
Optical Redox Imaging of Ex Vivo Hippocampal Tissue Reveals Age-Dependent Alterations in the 5XFAD Mouse Model of Alzheimer’s Disease

 ,
,  , , ,
, , , {kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
Abstract
:1. Introduction
2. Materials and Methods
2.1. Animals and Brain Tissue Dissection
2.1.1. Mice
2.1.2. Mouse Hippocampal Tissue Preparation
2.2. ORI of Fresh Hippocampal Tissue and Data Processing
2.3. Aβ42 Quantification
2.4. Statistical Analysis
3. Results
3.1. Confirmation of Redox Signals in Hippocampal Tissue
3.2. Redox Kinetics during Imaging in the Hippocampal Tissue Sections
3.3. Relationship between the Redox Indices and Age
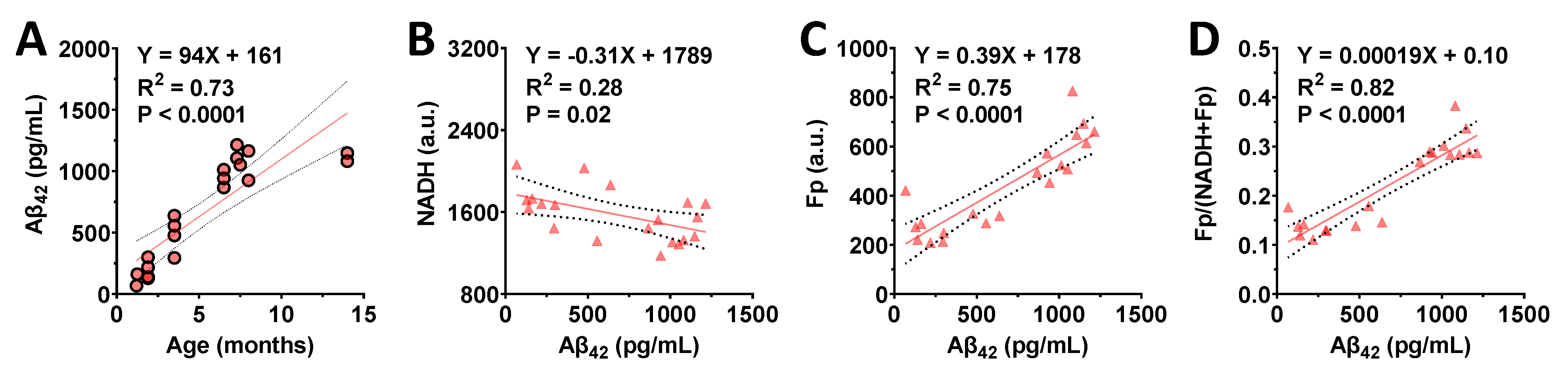
3.4. Correlations between the Redox Indices and Aβ42 Level in the Hippocampi of AD Mice
4. Discussion
4.1. The Hippocampal ORI Measures Differentiate Normal Aging from AD
4.2. The ORI Measures Correlate with Hippocampal Aβ42 Level in 5XFAD Mice
4.3. Limitations of Our Study and Open Questions
5. Conclusions
Supplementary Materials
Author Contributions
Funding
Institutional Review Board Statement
Informed Consent Statement
Data Availability Statement
Acknowledgments
Conflicts of Interest
References
- Shokouhi, S.; Claassen, D.; Riddle, W. Imaging Brain Metabolism and Pathology in Alzheimer’s Disease with Positron Emission Tomography. J. Alzheimer’s Dis. Parkinsonism 2014, 4, 143. [Google Scholar] [CrossRef]
- An, Y.; Varma, V.R.; Varma, S.; Casanova, R.; Dammer, E.; Pletnikova, O.; Chia, C.W.; Egan, J.M.; Ferrucci, L.; Troncoso, J.; et al. Evidence for brain glucose dysregulation in Alzheimer’s disease. Alzheimer’s Dement. 2018, 14, 318–329. [Google Scholar] [CrossRef] [PubMed]
- Reitz, C. Dyslipidemia and the risk of Alzheimer’s disease. Curr. Atheroscler. Rep. 2013, 15, 307. [Google Scholar] [CrossRef] [PubMed]
- Snowden, S.G.; Ebshiana, A.A.; Hye, A.; An, Y.; Pletnikova, O.; O’Brien, R.; Troncoso, J.; Legido-Quigley, C.; Thambisetty, M. Association between fatty acid metabolism in the brain and Alzheimer disease neuropathology and cognitive performance: A nontargeted metabolomic study. PLoS Med. 2017, 14, e1002266. [Google Scholar] [CrossRef]
- Atamna, H.; Frey, W.H., 2nd. Mechanisms of mitochondrial dysfunction and energy deficiency in Alzheimer’s disease. Mitochondrion 2007, 7, 297–310. [Google Scholar] [CrossRef]
- Zhang, C.; Rissman, R.A.; Feng, J. Characterization of ATP alternations in an Alzheimer’s disease transgenic mouse model. J. Alzheimer’s Dis. 2015, 44, 375–378. [Google Scholar] [CrossRef]
- Avetisyan, A.V.; Samokhin, A.N.; Alexandrova, I.Y.; Zinovkin, R.A.; Simonyan, R.A.; Bobkova, N.V. Mitochondrial Dysfunction in Neocortex and Hippocampus of Olfactory Bulbectomized Mice, a Model of Alzheimer’s Disease. Biochemistry 2016, 81, 615–623. [Google Scholar] [CrossRef]
- Cenini, G.; Lloret, A.; Cascella, R. Oxidative Stress in Neurodegenerative Diseases: From a Mitochondrial Point of View. Oxid. Med. Cell. Longev. 2019, 2019, 2105607. [Google Scholar] [CrossRef]
- Ghosh, D.; LeVault, K.R.; Barnett, A.J.; Brewer, G.J. A Reversible Early Oxidized Redox State That Precedes Macromolecular ROS Damage in Aging Nontransgenic and 3xTg-AD Mouse Neurons. J. Neurosci. 2012, 32, 5821–5832. [Google Scholar] [CrossRef]
- Long, A.N.; Owens, K.; Schlappal, A.E.; Kristian, T.; Fishman, P.S.; Schuh, R.A. Effect of nicotinamide mononucleotide on brain mitochondrial respiratory deficits in an Alzheimer’s disease-relevant murine model. BMC Neurol. 2015, 15, 19. [Google Scholar] [CrossRef] [Green Version]
- Dong, Y.; Brewer, G.J. Global Metabolic Shifts in Age and Alzheimer’s Disease Mouse Brains Pivot at NAD+/NADH Redox Sites. J. Alzheimer’s Dis. JAD 2019, 71, 119–140. [Google Scholar] [CrossRef] [PubMed]
- Zhang, Z.; Xu, H.N.; Li, S.; Davila, A., Jr.; Chellappa, K.; Davis, J.G.; Guan, Y.; Frederick, D.W.; Chu, W.; Zhao, H.; et al. Rapamycin maintains NAD+/NADH redox homeostasis in muscle cells. Aging 2020, 12, 17786–17799. [Google Scholar] [CrossRef] [PubMed]
- Lautrup, S.; Sinclair, D.A.; Mattson, M.P.; Fang, E.F. NAD+ in Brain Aging and Neurodegenerative Disorders. Cell Metab. 2019, 30, 630–655. [Google Scholar] [CrossRef] [PubMed]
- Dong, Y.; Digman, M.A.; Brewer, G.J. Age- and AD-related redox state of NADH in subcellular compartments by fluorescence lifetime imaging microscopy. Geroscience 2019, 41, 51–67. [Google Scholar] [CrossRef]
- Ghosh, D.; Levault, K.R.; Brewer, G.J. Relative importance of redox buffers GSH and NAD(P)H in age-related neurodegeneration and Alzheimer disease-like mouse neurons. Aging Cell 2014, 13, 631–640. [Google Scholar] [CrossRef]
- Hou, Y.; Lautrup, S.; Cordonnier, S.; Wang, Y.; Croteau, D.L.; Zavala, E.; Zhang, Y.; Moritoh, K.; O’Connell, J.F.; Baptiste, B.A.; et al. NAD+ supplementation normalizes key Alzheimer’s features and DNA damage responses in a new AD mouse model with introduced DNA repair deficiency. Proc. Natl. Acad. Sci. USA 2018, 115, e1876–e1885. [Google Scholar] [CrossRef]
- Gong, B.; Pan, Y.; Vempati, P.; Zhao, W.; Knable, L.; Ho, L.; Wang, J.; Sastre, M.; Ono, K.; Sauve, A.A.; et al. Nicotinamide riboside restores cognition through an upregulation of proliferator-activated receptor-γ coactivator 1α regulated β-secretase 1 degradation and mitochondrial gene expression in Alzheimer’s mouse models. Neurobiol. Aging 2013, 34, 1581–1588. [Google Scholar] [CrossRef]
- Liu, D.; Pitta, M.; Jiang, H.; Lee, J.H.; Zhang, G.; Chen, X.; Kawamoto, E.M.; Mattson, M.P. Nicotinamide forestalls pathology and cognitive decline in Alzheimer mice: Evidence for improved neuronal bioenergetics and autophagy procession. Neurobiol. Aging 2013, 34, 1564–1580. [Google Scholar] [CrossRef]
- Wang, X.; Hu, X.; Yang, Y.; Takata, T.; Sakurai, T. Nicotinamide mononucleotide protects against beta-amyloid oligomer-induced cognitive impairment and neuronal death. Brain Res. 2016, 1643, 1–9. [Google Scholar] [CrossRef]
- Xie, X.; Gao, Y.; Zeng, M.; Wang, Y.; Wei, T.F.; Lu, Y.B.; Zhang, W.P. Nicotinamide ribose ameliorates cognitive impairment of aged and Alzheimer’s disease model mice. Metab. Brain Dis. 2019, 34, 353–366. [Google Scholar] [CrossRef]
- Yao, Z.; Yang, W.; Gao, Z.; Jia, P. Nicotinamide mononucleotide inhibits JNK activation to reverse Alzheimer disease. Neurosci. Lett. 2017, 647, 133–140. [Google Scholar] [CrossRef] [PubMed]
- Heikal, A.A. Intracellular coenzymes as natural biomarkers for metabolic activities and mitochondrial anomalies. Biomark Med. 2010, 4, 241–263. [Google Scholar] [CrossRef] [PubMed]
- Chance, B.; Legallais, V.; Schoener, B. Metabolically Linked Changes in Fluorescence Emission Spectra of Cortex of Rat Brain, Kidney and Adrenal Gland. Nature 1962, 195, 1073–1075. [Google Scholar] [CrossRef] [PubMed]
- Chance, B.; Jamieson, D.; Coles, H. Energy-Linked Pyridine Nucleotide Reduction: Inhibitory Effects of Hyperbaric Oxygen In Vitro and In Vivo. Nature 1965, 206, 257–263. [Google Scholar] [CrossRef]
- Xu, H.N.; Lin, Z.; Gandhi, C.K.; Amatya, S.; Wang, Y.; Li, L.Z.; Floros, J. Sex and SP-A2 dependent NAD(H) Redox Alterations in Mouse Alveolar Macrophages in Response to Ozone Exposure: Potential Implications for COVID-19. Antioxidants 2020, 9, 915. [Google Scholar] [CrossRef]
- Podsednik, A.; Jacob, A.; Li, L.Z.; Xu, H.N. Relationship between optical redox status and reactive oxygen species in cancer cells. React. Oxyg. Species 2020, 9, 95–108. [Google Scholar] [CrossRef]
- Podsednik, A.; Jiang, J.; Jacob, A.; Li, L.Z.; Xu, H.N. Optical Redox Imaging of Treatment Responses to Nampt Inhibition and Combination Therapy in Triple-Negative Breast Cancer Cells. Int. J. Mol. Sci. 2021, 22, 5563. [Google Scholar] [CrossRef]
- Xu, H.N.; Floros, J.; Li, L.Z.; Amatya, S. Imaging NAD(H) Redox Alterations in Cryopreserved Alveolar Macrophages from Ozone-Exposed Mice and the Impact of Nutrient Starvation during Long Lag Times. Antioxidants 2021, 10, 767. [Google Scholar] [CrossRef]
- Garcia-Mesa, Y.; Xu, H.N.; Vance, P.; Gruenewald, A.L.; Garza, R.; Midkiff, C.; Alvarez-Hernandez, X.; Irwin, D.J.; Gill, A.J.; Kolson, D.L. Dimethyl Fumarate, an Approved Multiple Sclerosis Treatment, Reduces Brain Oxidative Stress in SIV-Infected Rhesus Macaques: Potential Therapeutic Repurposing for HIV Neuroprotection. Antioxidants 2021, 10, 416. [Google Scholar]
- Oakley, H.; Cole, S.L.; Logan, S.; Maus, E.; Shao, P.; Craft, J.; Guillozet-Bongaarts, A.; Ohno, M.; Disterhoft, J.; Van Eldik, L.; et al. Intraneuronal beta-amyloid aggregates, neurodegeneration, and neuron loss in transgenic mice with five familial Alzheimer’s disease mutations: Potential factors in amyloid plaque formation. J. Neurosci. Off. J. Soc. Neurosci. 2006, 26, 10129–10140. [Google Scholar] [CrossRef]
- Gourmaud, S.; Shou, H.; Irwin, D.J.; Sansalone, K.; Jacobs, L.M.; Lucas, T.H.; Marsh, E.D.; Davis, K.A.; Jensen, F.E.; Talos, D.M. Alzheimer-like amyloid and tau alterations associated with cognitive deficit in temporal lobe epilepsy. Brain 2019, 143, 191–209. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Xu, H.N.; Zhao, H.; Chellappa, K.; Davis, J.G.; Nioka, S.; Baur, J.A.; Li, L.Z. Optical redox imaging of fixed unstained muscle slides reveals useful biological information. Mol. Imaging Biol. 2019, 21, 417–425. [Google Scholar] [CrossRef] [PubMed]
- Xu, H.N.; Feng, M.; Nath, K.; Nelson, D.; Roman, J.; Zhao, H.; Lin, Z.; Glickson, J.; Li, L.Z. Optical redox imaging of lonidamine treatment response of melanoma cells and xenografts. Mol. Imaging Biol. 2019, 21, 426–435. [Google Scholar] [CrossRef] [PubMed]
- Bartolome, F.; Abramov, A.Y. Measurement of mitochondrial NADH and FAD autofluorescence in live cells. Methods Mol. Biol. 2015, 1264, 263–270. [Google Scholar] [CrossRef]
- Kim, B.Y.; Lim, H.S.; Kim, Y.; Kim, Y.J.; Koo, I.; Jeong, S.J. Evaluation of Animal Models by Comparison with Human Late-Onset Alzheimer’s Disease. Mol. Neurobiol. 2018, 55, 9234–9250. [Google Scholar] [CrossRef]
- Kanno, T.; Tsuchiya, A.; Nishizaki, T. Hyperphosphorylation of Tau at Ser396 occurs in the much earlier stage than appearance of learning and memory disorders in 5XFAD mice. Behav. Brain Res. 2014, 274, 302–306. [Google Scholar] [CrossRef]
- Landel, V.; Baranger, K.; Virard, I.; Loriod, B.; Khrestchatisky, M.; Rivera, S.; Benech, P.; Féron, F. Temporal gene profiling of the 5XFAD transgenic mouse model highlights the importance of microglial activation in Alzheimer’s disease. Mol. Neurodegener. 2014, 9, 33. [Google Scholar] [CrossRef]
- Cheignon, C.; Tomas, M.; Bonnefont-Rousselot, D.; Faller, P.; Hureau, C.; Collin, F. Oxidative stress and the amyloid beta peptide in Alzheimer’s disease. Redox Biol. 2018, 14, 450–464. [Google Scholar] [CrossRef]
- Hong, I.; Kang, T.; Yoo, Y.; Park, R.; Lee, J.; Lee, S.; Kim, J.; Song, B.; Kim, S.Y.; Moon, M.; et al. Quantitative proteomic analysis of the hippocampus in the 5XFAD mouse model at early stages of Alzheimer’s disease pathology. J. Alzheimer’s Dis. 2013, 36, 321–334. [Google Scholar] [CrossRef]
- Klaidman, L.K.; Leung, A.C.; Adams, J.D., Jr. High-performance liquid chromatography analysis of oxidized and reduced pyridine dinucleotides in specific brain regions. Anal. Biochem. 1995, 228, 312–317. [Google Scholar] [CrossRef]
- Quinn, K.P.; Sridharan, G.V.; Hayden, R.S.; Kaplan, D.L.; Lee, K.; Georgakoudi, I. Quantitative metabolic imaging using endogenous fluorescence to detect stem cell differentiation. Sci. Rep. 2013, 3, 3432. [Google Scholar] [CrossRef] [Green Version]
- Varone, A.; Xylas, J.; Quinn, K.P.; Pouli, D.; Sridharan, G.; McLaughlin-Drubin, M.E.; Alonzo, C.; Lee, K.; Munger, K.; Georgakoudi, I. Endogenous two-photon fluorescence imaging elucidates metabolic changes related to enhanced glycolysis and glutamine consumption in precancerous epithelial tissues. Cancer Res. 2014, 74, 3067–3075. [Google Scholar] [CrossRef] [PubMed]
- Santidrian, A.F.; LeBoeuf, S.E.; Wold, E.D.; Ritland, M.; Forsyth, J.S.; Felding, B.H. Nicotinamide phosphoribosyltransferase can affect metastatic activity and cell adhesive functions by regulating integrins in breast cancer. DNA Repair 2014, 23, 79–87. [Google Scholar] [CrossRef] [PubMed]
- Zhu, X.H.; Lu, M.; Lee, B.Y.; Ugurbil, K.; Chen, W. In vivo NAD assay reveals the intracellular NAD contents and redox state in healthy human brain and their age dependences. Proc. Natl. Acad. Sci. USA 2015, 112, 2876–2881. [Google Scholar] [CrossRef] [PubMed] [Green Version]





Publisher’s Note: MDPI stays neutral with regard to jurisdictional claims in published maps and institutional affiliations. |
© 2022 by the authors. Licensee MDPI, Basel, Switzerland. This article is an open access article distributed under the terms and conditions of the Creative Commons Attribution (CC BY) license (https://creativecommons.org/licenses/by/4.0/).
Share and Cite
Xu, H.N.; Gourmaud, S.; Podsednik, A.; Li, X.; Zhao, H.; Jensen, F.E.; Talos, D.M.; Li, L.Z. Optical Redox Imaging of Ex Vivo Hippocampal Tissue Reveals Age-Dependent Alterations in the 5XFAD Mouse Model of Alzheimer’s Disease. Metabolites 2022, 12, 786. https://doi.org/10.3390/metabo12090786
Xu HN, Gourmaud S, Podsednik A, Li X, Zhao H, Jensen FE, Talos DM, Li LZ. Optical Redox Imaging of Ex Vivo Hippocampal Tissue Reveals Age-Dependent Alterations in the 5XFAD Mouse Model of Alzheimer’s Disease. Metabolites. 2022; 12(9):786. https://doi.org/10.3390/metabo12090786
Chicago/Turabian StyleXu, He N., Sarah Gourmaud, Allison Podsednik, Xiaofan Li, Huaqing Zhao, Frances E. Jensen, Delia M. Talos, and Lin Z. Li. 2022. "Optical Redox Imaging of Ex Vivo Hippocampal Tissue Reveals Age-Dependent Alterations in the 5XFAD Mouse Model of Alzheimer’s Disease" Metabolites 12, no. 9: 786. https://doi.org/10.3390/metabo12090786
APA StyleXu, H. N., Gourmaud, S., Podsednik, A., Li, X., Zhao, H., Jensen, F. E., Talos, D. M., & Li, L. Z. (2022). Optical Redox Imaging of Ex Vivo Hippocampal Tissue Reveals Age-Dependent Alterations in the 5XFAD Mouse Model of Alzheimer’s Disease. Metabolites, 12(9), 786. https://doi.org/10.3390/metabo12090786