


Site-Differentiated Iron–Sulfur Cluster Ligation Affects Flavin-Based Electron Bifurcation Activity

Abstract
:1. Introduction
2. Materials and Methods
2.1. Plasmid Construction
2.2. NfnSL Protein Overexpression and Purification
2.3. Cofactor Reconstitution and Protein Preparation
2.4. Biochemical Characterizations
3. Results
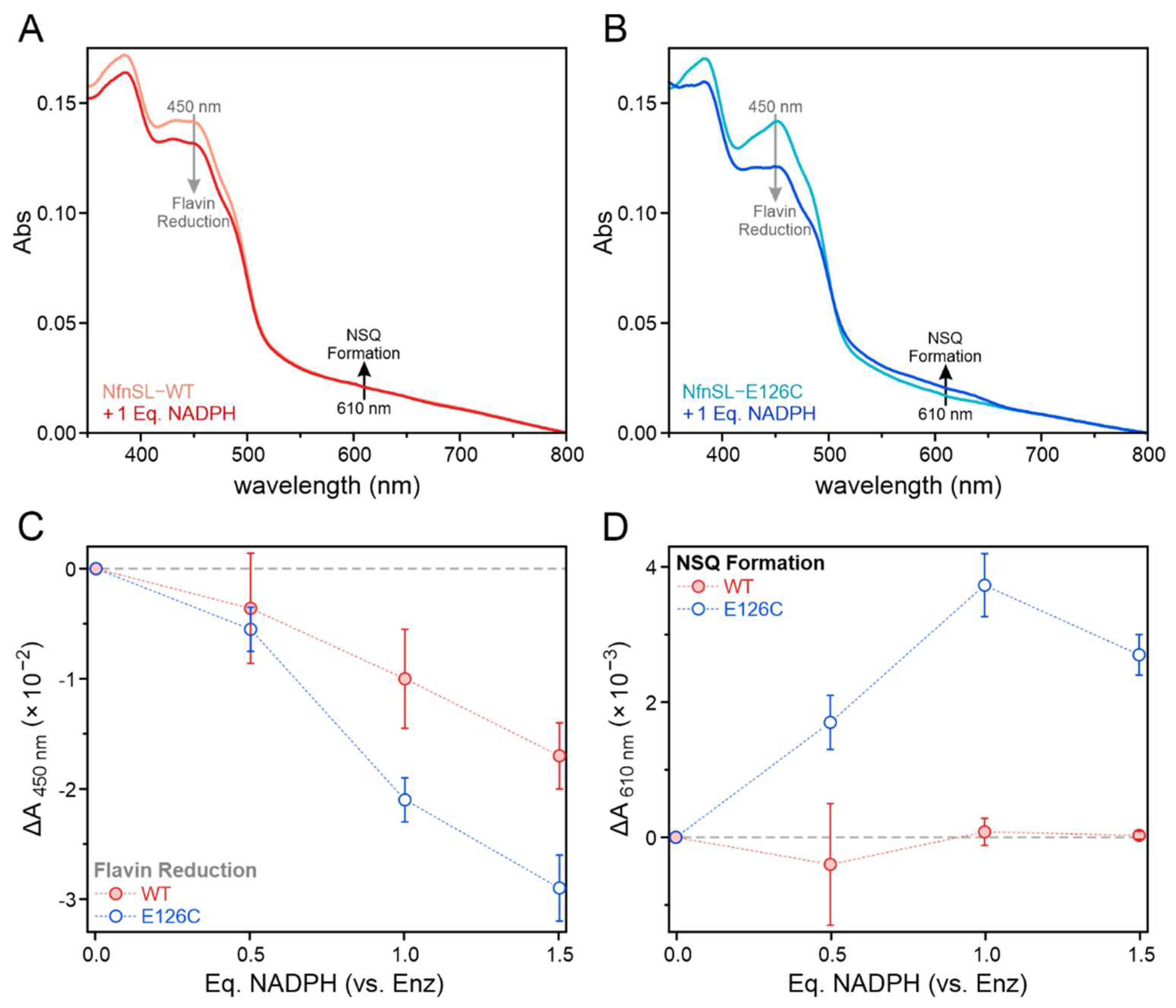
3.1. Protein Expression, Purification, and Reconstitution
3.2. Dye-Based Steady-State Kinetics
3.3. Binding of NADP(H) to NfnL-E126C
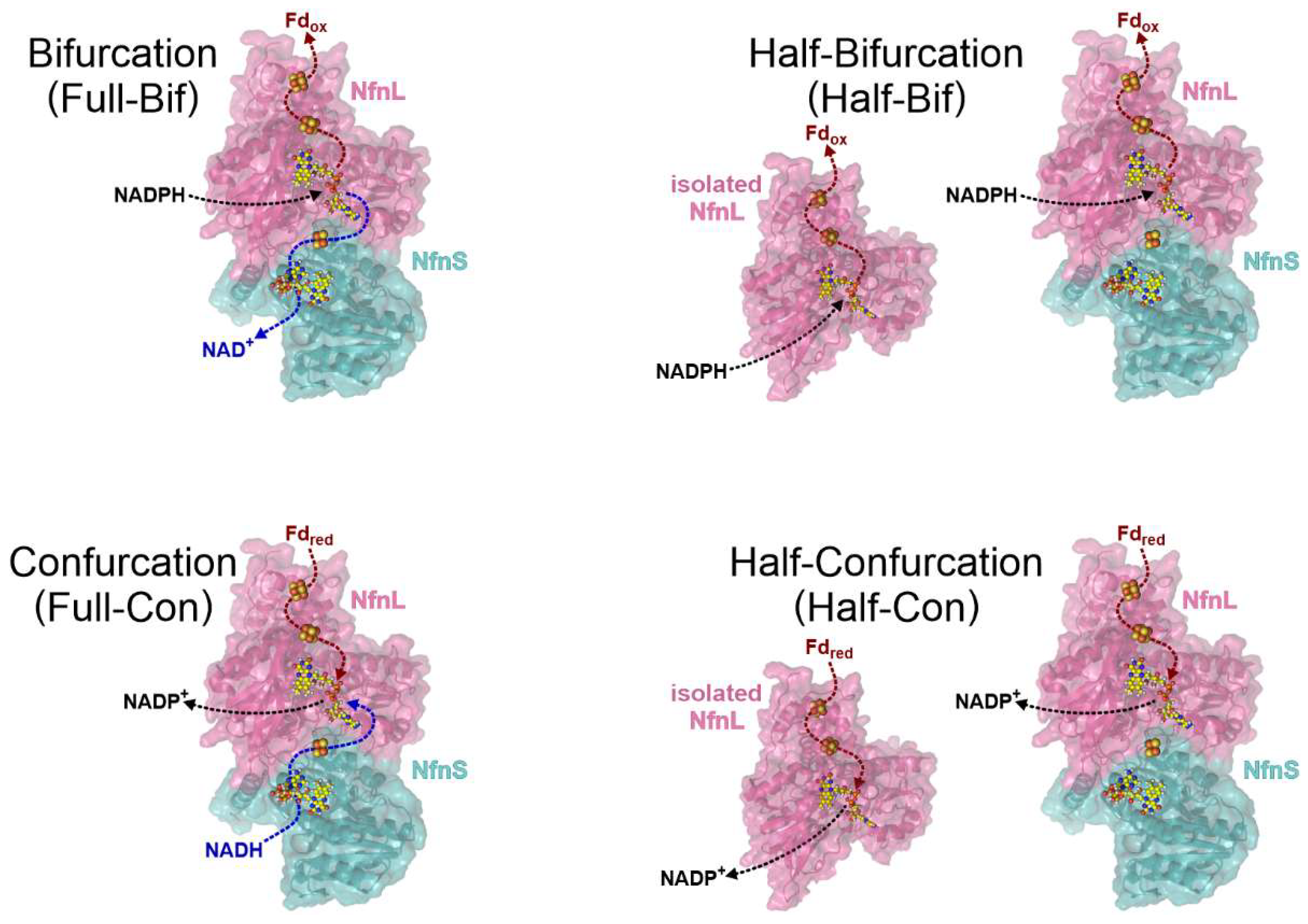
3.4. Full-Bifurcation and Full-Confurcation Activity Assays
3.5. Half-Bifurcation and Half-Confurcation Activity Assays
3.6. Electron Distribution along the High-Potential Branch of Nfn
4. Discussion
5. Conclusions
Author Contributions
Funding
Institutional Review Board Statement
Informed Consent Statement
Data Availability Statement
Acknowledgments
Conflicts of Interest
References
- Wise, C.E.; Ledinina, A.E.; Yuly, J.L.; Artz, J.H.; Lubner, C.E. The role of thermodynamic features on the functional activity of electron bifurcating enzymes. Biochim. Biophys. Acta Bioenerg. 2021, 1862, 148377. [Google Scholar] [CrossRef]
- Buckel, W.; Thauer, R.K. Flavin-Based Electron Bifurcation, Ferredoxin, Flavodoxin, and Anaerobic Respiration With Protons (Ech) or NAD(+) (Rnf) as Electron Acceptors: A Historical Review. Front. Microbiol. 2018, 9, 401. [Google Scholar] [CrossRef] [PubMed]
- Poudel, S.; Dunham, E.C.; Lindsay, M.R.; Amenabar, M.J.; Fones, E.M.; Colman, D.R.; Boyd, E.S. Origin and Evolution of Flavin-Based Electron Bifurcating Enzymes. Front. Microbiol. 2018, 9, 1762. [Google Scholar] [CrossRef] [PubMed]
- Liang, J.; Huang, H.; Wang, S. Distribution, Evolution, Catalytic Mechanism, and Physiological Functions of the Flavin-Based Electron-Bifurcating NADH-Dependent Reduced Ferredoxin: NADP(+) Oxidoreductase. Front. Microbiol. 2019, 10, 373. [Google Scholar] [CrossRef] [PubMed]
- Jay, Z.J.; Hunt, K.A.; Chou, K.J.; Schut, G.J.; Maness, P.C.; Adams, M.W.W.; Carlson, R.P. Integrated thermodynamic analysis of electron bifurcating [FeFe]-hydrogenase to inform anaerobic metabolism and H2 production. Biochim. Biophys. Acta Bioenerg. 2020, 1861, 148087. [Google Scholar] [CrossRef]
- Kayastha, K.; Vitt, S.; Buckel, W.; Ermler, U. Flavins in the electron bifurcation process. Arch. Biochem. Biophys. 2021, 701, 108796. [Google Scholar] [CrossRef]
- Appel, L.; Willistein, M.; Dahl, C.; Ermler, U.; Boll, M. Functional diversity of prokaryotic HdrA(BC) modules: Role in flavin-based electron bifurcation processes and beyond. Biochim. Biophys. Acta Bioenerg. 2021, 1862, 148379. [Google Scholar] [CrossRef]
- Yuly, J.L.; Zhang, P.; Lubner, C.E.; Peters, J.W.; Beratan, D.N. Universal free-energy landscape produces efficient and reversible electron bifurcation. Proc. Natl. Acad. Sci. USA 2020, 117, 21045–21051. [Google Scholar] [CrossRef]
- Wise, C.E.; Ledinina, A.E.; Mulder, D.W.; Chou, K.J.; Peters, J.W.; King, P.W.; Lubner, C.E. An uncharacteristically low-potential flavin governs the energy landscape of electron bifurcation. Proc. Natl. Acad. Sci. USA 2022, 119, e2117882119. [Google Scholar] [CrossRef]
- Lubner, C.E.; Jennings, D.P.; Mulder, D.W.; Schut, G.J.; Zadvornyy, O.A.; Hoben, J.P.; Tokmina-Lukaszewska, M.; Berry, L.; Nguyen, D.M.; Lipscomb, G.L.; et al. Mechanistic insights into energy conservation by flavin-based electron bifurcation. Nat. Chem. Biol. 2017, 13, 655–659. [Google Scholar] [CrossRef]
- Zhang, P.; Yuly, J.L.; Lubner, C.E.; Mulder, D.W.; King, P.W.; Peters, J.W.; Beratan, D.N. Electron Bifurcation: Thermodynamics and Kinetics of Two-Electron Brokering in Biological Redox Chemistry. Acc. Chem. Res. 2017, 50, 2410–2417. [Google Scholar] [CrossRef] [PubMed]
- Lubner, C.E.; Artz, J.H.; Mulder, D.W.; Oza, A.; Ward, R.J.; Williams, S.G.; Jones, A.K.; Peters, J.W.; Smalyukh, I.I.; Bharadwaj, V.S.; et al. A site-differentiated [4Fe-4S] cluster controls electron transfer reactivity of Clostridium acetobutylicum [FeFe]-hydrogenase I. Chem. Sci. 2022, 13, 4581–4588. [Google Scholar] [CrossRef]
- Bak, D.W.; Elliott, S.J. Alternative FeS cluster ligands: Tuning redox potentials and chemistry. Curr. Opin. Chem. Biol. 2014, 19, 50–58. [Google Scholar] [CrossRef] [PubMed]
- Adrover, M.; Howes, B.D.; Iannuzzi, C.; Smulevich, G.; Pastore, A. Anatomy of an iron-sulfur cluster scaffold protein: Understanding the determinants of [2Fe-2S] cluster stability on IscU. Biochim. Biophys. Acta 2015, 1853, 1448–1456. [Google Scholar] [CrossRef]
- Forzi, L.; Koch, J.; Guss, A.M.; Radosevich, C.G.; Metcalf, W.W.; Hedderich, R. Assignment of the [4Fe-4S] clusters of Ech hydrogenase from Methanosarcina barkeri to individual subunits via the characterization of site-directed mutants. FEBS J. 2005, 272, 4741–4753. [Google Scholar] [CrossRef] [PubMed]
- Dementin, S.; Belle, V.; Bertrand, P.; Guigliarelli, B.; Adryanczyk-Perrier, G.; De Lacey, A.L.; Fernandez, V.M.; Rousset, M.; Leger, C. Changing the ligation of the distal [4Fe4S] cluster in NiFe hydrogenase impairs inter- and intramolecular electron transfers. J. Am. Chem. Soc. 2006, 128, 5209–5218. [Google Scholar] [CrossRef]
- Yu, L.; Bryant, D.A.; Golbeck, J.H. Evidence for a mixed-ligand [4Fe-4S] cluster in the C14D mutant of PsaC. Altered reduction potentials and EPR spectral properties of the FA and FB clusters on rebinding to the P700-FX core. Biochemistry 1995, 34, 7861–7868. [Google Scholar] [CrossRef]
- Dicus, M.M.; Conlan, A.; Nechushtai, R.; Jennings, P.A.; Paddock, M.L.; Britt, R.D.; Stoll, S. Binding of histidine in the (Cys)3(His)1-coordinated [2Fe-2S] cluster of human mitoNEET. J. Am. Chem. Soc. 2010, 132, 2037–2049. [Google Scholar] [CrossRef]
- Nakamaru-Ogiso, E.; Matsuno-Yagi, A.; Yoshikawa, S.; Yagi, T.; Ohnishi, T. Iron-sulfur cluster N5 is coordinated by an HXXXCXXCXXXXXC motif in the NuoG subunit of Escherichia coli NADH:quinone oxidoreductase (complex I). J. Biol. Chem. 2008, 283, 25979–25987. [Google Scholar] [CrossRef]
- Gasteiger, E.; Hoogland, C.; Gattiker, A.; Duvaud, S.; Wilkins, M.R.; Appel, R.D.; Bairoch, A. Protein Identification and Analysis Tools on the ExPASy Server. In The Proteomics Protocols Handbook, 1st ed.; Walker, J.M., Ed.; Humana Press: Totowa, NJ, USA, 2005; pp. 571–607. [Google Scholar]
- Conover, R.C.; Kowal, A.T.; Fu, W.G.; Park, J.B.; Aono, S.; Adams, M.W.; Johnson, M.K. Spectroscopic characterization of the novel iron-sulfur cluster in Pyrococcus furiosus ferredoxin. J. Biol. Chem. 1990, 265, 8533–8541. [Google Scholar] [CrossRef]
- Brereton, P.S.; Verhagen, M.F.; Zhou, Z.H.; Adams, M.W. Effect of iron-sulfur cluster environment in modulating the thermodynamic properties and biological function of ferredoxin from Pyrococcus furiosus. Biochemistry 1998, 37, 7351–7362. [Google Scholar] [CrossRef] [PubMed]
- Demmer, J.K.; Huang, H.; Wang, S.; Demmer, U.; Thauer, R.K.; Ermler, U. Insights into Flavin-based Electron Bifurcation via the NADH-dependent Reduced Ferredoxin:NADP Oxidoreductase Structure. J. Biol. Chem. 2015, 290, 21985–21995. [Google Scholar] [CrossRef] [PubMed]
- Huang, H.; Wang, S.; Moll, J.; Thauer, R.K. Electron bifurcation involved in the energy metabolism of the acetogenic bacterium Moorella thermoacetica growing on glucose or H2 plus CO2. J. Bacteriol. 2012, 194, 3689–3699. [Google Scholar] [CrossRef] [PubMed]
- Wang, S.; Huang, H.; Moll, J.; Thauer, R.K. NADP+ reduction with reduced ferredoxin and NADP+ reduction with NADH are coupled via an electron-bifurcating enzyme complex in Clostridium kluyveri. J. Bacteriol. 2010, 192, 5115–5123. [Google Scholar] [CrossRef] [PubMed]
- Rodkey, F.L.; Donovan, J.A., Jr. Oxidation-reduction potentials of the triphosphopyridine nucleotide system. J. Biol. Chem. 1959, 234, 677–680. [Google Scholar] [CrossRef]
- Spaans, S.K.; Weusthuis, R.A.; van der Oost, J.; Kengen, S.W. NADPH-generating systems in bacteria and archaea. Front. Microbiol. 2015, 6, 742. [Google Scholar] [CrossRef]
- Yuly, J.L.; Lubner, C.E.; Zhang, P.; Beratan, D.N.; Peters, J.W. Electron bifurcation: Progress and grand challenges. Chem. Commun. 2019, 55, 11823–11832. [Google Scholar] [CrossRef]
- Peters, J.W.; Beratan, D.N.; Bothner, B.; Dyer, R.B.; Harwood, C.S.; Heiden, Z.M.; Hille, R.; Jones, A.K.; King, P.W.; Lu, Y.; et al. A new era for electron bifurcation. Curr. Opin. Chem. Biol. 2018, 47, 32–38. [Google Scholar] [CrossRef]
- Berry, L.; Poudel, S.; Tokmina-Lukaszewska, M.; Colman, D.R.; Nguyen, D.M.N.; Schut, G.J.; Adams, M.W.W.; Peters, J.W.; Boyd, E.S.; Bothner, B. H/D exchange mass spectrometry and statistical coupling analysis reveal a role for allostery in a ferredoxin-dependent bifurcating transhydrogenase catalytic cycle. Biochim. Biophys. Acta 2018, 1862, 9–17. [Google Scholar] [CrossRef]
- Hoben, J.P.; Lubner, C.E.; Ratzloff, M.W.; Schut, G.J.; Nguyen, D.M.N.; Hempel, K.W.; Adams, M.W.W.; King, P.W.; Miller, A.F. Equilibrium and ultrafast kinetic studies manipulating electron transfer: A short-lived flavin semiquinone is not sufficient for electron bifurcation. J. Biol. Chem. 2017, 292, 14039–14049. [Google Scholar] [CrossRef] [Green Version]
- Marcus, R.A.; Sutin, N. Electron transfers in chemistry and biology. Biochim. Biophys. Acta 1985, 811, 265–322. [Google Scholar] [CrossRef]
- Gao, X.; Wen, X.; Esser, L.; Quinn, B.; Yu, L.; Yu, C.A.; Xia, D. Structural basis for the quinone reduction in the bc1 complex: A comparative analysis of crystal structures of mitochondrial cytochrome bc1 with bound substrate and inhibitors at the Qi site. Biochemistry 2003, 42, 9067–9080. [Google Scholar] [CrossRef] [PubMed]
- Feng, X.; Schut, G.J.; Haja, D.K.; Adams, M.W.W.; Li, H. Structure and electron transfer pathways of an electron-bifurcating NiFe-hydrogenase. Sci. Adv. 2022, 8, eabm7546. [Google Scholar] [CrossRef]
- Nguyen, D.M.N.; Schut, G.J.; Zadvornyy, O.A.; Tokmina-Lukaszewska, M.; Poudel, S.; Lipscomb, G.L.; Adams, L.A.; Dinsmore, J.T.; Nixon, W.J.; Boyd, E.S.; et al. Two functionally distinct NADP(+)-dependent ferredoxin oxidoreductases maintain the primary redox balance of Pyrococcus furiosus. J. Biol. Chem. 2017, 292, 14603–14616. [Google Scholar] [CrossRef] [PubMed] [Green Version]







{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
| NfnL | ±NfnS | KM (mM) | Vmax (mM·s−1) | kcat (s−1) | kcat/KM (mM−1·s−1) |
|---|---|---|---|---|---|
| WT | − | 1.3 ± 0.1 | 0.05 ± 0.01 | 2.3 ± 0.3 | 1.7 ± 0.3 |
| + | 0.37 ± 0.02 | 0.11 ± 0.02 | 5.7 ± 0.8 | 12 ± 3 | |
| E126C | − | 0.30 ± 0.03 | 0.07 ± 0.01 | 3.5 ± 0.7 | 15 ± 2 |
| + | 0.37 ± 0.01 | 0.12 ± 0.01 | 5.9 ± 0.6 | 16 ± 2 |
Publisher’s Note: MDPI stays neutral with regard to jurisdictional claims in published maps and institutional affiliations. |
© 2022 by the authors. Licensee MDPI, Basel, Switzerland. This article is an open access article distributed under the terms and conditions of the Creative Commons Attribution (CC BY) license (https://creativecommons.org/licenses/by/4.0/).
Share and Cite
Wise, C.E.; Ledinina, A.E.; Lubner, C.E. Site-Differentiated Iron–Sulfur Cluster Ligation Affects Flavin-Based Electron Bifurcation Activity. Metabolites 2022, 12, 823. https://doi.org/10.3390/metabo12090823
Wise CE, Ledinina AE, Lubner CE. Site-Differentiated Iron–Sulfur Cluster Ligation Affects Flavin-Based Electron Bifurcation Activity. Metabolites. 2022; 12(9):823. https://doi.org/10.3390/metabo12090823
Chicago/Turabian StyleWise, Courtney E., Anastasia E. Ledinina, and Carolyn E. Lubner. 2022. "Site-Differentiated Iron–Sulfur Cluster Ligation Affects Flavin-Based Electron Bifurcation Activity" Metabolites 12, no. 9: 823. https://doi.org/10.3390/metabo12090823
APA StyleWise, C. E., Ledinina, A. E., & Lubner, C. E. (2022). Site-Differentiated Iron–Sulfur Cluster Ligation Affects Flavin-Based Electron Bifurcation Activity. Metabolites, 12(9), 823. https://doi.org/10.3390/metabo12090823