
Host–Gut Microbiome Metabolic Interactions in PFAS-Impacted Freshwater Turtles (Emydura macquarii macquarii)

 ,
, 
 ,
,  , and
, and
Abstract
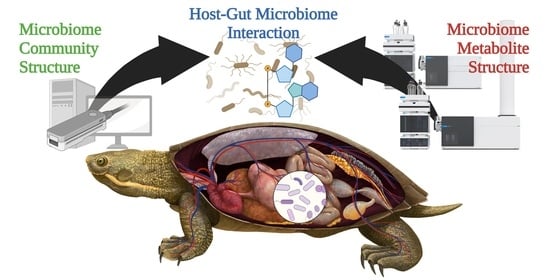
:
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
1. Introduction
2. Results
2.1. Turtle Physiology and Biochemistry
2.2. Community Metabolomics Profile
2.3. Microbiome Community Profile
2.4. Metabolite Origin Analysis
2.5. Host-Microbiome Function Analysis
2.6. Metabolite–Microbiome–Host Correlation Analysis
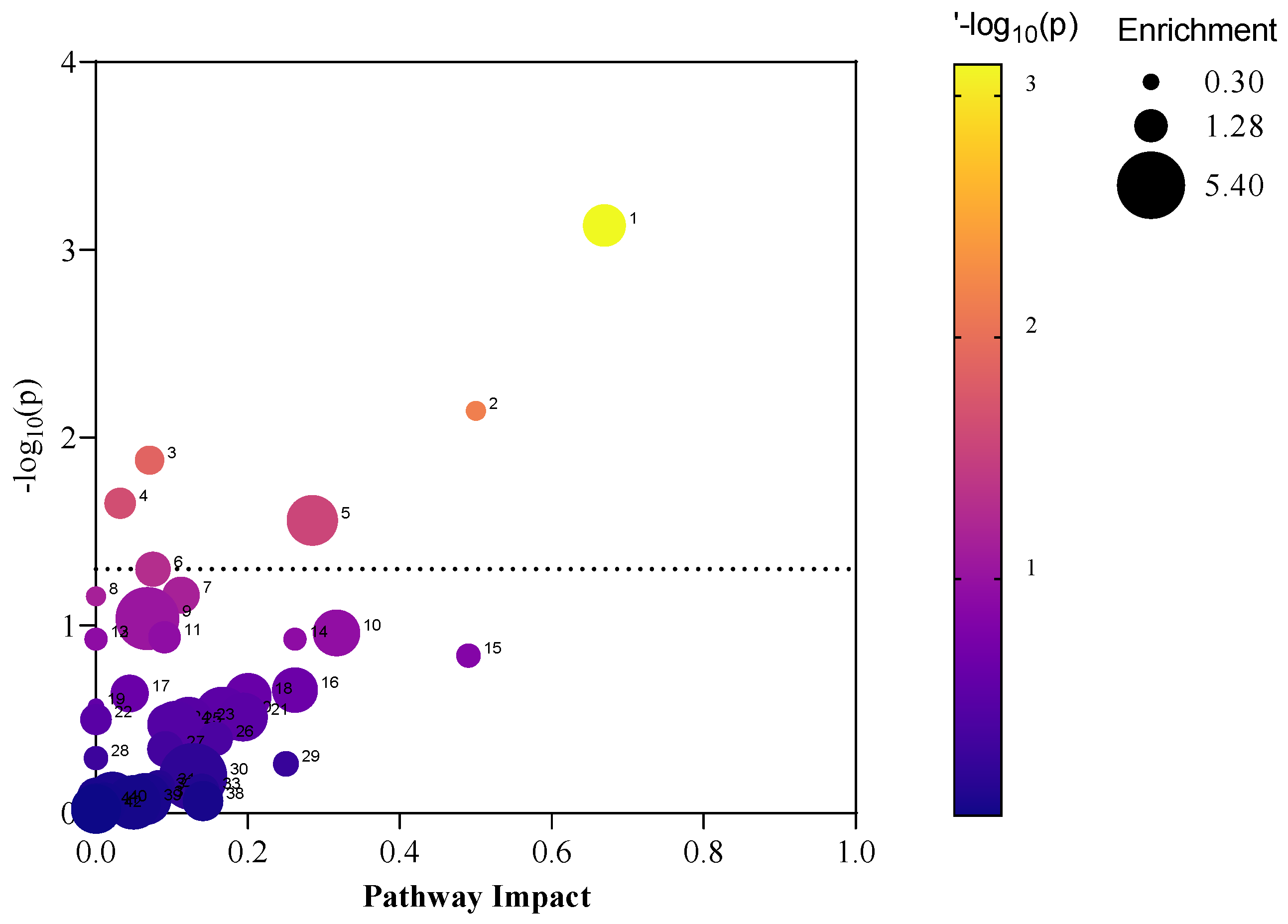
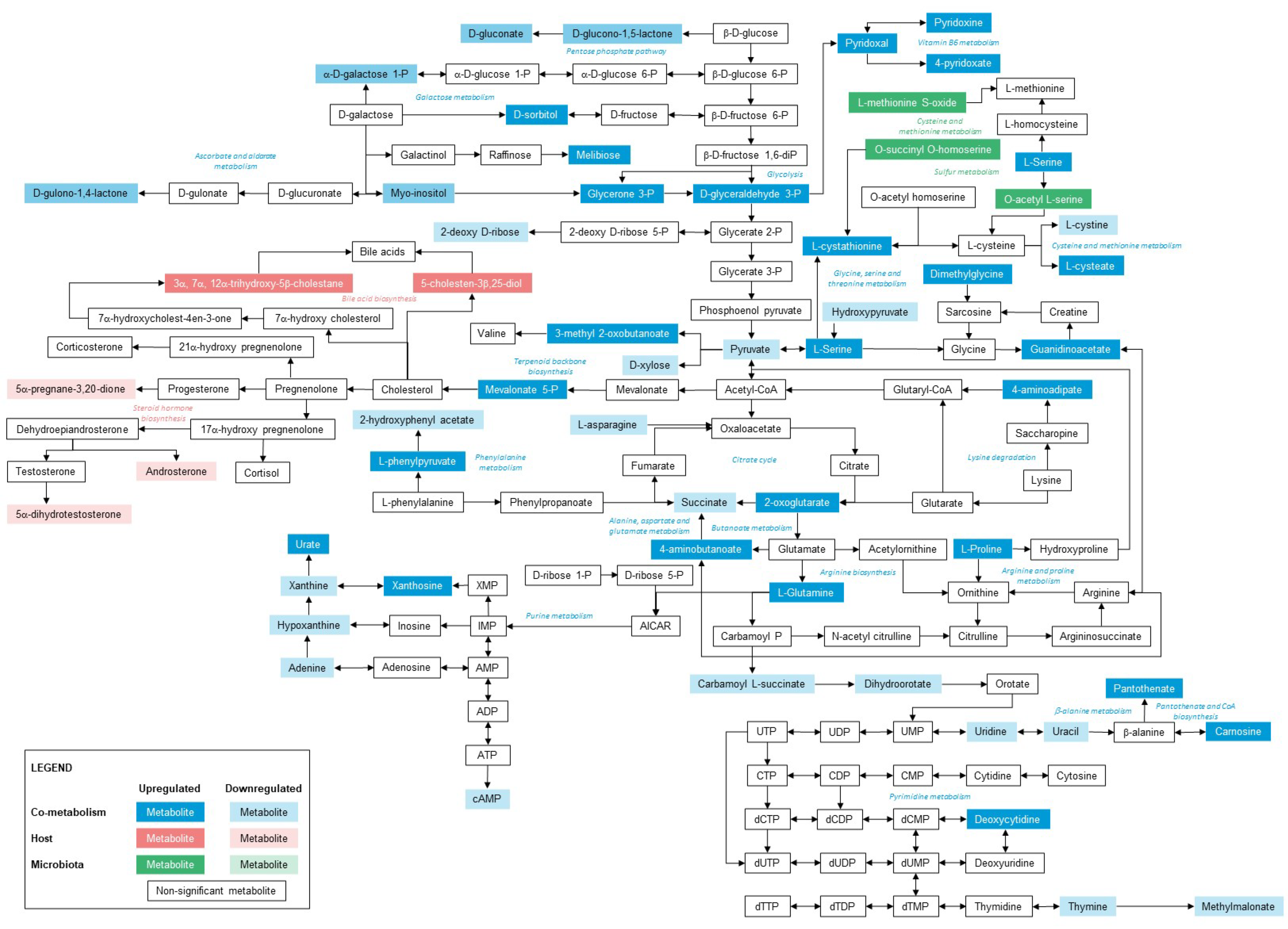
2.7. Metabolic Pathway Mapping
3. Discussion
3.1. Community Composition
3.2. Community Metabolic Function
3.3. Host–Gut Microbiome Metabolism
4. Materials and Methods
4.1. Animal Ethics
4.2. Turtle Collection from a Known PFAS-Impacted Site
4.3. Turtle Faecal Samples
4.4. Turtle Faecal Microbiome Analysis
4.5. Turtle Faecal Community Metabolome Analysis
4.6. Statistical Analysis and Host–Microbiome Data Analysis
5. Conclusions
Supplementary Materials
Author Contributions
Funding
Institutional Review Board Statement
Informed Consent Statement
Data Availability Statement
Acknowledgments
Conflicts of Interest
Abbreviations
References
- Georges, A. Setting conservation priorities for Australian freshwater turtles. In Herpetology in Australia: A Diverse Discipline; Lunney, D., Ayers, D., Eds.; Royal Zoological Society of New South Wales: Sydney, Australia, 1993; pp. 49–58. [Google Scholar]
- Georges, A.; Thomson, S. Diversity of Australasian freshwater turtles, with an annotated synonymy and keys to species. Zootaxa 2010, 2496, 1–37. [Google Scholar] [CrossRef]
- Government, Q. Species Profile—Emydura macquarii macquarii (Murray Turtle). 2022. Available online: https://apps.des.qld.gov.au/species-search/details/?id=43 (accessed on 10 May 2022).
- Chessman, B.C. Declines of freshwater turtles associated with climatic drying in Australia’s Murray–Darling Basin. Wildl. Res. 2011, 38, 664–671. [Google Scholar] [CrossRef]
- Van Dyke, J.U.; Spencer, R.J.; Thompson, M.B.; Chessman, B.; Howard, K.; Georges, A. Conservation implications of turtle declines in Australia’s Murray River system. Sci. Rep. 2019, 9, 1998. [Google Scholar] [CrossRef] [PubMed]
- Basin, D. Ecology of the Macquarie Turtle (Emydura macquarii macquarii) Downstream of a Large Hypolimnetic-Releasing Impoundment in Australia’s Southern Murray. Ph.D. Thesis, Charles Sturt University, Bathurst, Australia, 2018. [Google Scholar]
- Hopkins, B.C.; Willson, J.D.; Hopkins, W.A. Mercury exposure is associated with negative effects on turtle reproduction. Environ. Sci. Technol. 2013, 47, 2416–2422. [Google Scholar] [CrossRef] [PubMed]
- Van Dyke, J.U.; Hopkins, W.A.; Jackson, B.P. Influence of relative trophic position and carbon source on selenium bioaccumulation in turtles from a coal fly-ash spill site. Environ. Pollut. 2013, 182, 45–52. [Google Scholar] [CrossRef]
- Ferronato, B.O.; Roe, J.H.; Georges, A. Reptile bycatch in a pest-exclusion fence established for wildlife reintroductions. J. Nat. Conserv. 2014, 22, 577–585. [Google Scholar] [CrossRef]
- Ferronato, B.O.; Roe, J.H.; Georges, A. Urban hazards: Spatial ecology and survivorship of a turtle in an expanding suburban environment. Urban Ecosyst. 2016, 19, 415–428. [Google Scholar] [CrossRef]
- Kissa, E. Fluorinated Surfactants and Repellents; CRC Press: Boca Raton, FL, USA, 2001; Volume 97. [Google Scholar]
- Glüge, J.; Scheringer, M.; Cousins, I.T.; DeWitt, J.C.; Goldenman, G.; Herzke, D.; Lohmann, R.; Ng, C.A.; Trier, X.; Wang, Z. An overview of the uses of per-and polyfluoroalkyl substances (PFAS). Environ. Sci. Process. Impacts 2020, 22, 2345–2373. [Google Scholar] [CrossRef] [PubMed]
- Abunada, Z.; Alazaiza, M.Y.D.; Bashir, M.J.K. An Overview of Per- and Polyfluoroalkyl Substances (PFAS) in the Environment: Source, Fate, Risk and Regulations. Water 2020, 12, 3590. [Google Scholar] [CrossRef]
- Giesy, J.P.; Kannan, K. Global Distribution of Perfluorooctane Sulfonate in Wildlife. Environ. Sci. Technol. 2001, 35, 1339–1342. [Google Scholar] [CrossRef]
- Houde, M.; De Silva, A.O.; Muir, D.C.; Letcher, R.J. Monitoring of perfluorinated compounds in aquatic biota: An updated review: PFCs in aquatic biota. Environ. Sci. Technol. 2011, 45, 7962–7973. [Google Scholar] [CrossRef] [PubMed]
- Burkhard, L.P. Evaluation of published bioconcentration factor (BCF) and bioaccumulation factor (BAF) data for per-and polyfluoroalkyl substances across aquatic species. Environ. Toxicol. Chem. 2021, 40, 1530–1543. [Google Scholar] [CrossRef] [PubMed]
- Kannan, K.; Tao, L.; Sinclair, E.; Pastva, S.D.; Jude, D.J.; Giesy, J.P. Perfluorinated compounds in aquatic organisms at various trophic levels in a Great Lakes food chain. Arch. Environ. Contam. Toxicol. 2005, 48, 559–566. [Google Scholar] [CrossRef] [PubMed]
- De Solla, S.; De Silva, A.; Letcher, R. Highly elevated levels of perfluorooctane sulfonate and other perfluorinated acids found in biota and surface water downstream of an international airport, Hamilton, Ontario, Canada. Environ. Int. 2012, 39, 19–26. [Google Scholar] [CrossRef]
- Guerranti, C.; Ancora, S.; Bianchi, N.; Perra, G.; Fanello, E.L.; Corsolini, S.; Fossi, M.C.; Focardi, S.E. Perfluorinated compounds in blood of Caretta caretta from the Mediterranean Sea. Mar. Pollut. Bull. 2013, 73, 98–101. [Google Scholar] [CrossRef]
- Keller, J.M.; Ngai, L.; McNeill, J.B.; Wood, L.D.; Stewart, K.R.; O’Connell, S.G.; Kucklick, J.R. Perfluoroalkyl contaminants in plasma of five sea turtle species: Comparisons in concentration and potential health risks. Environ. Toxicol. Chem. 2012, 31, 1223–1230. [Google Scholar] [CrossRef] [PubMed]
- Morikawa, A.; Kamei, N.; Harada, K.; Inoue, K.; Yoshinaga, T.; Saito, N.; Koizumi, A. The bioconcentration factor of perfluorooctane sulfonate is significantly larger than that of perfluorooctanoate in wild turtles (Trachemys scripta elegans and Chinemys reevesii): An Ai river ecological study in Japan. Ecotoxicol. Environ. Saf. 2006, 65, 14–21. [Google Scholar] [CrossRef]
- Beale, D.J.; Hillyer, K.; Nilsson, S.; Limpus, D.; Bose, U.; Broadbent, J.A.; Vardy, S. Bioaccumulation and metabolic response of PFAS mixtures in wild-caught freshwater turtles (Emydura macquarii macquarii) using omics-based ecosurveillance techniques. Sci. Total Environ. 2022, 806, 151264. [Google Scholar] [CrossRef]
- Chen, Z.; Yang, T.; Walker, D.I.; Thomas, D.C.; Qiu, C.; Chatzi, L.; Alderete, T.L.; Kim, J.S.; Conti, D.V.; Breton, C.V. Dysregulated lipid and fatty acid metabolism link perfluoroalkyl substances exposure and impaired glucose metabolism in young adults. Environ. Int. 2020, 145, 106091. [Google Scholar] [CrossRef]
- Fan, Y.; Li, X.; Xu, Q.; Zhang, Y.; Yang, X.; Han, X.; Du, G.; Xia, Y.; Wang, X.; Lu, C. Serum albumin mediates the effect of multiple per-and polyfluoroalkyl substances on serum lipid levels. Environ. Pollut. 2020, 266, 115138. [Google Scholar] [CrossRef]
- Kobayashi, S.; Sata, F.; Goudarzi, H.; Araki, A.; Miyashita, C.; Sasaki, S.; Okada, E.; Iwasaki, Y.; Nakajima, T.; Kishi, R. Associations among perfluorooctanesulfonic/perfluorooctanoic acid levels, nuclear receptor gene polymorphisms, and lipid levels in pregnant women in the Hokkaido study. Sci. Rep. 2021, 11, 9994. [Google Scholar] [CrossRef] [PubMed]
- Ortiz-Villanueva, E.; Jaumot, J.; Martínez, R.; Navarro-Martín, L.; Piña, B.; Tauler, R. Assessment of endocrine disruptors effects on zebrafish (Danio rerio) embryos by untargeted LC-HRMS metabolomic analysis. Sci. Total Environ. 2018, 635, 156–166. [Google Scholar] [CrossRef] [PubMed]
- Seyoum, A.; Pradhan, A.; Jass, J.; Olsson, P.-E. Perfluorinated alkyl substances impede growth, reproduction, lipid metabolism and lifespan in Daphnia magna. Sci. Total Environ. 2020, 737, 139682. [Google Scholar] [CrossRef] [PubMed]
- Taylor, M.D.; Bräunig, J.; Mueller, J.F.; Crompton, M.; Dunstan, R.H.; Nilsson, S. Metabolomic profiles associated with exposure to per-and polyfluoroalkyl substances (PFASs) in aquatic environments. Environ. Sci. Process. Impacts 2019, 21, 1980–1990. [Google Scholar] [CrossRef] [PubMed]
- Pfohl, M.; Marques, E.; Auclair, A.; Barlock, B.; Jamwal, R.; Goedken, M.; Akhlaghi, F.; Slitt, A.L. An ‘omics approach to unraveling the paradoxical effect of diet on perfluorooctanesulfonic acid (PFOS) and perfluorononanoic acid (PFNA)-induced hepatic steatosis. Toxicol. Sci. 2021, 180, 277–294. [Google Scholar] [CrossRef]
- Beale, D.J.; Nilsson, S.; Bose, U.; Bourne, N.; Stockwell, S.; Broadbent, J.A.; Gonzalez-Astudillo, V.; Braun, C.; Baddiley, B.; Limpus, D.; et al. Bioaccumulation and impact of maternal PFAS offloading on egg biochemistry from wild-caught freshwater turtles (Emydura macquarii macquarii). Sci. Total Environ. 2022, 817, 153019. [Google Scholar] [CrossRef]
- Beale, D.J.; Bisset, A.; Nilsson, S.; Bose, U.; Nelis, J.; Nahar, A.; Smith, M.; Gonzalez-Astudillo, V.; Braun, C.; Baddiley, B.; et al. Perturbation of the gut microbiome in wild-caught freshwater turtles (Emydura macquarii macquarii) exposed to elevated PFAS levels. Sci. Total Environ. 2022, 838, 156324. [Google Scholar] [CrossRef]
- Yu, G.; Xu, C.; Zhang, D.; Ju, F.; Ni, Y. MetOrigin: Discriminating the origins of microbial metabolites for integrative analysis of the gut microbiome and metabolome. iMeta 2022, 1, e10. [Google Scholar] [CrossRef]
- Ahasan, M.S.; Waltzek, T.B.; Huerlimann, R.; Ariel, E. Comparative analysis of gut bacterial communities of green turtles (Chelonia mydas) pre-hospitalization and post-rehabilitation by high-throughput sequencing of bacterial 16S rRNA gene. Microbiol. Res. 2018, 207, 91–99. [Google Scholar] [CrossRef]
- Bloodgood, J.C.G.; Hernandez, S.M.; Isaiah, A.; Suchodolski, J.S.; Hoopes, L.A.; Thompson, P.M.; Waltzek, T.B.; Norton, T.M. The effect of diet on the gastrointestinal microbiome of juvenile rehabilitating green turtles (Chelonia mydas). PLoS ONE 2020, 15, e0227060. [Google Scholar] [CrossRef]
- Pan, Z.; Yuan, X.; Tu, W.; Fu, Z.; Jin, Y. Subchronic exposure of environmentally relevant concentrations of F-53B in mice resulted in gut barrier dysfunction and colonic inflammation in a sex-independent manner. Environ. Pollut. 2019, 253, 268–277. [Google Scholar] [CrossRef] [PubMed]
- Biagi, E.; Nylund, L.; Candela, M.; Ostan, R.; Bucci, L.; Pini, E.; Nikkïla, J.; Monti, D.; Satokari, R.; Franceschi, C. Through ageing, and beyond: Gut microbiota and inflammatory status in seniors and centenarians. PLoS ONE 2010, 5, e10667. [Google Scholar] [CrossRef]
- Magne, F.; Gotteland, M.; Gauthier, L.; Zazueta, A.; Pesoa, S.; Navarrete, P.; Balamurugan, R. The firmicutes/bacteroidetes ratio: A relevant marker of gut dysbiosis in obese patients? Nutrients 2020, 12, 1474. [Google Scholar] [CrossRef] [PubMed]
- Guo, P.; Furnary, T.; Vasiliou, V.; Yan, Q.; Nyhan, K.; Jones, D.P.; Johnson, C.H.; Liew, Z. Non-targeted metabolomics and associations with per-and polyfluoroalkyl substances (PFAS) exposure in humans: A scoping review. Environ. Int. 2022, 162, 107159. [Google Scholar] [CrossRef] [PubMed]
- Lankadurai, B.P.; Furdui, V.I.; Reiner, E.J.; Simpson, A.J.; Simpson, M.J. 1H NMR-Based Metabolomic Analysis of Sub-Lethal Perfluorooctane Sulfonate Exposure to the Earthworm, Eisenia fetida, in Soil. Metabolites 2013, 3, 718–740. [Google Scholar] [CrossRef]
- Kariuki, M.N.; Nagato, E.G.; Lankadurai, B.P.; Simpson, A.J.; Simpson, M.J. Analysis of Sub-Lethal Toxicity of Perfluorooctane Sulfonate (PFOS) to Daphnia magna Using 1H Nuclear Magnetic Resonance-Based Metabolomics. Metabolites 2017, 7, 15. [Google Scholar] [CrossRef]
- Louis, P.; Hold, G.L.; Flint, H.J. The gut microbiota, bacterial metabolites and colorectal cancer. Nat. Rev. Microbiol. 2014, 12, 661–672. [Google Scholar] [CrossRef]
- Kingsley, S.L.; Walker, D.I.; Calafat, A.M.; Chen, A.; Papandonatos, G.D.; Xu, Y.; Jones, D.P.; Lanphear, B.P.; Pennell, K.D.; Braun, J.M. Metabolomics of childhood exposure to perfluoroalkyl substances: A cross-sectional study. Metabolomics 2019, 15, 95. [Google Scholar] [CrossRef]
- Chang, C.-J.; Barr, D.B.; Ryan, P.B.; Panuwet, P.; Smarr, M.M.; Liu, K.; Kannan, K.; Yakimavets, V.; Tan, Y.; Ly, V.; et al. Per- and polyfluoroalkyl substance (PFAS) exposure, maternal metabolomic perturbation, and fetal growth in African American women: A meet-in-the-middle approach. Environ. Int. 2022, 158, 106964. [Google Scholar] [CrossRef]
- Nyhan, W.L. Disorders of purine and pyrimidine metabolism. Mol. Genet. Metab. 2005, 86, 25–33. [Google Scholar] [CrossRef]
- Hubert, L.; Sutton, V.R. Chapter 12—Disorders of purine and pyrimidine metabolism. In Biomarkers in Inborn Errors of Metabolism; Garg, U., Smith, L.D., Eds.; Elsevier: San Diego, CA, USA, 2017; pp. 283–299. [Google Scholar] [CrossRef]
- Kamatani, N.; Jinnah, H.A.; Hennekam, R.C.M.; van Kuilenburg, A.B.P. Purine and Pyrimidine Metabolism. In Reference Module in Biomedical Sciences; Elsevier: Amsterdam, The Netherlands, 2014. [Google Scholar] [CrossRef]
- Nuki, G. Disorders of purine and pyrimidine metabolism. In Oxford Textbook of Medicine, 6th ed.; Oxford University Press: Oxford, UK, 2020; Volume 3, pp. 1376–1388. [Google Scholar]
- Wang, C.; Zhao, Y.; Jin, Y. The emerging PFOS alternative OBS exposure induced gut microbiota dysbiosis and hepatic metabolism disorder in adult zebrafish. Comp. Biochem. Physiol. Part C Toxicol. Pharmacol. 2020, 230, 108703. [Google Scholar] [CrossRef] [PubMed]
- Marcobal, A.; Kashyap, P.; Nelson, T.; Aronov, P.; Donia, M.; Spormann, A.; Fischbach, M.; Sonnenburg, J. A metabolomic view of how the human gut microbiota impacts the host metabolome using humanized and gnotobiotic mice. ISME J. 2013, 7, 1933–1943. [Google Scholar] [CrossRef] [PubMed]
- Zierer, J.; Jackson, M.A.; Kastenmüller, G.; Mangino, M.; Long, T.; Telenti, A.; Mohney, R.P.; Small, K.S.; Bell, J.T.; Steves, C.J.; et al. The faecal metabolome as a functional readout of the gut microbiome. Nat. Genet. 2018, 50, 790–795. [Google Scholar] [CrossRef]
- Pedersen, H.K.; Gudmundsdottir, V.; Nielsen, H.B.; Hyotylainen, T.; Nielsen, T.; Jensen, B.A.; Forslund, K.; Hildebrand, F.; Prifti, E.; Falony, G. Human gut microbes impact host serum metabolome and insulin sensitivity. Nature 2016, 535, 376–381. [Google Scholar] [CrossRef]
- Clarke, G.; Stilling, R.M.; Kennedy, P.J.; Stanton, C.; Cryan, J.F.; Dinan, T.G. Minireview: Gut microbiota: The neglected endocrine organ. Mol. Endocrinol. 2014, 28, 1221–1238. [Google Scholar] [CrossRef] [PubMed]
- Neis, E.P.J.G.; Dejong, C.H.C.; Rensen, S.S. The role of microbial amino acid metabolism in host metabolism. Nutrients 2015, 7, 2930–2946. [Google Scholar] [CrossRef] [PubMed]
- Honda, K.; Littman, D.R. The microbiota in adaptive immune homeostasis and disease. Nature 2016, 535, 75–84. [Google Scholar] [CrossRef] [PubMed]
- Lu, L.; Luo, T.; Zhao, Y.; Cai, C.; Fu, Z.; Jin, Y. Interaction between microplastics and microorganism as well as gut microbiota: A consideration on environmental animal and human health. Sci. Total Environ. 2019, 667, 94–100. [Google Scholar] [CrossRef]
- Biegel, L.B.; Liu, R.C.; Hurtt, M.E.; Cook, J.C. Effects of ammonium perfluorooctanoate on Leydig cell function: In Vitro, In Vivo, and ex vivo studies. Toxicol. Appl. Pharmacol. 1995, 134, 18–25. [Google Scholar] [CrossRef]
- Wang, Z.; Zhang, T.; Wu, J.; Wei, X.; Xu, A.; Wang, S.; Wang, Z. Male reproductive toxicity of perfluorooctanoate (PFOA): Rodent studies. Chemosphere 2021, 270, 128608. [Google Scholar] [CrossRef]
- Zhao, B.; Li, L.; Liu, J.; Li, H.; Zhang, C.; Han, P.; Zhang, Y.; Yuan, X.; Ge, R.S.; Chu, Y. Exposure to Perfluorooctane Sulfonate In Utero Reduces Testosterone Production in Rat Fetal Leydig Cells. PLoS ONE 2014, 9, e78888. [Google Scholar] [CrossRef] [PubMed]
- Ding, N.; Harlow, S.D.; Randolph, J.F., Jr.; Loch-Caruso, R.; Park, S.K. Perfluoroalkyl and polyfluoroalkyl substances (PFAS) and their effects on the ovary. Hum. Reprod. Update 2020, 26, 724–752. [Google Scholar] [CrossRef] [PubMed]
- Behr, A.-C.; Kwiatkowski, A.; Ståhlman, M.; Schmidt, F.F.; Luckert, C.; Braeuning, A.; Buhrke, T. Impairment of bile acid metabolism by perfluorooctanoic acid (PFOA) and perfluorooctanesulfonic acid (PFOS) in human HepaRG hepatoma cells. Arch. Toxicol. 2020, 94, 1673–1686. [Google Scholar] [CrossRef] [PubMed]
- Fenton, S.E.; Ducatman, A.; Boobis, A.; DeWitt, J.C.; Lau, C.; Ng, C.; Smith, J.S.; Roberts, S.M. Per- and Polyfluoroalkyl Substance Toxicity and Human Health Review: Current State of Knowledge and Strategies for Informing Future Research. Environ. Toxicol. Chem. 2021, 40, 606–630. [Google Scholar] [CrossRef] [PubMed]
- Labine, L.M.; Oliveira Pereira, E.A.; Kleywegt, S.; Jobst, K.J.; Simpson, A.J.; Simpson, M.J. Comparison of sub-lethal metabolic perturbations of select legacy and novel perfluorinated alkyl substances (PFAS) in Daphnia magna. Environ. Res. 2022, 212, 113582. [Google Scholar] [CrossRef] [PubMed]
- Wang, C.; Fang, C.; Wang, C.; Jin, C.; Qian, M.; Jin, Y. Maternal Sodium p-Perfluorous Nonenoxybenzene Sulfonate Exposure Disturbed Lipid Metabolism and Induced an Imbalance in Tyrosine Metabolism in the F1 Generation of Mice. Chem. Res. Toxicol. 2022, 35, 651–662. [Google Scholar] [CrossRef]
- Engsbro, A.L.; Nielsen, K.L.; Hornum, M.; Andersen, L.P. Laribacter hongkongensis: Clinical presentation, epidemiology and treatment. A review of the literature and report of the first case in Denmark. Infect. Dis. 2018, 50, 417–422. [Google Scholar] [CrossRef]
- Meyburgh, C.M.; Bragg, R.R.; Boucher, C.E. Lactococcus garvieae: An emerging bacterial pathogen of fish. Dis. Aquat. Org. 2017, 123, 67–79. [Google Scholar] [CrossRef]
- Lima, C.O.G.X.; da Rocha, V.M.; Ferreira, E.d.O.; Filho, J.S.; Serradas, L.R.; Silva, R.O.S.; Lobato, F.C.F.; Domingues, R.M.C.P. Clostridium baratii: A rare case of pneumonia associated with an Alzheimer patient in Rio de Janeiro, Brazil. JMM Case Rep. 2016, 3, e005041. [Google Scholar] [CrossRef]
- Patil, S.; Willett, O.; Thompkins, T.; Hermann, R.; Ramanathan, S.; Cornett, E.M.; Fox, C.J.; Kaye, A.D. Botulinum Toxin: Pharmacology and Therapeutic Roles in Pain States. Curr. Pain Headache Rep. 2016, 20, 15. [Google Scholar] [CrossRef]
- Wambui, J.; Cernela, N.; Corti, S.; Stephan, R. Comparative Genome Analysis and Phenotypic Characterization of Clostridium gasigenes CGAS001 Isolated From Chilled Vacuum-Packed Lamb Meat. Front. Microbiol. 2020, 11, 2048. [Google Scholar] [CrossRef] [PubMed]
- Harrell, L.J.; Cameron, M.L.; O’Hara, C.M. Rahnella aquatilis, an unusual gram-negative rod isolated from the bronchial washing of a patient with acquired immunodeficiency syndrome. J. Clin. Microbiol. 1989, 27, 1671–1672. [Google Scholar] [CrossRef] [PubMed]
- Brenner, D.J.; Müller, H.E.; Steigerwalt, A.G.; Whitney, A.M.; O’hara, C.M.; Kämpfer, P. Two new Rahnella genomospecies that cannot be phenotypically differentiated from Rahnella aquatilis. Int. J. Syst. Bacteriol. 1998, 48 Pt 1, 141–149. [Google Scholar] [CrossRef] [PubMed]
- Cassir, N.; Benamar, S.; La Scola, B. Clostridium butyricum: From beneficial to a new emerging pathogen. Clin. Microbiol. Infect. 2016, 22, 37–45. [Google Scholar] [CrossRef]
- Zhao, L.; Lou, H.; Peng, Y.; Chen, S.; Zhang, Y.; Li, X. Comprehensive relationships between gut microbiome and faecal metabolome in individuals with type 2 diabetes and its complications. Endocrine 2019, 66, 526–537. [Google Scholar] [CrossRef] [PubMed]
- Vandeputte, D.; Falony, G.; Vieira-Silva, S.; Tito, R.Y.; Joossens, M.; Raes, J. Stool consistency is strongly associated with gut microbiota richness and composition, enterotypes and bacterial growth rates. Gut 2016, 65, 57–62. [Google Scholar] [CrossRef] [PubMed]
- Tigchelaar, E.; Bonder, M.; Jankipersadsing, S.; Fu, J.; Wijmenga, C.; Zhernakova, A. Gut microbiota composition associated with stool consistency. Gut 2016, 65, 540–542. [Google Scholar] [CrossRef]
- Lee-Sarwar, K.A.; Lasky-Su, J.; Kelly, R.S.; Litonjua, A.A.; Weiss, S.T. Metabolome–Microbiome Crosstalk and Human Disease. Metabolites 2020, 10, 181. [Google Scholar] [CrossRef]
- McHardy, I.H.; Goudarzi, M.; Tong, M.; Ruegger, P.M.; Schwager, E.; Weger, J.R.; Graeber, T.G.; Sonnenburg, J.L.; Horvath, S.; Huttenhower, C. Integrative analysis of the microbiome and metabolome of the human intestinal mucosal surface reveals exquisite inter-relationships. Microbiome 2013, 1, 17. [Google Scholar] [CrossRef] [PubMed]
- ANZECC; ARMCANZ. Australian and New Zealand Guidelines for Fresh and Marine Water Quality. 2018. Available online: https://www.waterquality.gov.au/anz-guidelines (accessed on 15 July 2022).
- Douglas, G.M.; Maffei, V.J.; Zaneveld, J.R.; Yurgel, S.N.; Brown, J.R.; Taylor, C.M.; Huttenhower, C.; Langille, M.G.I. PICRUSt2 for prediction of metagenome functions. Nat. Biotechnol. 2020, 38, 685–688. [Google Scholar] [CrossRef]
- Sartain, M. The Agilent Metabolomics Dynamic MRM Database and Method; Technical Overview Publication Number 5991-6482EN; Agilent Technologies: Santa Clara, CA, USA, 2016. [Google Scholar]
- Gyawali, P.; Karpe, A.V.; Hillyer, K.E.; Nguyen, T.V.; Hewitt, J.; Beale, D.J. A multi-platform metabolomics approach to identify possible biomarkers for human faecal contamination in Greenshell™ mussels (Perna canaliculus). Sci. Total Environ. 2021, 771, 145363. [Google Scholar] [CrossRef] [PubMed]
- Pang, Z.; Chong, J.; Zhou, G.; de Lima Morais, D.A.; Chang, L.; Barrette, M.; Gauthier, C.; Jacques, P.; Li, S.; Xia, J. MetaboAnalyst 5.0: Narrowing the gap between raw spectra and functional insights. Nucleic Acids Res. 2021, 49, W388–W396. [Google Scholar] [CrossRef] [PubMed]







Publisher’s Note: MDPI stays neutral with regard to jurisdictional claims in published maps and institutional affiliations. |
© 2022 by the authors. Licensee MDPI, Basel, Switzerland. This article is an open access article distributed under the terms and conditions of the Creative Commons Attribution (CC BY) license (https://creativecommons.org/licenses/by/4.0/).
Share and Cite
Beale, D.J.; Nguyen, T.V.; Shah, R.M.; Bissett, A.; Nahar, A.; Smith, M.; Gonzalez-Astudillo, V.; Braun, C.; Baddiley, B.; Vardy, S. Host–Gut Microbiome Metabolic Interactions in PFAS-Impacted Freshwater Turtles (Emydura macquarii macquarii). Metabolites 2022, 12, 747. https://doi.org/10.3390/metabo12080747
Beale DJ, Nguyen TV, Shah RM, Bissett A, Nahar A, Smith M, Gonzalez-Astudillo V, Braun C, Baddiley B, Vardy S. Host–Gut Microbiome Metabolic Interactions in PFAS-Impacted Freshwater Turtles (Emydura macquarii macquarii). Metabolites. 2022; 12(8):747. https://doi.org/10.3390/metabo12080747
Chicago/Turabian StyleBeale, David J., Thao V. Nguyen, Rohan M. Shah, Andrew Bissett, Akhikun Nahar, Matthew Smith, Viviana Gonzalez-Astudillo, Christoph Braun, Brenda Baddiley, and Suzanne Vardy. 2022. "Host–Gut Microbiome Metabolic Interactions in PFAS-Impacted Freshwater Turtles (Emydura macquarii macquarii)" Metabolites 12, no. 8: 747. https://doi.org/10.3390/metabo12080747
APA StyleBeale, D. J., Nguyen, T. V., Shah, R. M., Bissett, A., Nahar, A., Smith, M., Gonzalez-Astudillo, V., Braun, C., Baddiley, B., & Vardy, S. (2022). Host–Gut Microbiome Metabolic Interactions in PFAS-Impacted Freshwater Turtles (Emydura macquarii macquarii). Metabolites, 12(8), 747. https://doi.org/10.3390/metabo12080747