhering to Guidelines Proposed for Metabolite Identification in LC-MS Untargeted Metabolomics? A Meta-Analysis of the Literature. J. Proteome Res. 2022, 21, 590–598. [Google Scholar] [CrossRef]
- Wishart, D.S.; Feunang, Y.D.; Marcu, A.; Guo, A.C.; Liang, K.; Vázquez-Fresno, R.; Sajed, T.; Johnson, D.; Li, C.; Karu, N.; et al. HMDB 4.0: The human metabolome database for 2018. Nucleic Acids Res. 2018, 46, D608–D617. [Google Scholar] [CrossRef] [PubMed]
- Wishart, D.S.; Knox, C.; Guo, A.C.; Eisner, R.; Young, N.; Gautam, B.; Hau, D.D.; Psychogios, N.; Dong, E.; Bouatra, S.; et al. HMDB: A knowledgebase for the human metabolome. Nucleic Acids Res. 2009, 37, D603–D610. [Google Scholar] [CrossRef]
- Hirschman, L.; Yeh, A.; Blaschke, C.; Valencia, A. Overview of BioCreAtIvE: Critical assessment of information extraction for biology. BMC Bioinform. 2005, 6, S1. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Krallinger, M.; Leitner, F.; Rabal, O.; Vazquez, M.; Oyarzabal, J.; Valencia, A. CHEMDNER: The drugs and chemical names extraction challenge. J. Cheminform. 2015, 7, S1. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Wang, Q.; S Abdul, S.; Almeida, L.; Ananiadou, S.; Balderas-Martínez, Y.I.; Batista-Navarro, R.; Campos, D.; Chilton, L.; Chou, H.J.; Contreras, G.; et al. Overview of the interactive task in BioCreative V. Database 2016, 2016, baw119. [Google Scholar] [CrossRef] [Green Version]
- Leaman, R.; Wei, C.H.; Lu, Z. tmChem: A high performance approach for chemical named entity recognition and normalization. J. Cheminform. 2015, 7, S3. [Google Scholar] [CrossRef] [PubMed]
- Yoon, W.; So, C.H.; Lee, J.; Kang, J. CollaboNet: Collaboration of deep neural networks for biomedical named entity recognition. BMC Bioinform. 2019, 20, 85. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Hemati, W.; Mehler, A. LSTMVoter: Chemical named entity recognition using a conglomerate of sequence labeling tools. J. Cheminform. 2019, 11, 3. [Google Scholar] [CrossRef]
- Luo, L.; Yang, Z.; Yang, P.; Zhang, Y.; Wang, L.; Lin, H.; Wang, J. An attention-based BiLSTM-CRF approach to document-level chemical named entity recognition. Bioinformatics 2017, 34, 1381–1388. [Google Scholar] [CrossRef] [Green Version]
- Corbett, P.; Boyle, J. Chemlistem: Chemical named entity recognition using recurrent neural networks. J. Cheminform. 2018, 10, 59. [Google Scholar] [CrossRef] [Green Version]
- Habibi, M.; Weber, L.; Neves, M.; Wiegandt, D.L.; Leser, U. Deep learning with word embeddings improves biomedical named entity recognition. Bioinformatics 2017, 33, i37–i48. [Google Scholar] [CrossRef]
- Pennington, J.; Socher, R.; Manning, C. Glove: Global Vectors for Word Representation. In Proceedings of the 2014 Conference on Empirical Methods in Natural Language Processing (EMNLP), Doha, Qatar, 25–29 October 2014; pp. 1532–1543. [Google Scholar] [CrossRef]
- Devlin, J.; Chang, M.W.; Lee, K.; Toutanova, K. BERT: Pre-training of Deep Bidirectional Transformers for Language Understanding. In Proceedings of the 2019 Conference of the North, Minneapolis, MN, USA, 19 June 2019; pp. 4171–4186. [Google Scholar] [CrossRef]
- Lee, J.; Yoon, W.; Kim, S.; Kim, D.; Kim, S.; So, C.H.; Kang, J. BioBERT: A pre-trained biomedical language representation model for biomedical text mining. Bioinformatics 2019, 36, 1234–1240. [Google Scholar] [CrossRef] [PubMed]
- Beck, J. Report from the Field: PubMed Central, an XML-based Archive of Life Sciences Journal Articles. In Proceedings of the International Symposium on XML for the Long Haul: Issues in the Long-term Preservation of XML, Montréal, QC, Canada, 2 August 2010. [Google Scholar]
- Sun, C.; Yang, Z.; Wang, L.; Zhang, Y.; Lin, H.; Wang, J. Biomedical named entity recognition using BERT in the machine reading comprehension framework. J. Biomed. Inform. 2021, 118, 103799. [Google Scholar] [CrossRef] [PubMed]
- Islamaj, R.; Leaman, R.; Kim, S.; Kwon, D.; Wei, C.H.; Comeau, D.C.; Peng, Y.; Cissel, D.; Coss, C.; Fisher, C.; et al. NLM-Chem, a new resource for chemical entity recognition in PubMed full text literature. Sci. Data 2021, 8, 91. [Google Scholar] [CrossRef] [PubMed]
- Beck, T.; Shorter, T.; Hu, Y.; Li, Z.; Sun, S.; Popovici, C.M.; McQuibban, N.A.R.; Makraduli, F.; Yeung, C.S.; Rowlands, T.; et al. Auto-CORPus: A Natural Language Processing Tool for Standardising and Reusing Biomedical Literature. Front. Digit. Health 2022, 4, 788124. [Google Scholar] [CrossRef]
- Williams, A.J. Chemspider: A Platform for Crowdsourced Collaboration to Curate Data Derived From Public Compound Databases. In Collaborative Computational Technologies for Biomedical Research; Ekins, S., Hupcey, M.A.Z., Williams, A.J., Eds.; John Wiley & Sons, Inc.: Hoboken, NJ, USA, 2011; pp. 363–386. [Google Scholar] [CrossRef]
- De Matos, P.; Alcántara, R.; Dekker, A.; Ennis, M.; Hastings, J.; Haug, K.; Spiteri, I.; Turner, S.; Steinbeck, C. Chemical Entities of Biological Interest: An update. Nucleic Acids Res. 2010, 38, D249–D254. [Google Scholar] [CrossRef] [Green Version]
- Fundel, K.; Küffner, R.; Zimmer, R. RelEx—Relation extraction using dependency parse trees. Bioinformatics 2006, 23, 365–371. [Google Scholar] [CrossRef]
- Zhou, G.; Zhang, M.; Ji, D.; Zhu, Q. Tree Kernel-Based Relation Extraction with Context-Sensitive Structured Parse Tree Information. In Proceedings of the 2007 Joint Conference on Empirical Methods in Natural Language Processing and Computational Natural Language Learning, Prague, Czech Republic, 7 June 2007; pp. 728–736. [Google Scholar]
- Erhardt, R.A.A.; Schneider, R.; Blaschke, C. Status of text-mining techniques applied to biomedical text. Drug Discov. Today 2006, 11, 315–325. [Google Scholar] [CrossRef]
- Gridach, M. Character-level neural network for biomedical named entity recognition. J. Biomed. Inform. 2017, 70, 85–91. [Google Scholar] [CrossRef]
- Vander Heiden, M.G.; Cantley, L.C.; Thompson, C.B. Understanding the Warburg Effect: The Metabolic Requirements of Cell Proliferation. Science 2009, 324, 1029–1033. [Google Scholar] [CrossRef] [Green Version]
- Lapin, I.; Oxenkrug, G. Intensification Of The Central Serotoninergic Processes As A Possible Determinant Of The Thymoleptic Effect. Lancet 1969, 293, 132–136. [Google Scholar] [CrossRef]
- Noack, A. Modularity clustering is force-directed layout. Phys. Rev. E 2009, 79, 026102. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Ceusters, W. An information artifact ontology perspective on data collections and associated representational artifacts. Stud. Health Technol. Inform. 2012, 180, 68–72. [Google Scholar]
- Wolf, T.; Debut, L.; Sanh, V.; Chaumond, J.; Delangue, C.; Moi, A.; Cistac, P.; Rault, T.; Louf, R.; Funtowicz, M.; et al. Transformers: State-of-the-Art Natural Language Processing. In Proceedings of the 2020 Conference on Empirical Methods in Natural Language Processing: System Demonstrations, Online Conference, 16–20 November 2020; pp. 38–45. Available online: https://aclanthology.org/2020.emnlp-demos.6/ (accessed on 24 February 2022). [CrossRef]
- Hand, D.J.; Christen, P.; Kirielle, N. F*: An interpretable transformation of the F-measure. Mach. Learn. 2021, 110, 451–456. [Google Scholar] [CrossRef]
- Jessop, D.M.; Adams, S.E.; Willighagen, E.L.; Hawizy, L.; Murray-Rust, P. OSCAR4: A flexible architecture for chemical text-mining. J. Cheminform. 2011, 3, 41. [Google Scholar] [CrossRef] [Green Version]
- Gal, Y.; Ghahramani, Z. A Theoretically Grounded Application of Dropout in Recurrent Neural Networks. In Advances in Neural Information Processing Systems 29: Annual Conference on Neural Information Processing Systems 2016, Barcelona, Spain, 5–10 December 2016; Lee, D.D., Sugiyama, M., von Luxburg, U., Guyon, I., Garnett, R., Eds.; Curran Associates Inc.: Red Hook, NY, USA, 2016; pp. 1019–1027. [Google Scholar]
- Bastian, M.; Heymann, S.; Jacomy, M. Gephi: An Open Source Software for Exploring and Manipulating Networks. In Proceedings of the Third International AAAI Conference on Weblogs and Social Media, San Jose, CA, USA, 17–20 May 2009. [Google Scholar] [CrossRef]
- Jacomy, M.; Venturini, T.; Heymann, S.; Bastian, M. ForceAtlas2, a Continuous Graph Layout Algorithm for Handy Network Visualization Designed for the Gephi Software. PLoS ONE 2014, 9, e98679. [Google Scholar] [CrossRef] [PubMed]






| Journal | # of Metabolites | # of Articles | Average # of (Unique) Metabolites per Article |
|---|---|---|---|
| Analytical and Bioanalytical Chemistry | 716 | 34 | 21.1 |
| Analytical Chemistry | 863 | 48 | 18.0 |
| Journal of Chromatography A | 220 | 9 | 24.4 |
| Journal of Proteome Research | 694 | 24 | 28.9 |
| Metabolites | 1279 | 50 | 25.6 |
| Metabolomics | 1267 | 43 | 29.5 |
| PLOS One | 5400 | 175 | 30.9 |
| PNAS USA | 429 | 14 | 30.6 |
| Scientific Reports | 4278 | 126 | 34.0 |
| Trait | # of Metabolites | # of Articles | Average # of (Unique) Metabolites per Article |
|---|---|---|---|
| cancer | 12,662 | 492 | 25.7 |
| gastrointestinal | 876 | 37 | 23.7 |
| liver disease | 1959 | 121 | 16.2 |
| metabolic syndrome | 7081 | 286 | 24.8 |
| neurodegenerative, psychiatric, and brain illnesses | 2546 | 113 | 22.5 |
| respiratory diseases | 398 | 37 | 10.8 |
| sepsis | 552 | 22 | 25.1 |
| smoking | 1360 | 124 | 11.0 |
| Model | Training Data | Embedding | F1-Score | Precision | Recall | F*-Score | Model Size |
|---|---|---|---|---|---|---|---|
| Annotation pipeline | 0.8893 | 0.8850 | 0.8936 | 0.8006 | |||
| ChemListem [21] | CEMP BioCreative V.5 | GloVe | 0.7669 | 0.7301 | 0.8075 | 0.6219 | 26 MB |
| MetaboListem | metabolomics corpus | GloVe | 0.8900 | 0.8923 | 0.8877 | 0.8018 | 26 MB |
| TABoLiSTM | metabolomics corpus | BERT | 0.9004 | 0.9187 | 0.8829 | 0.8189 | 827 MB |
| TABoLiSTM | metabolomics corpus | BioBERT | 0.9089 | 0.9255 | 0.8928 | 0.8329 | 827 MB |
| Cancer | Gastrointestinal | Liver Disease | Metabolic Syndrome | ||||
|---|---|---|---|---|---|---|---|
| glucose | 142 (29%) | glucose | 12 (32%) | glucose | 29 (24%) | glucose | 178 (62%) |
| glutamine | 121 (25%) | tyrosine | 12 (32%) | cholesterol | 25 (21%) | cholesterol | 108 (38%) |
| lactate | 117 (24%) | lactate | 11 (30%) | creatinine | 21 (17%) | valine | 83 (29%) |
| alanine | 111 (23%) | acetate | 10 (27%) | tyrosine | 20 (17%) | leucine | 73 (26%) |
| glutamate | 109 (22%) | phenylalanine | 9 (24%) | glycine | 18 (15%) | tyrosine | 71 (25%) |
| tyrosine | 105 (21%) | leucine | 9 (24%) | phenylalanine | 17 (14%) | triglycerides | 70 (24%) |
| glycine | 105 (21%) | tryptophan | 9 (24%) | lactate | 16 (13%) | alanine | 67 (23%) |
| valine | 104 (21%) | alanine | 8 (22%) | bilirubin | 16 (13%) | isoleucine | 65 (23%) |
| choline | 103 (21%) | arginine | 8 (22%) | pyruvate | 15 (12%) | phenylalanine | 62 (22%) |
| creatinine | 95 (19%) | citrate | 8 (22%) | valine | 15 (12%) | glycine | 59 (21%) |
| Neurodegenerative, Psychiatric, and Brain Illnesses | Respiratory | Sepsis | Smoking | ||||
| tryptophan | 31 (27%) | lactate | 8 (22%) | phenylalanine | 11 (50%) | creatinine | 24 (19%) |
| glucose | 26 (23%) | glycine | 6 (16%) | glucose | 9 (41%) | cotinine | 15 (12%) |
| glycine | 25 (22%) | leucine | 6 (16%) | lactate | 9 (41%) | nicotine | 14 (11%) |
| glutamate | 22 (19%) | pyruvate | 6 (16%) | arginine | 8 (36%) | glucose | 14 (11%) |
| kynurenine | 22 (19%) | glucose | 6 (16%) | urea | 6 (27%) | leucine | 11 (9%) |
| phenylalanine | 21 (19%) | lysine | 5 (14%) | nitric oxide | 6 (27%) | isoleucine | 11 (9%) |
| serotonin | 18 (16%) | creatine | 5 (14%) | methionine | 6 (27%) | valine | 11 (9%) |
| tyrosine | 18 (16%) | isoleucine | 5 (14%) | pyruvate | 6 (27%) | cholesterol | 11 (9%) |
| creatinine | 18 (16%) | glutamine | 5 (14%) | creatinine | 6 (27%) | lactate | 10 (8%) |
| lactate | 17 (15%) | acetate | 4 (11%) | ATP | 5 (23%) | glutamate | 9 (7%) |
Publisher’s Note: MDPI stays neutral with regard to jurisdictional claims in published maps and institutional affiliations. |
© 2022 by the authors. Licensee MDPI, Basel, Switzerland. This article is an open access article distributed under the terms and conditions of the Creative Commons Attribution (CC BY) license (https://creativecommons.org/licenses/by/4.0/).
Share and Cite
Yeung, C.S.; Beck, T.; Posma, J.M. MetaboListem and TABoLiSTM: Two Deep Learning Algorithms for Metabolite Named Entity Recognition. Metabolites 2022, 12, 276. https://doi.org/10.3390/metabo12040276
Yeung CS, Beck T, Posma JM. MetaboListem and TABoLiSTM: Two Deep Learning Algorithms for Metabolite Named Entity Recognition. Metabolites. 2022; 12(4):276. https://doi.org/10.3390/metabo12040276
Chicago/Turabian StyleYeung, Cheng S., Tim Beck, and Joram M. Posma. 2022. "MetaboListem and TABoLiSTM: Two Deep Learning Algorithms for Metabolite Named Entity Recognition" Metabolites 12, no. 4: 276. https://doi.org/10.3390/metabo12040276
APA StyleYeung, C. S., Beck, T., & Posma, J. M. (2022). MetaboListem and TABoLiSTM: Two Deep Learning Algorithms for Metabolite Named Entity Recognition. Metabolites, 12(4), 276. https://doi.org/10.3390/metabo12040276