
Context-Specific Genome-Scale Metabolic Modelling and Its Application to the Analysis of COVID-19 Metabolic Signatures



Abstract
:1. Introduction
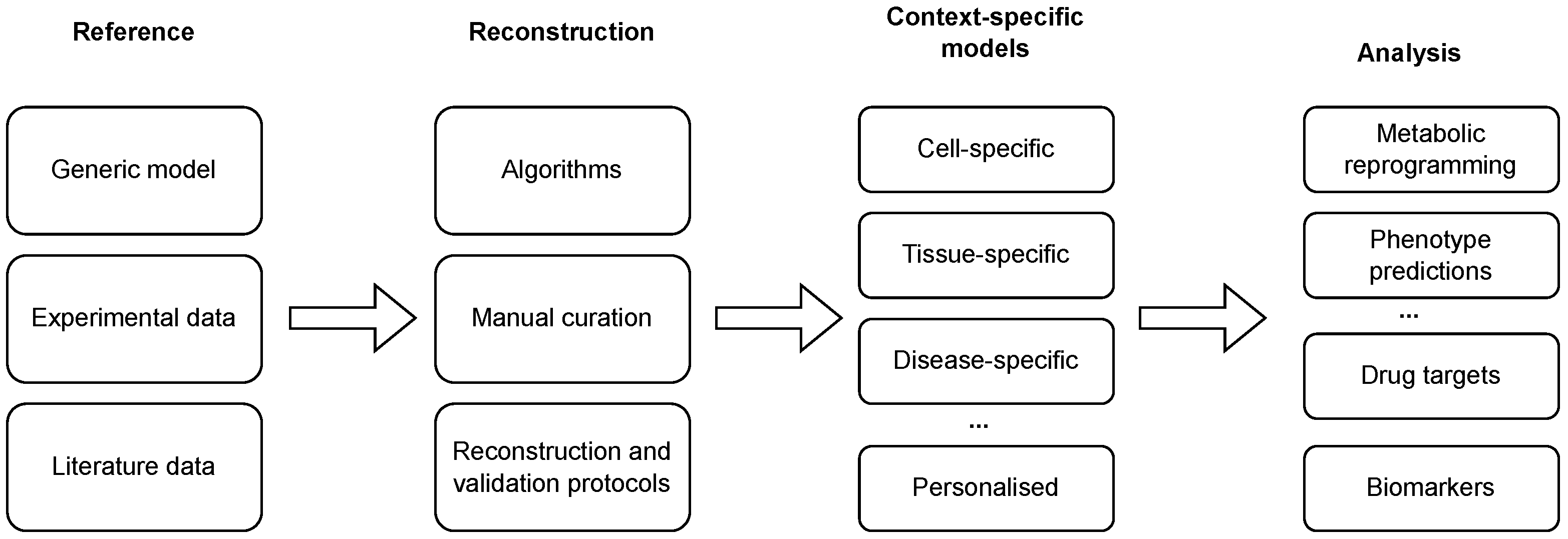
2. Genome-Scale Metabolic Modelling
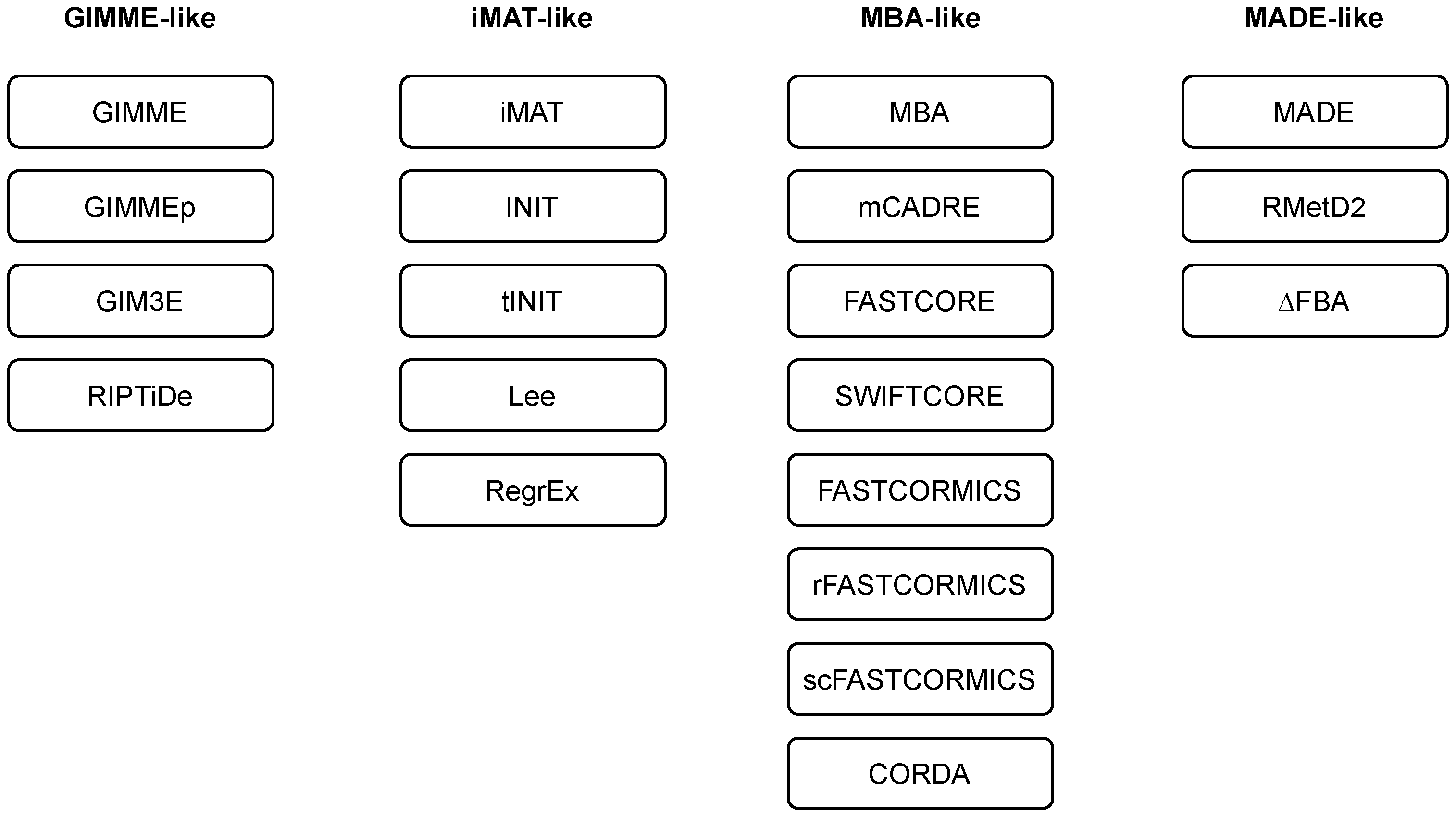
3. Algorithms and Tools for Reconstruction of Context-Specific Models
3.1. GIMME-like Family
3.2. iMAT-like Family
3.3. MBA-like Family
3.4. MADE-like Family
4. Data for Model Construction and Validation
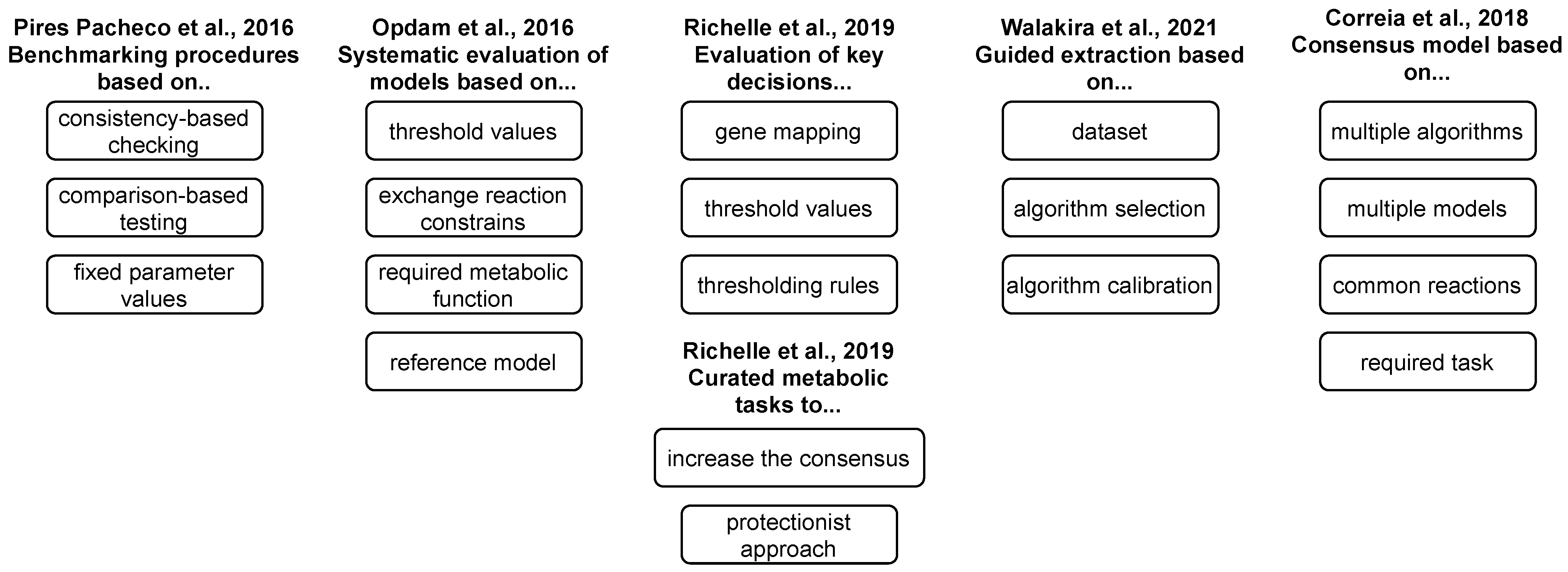
5. Reconstruction and Validation Protocols

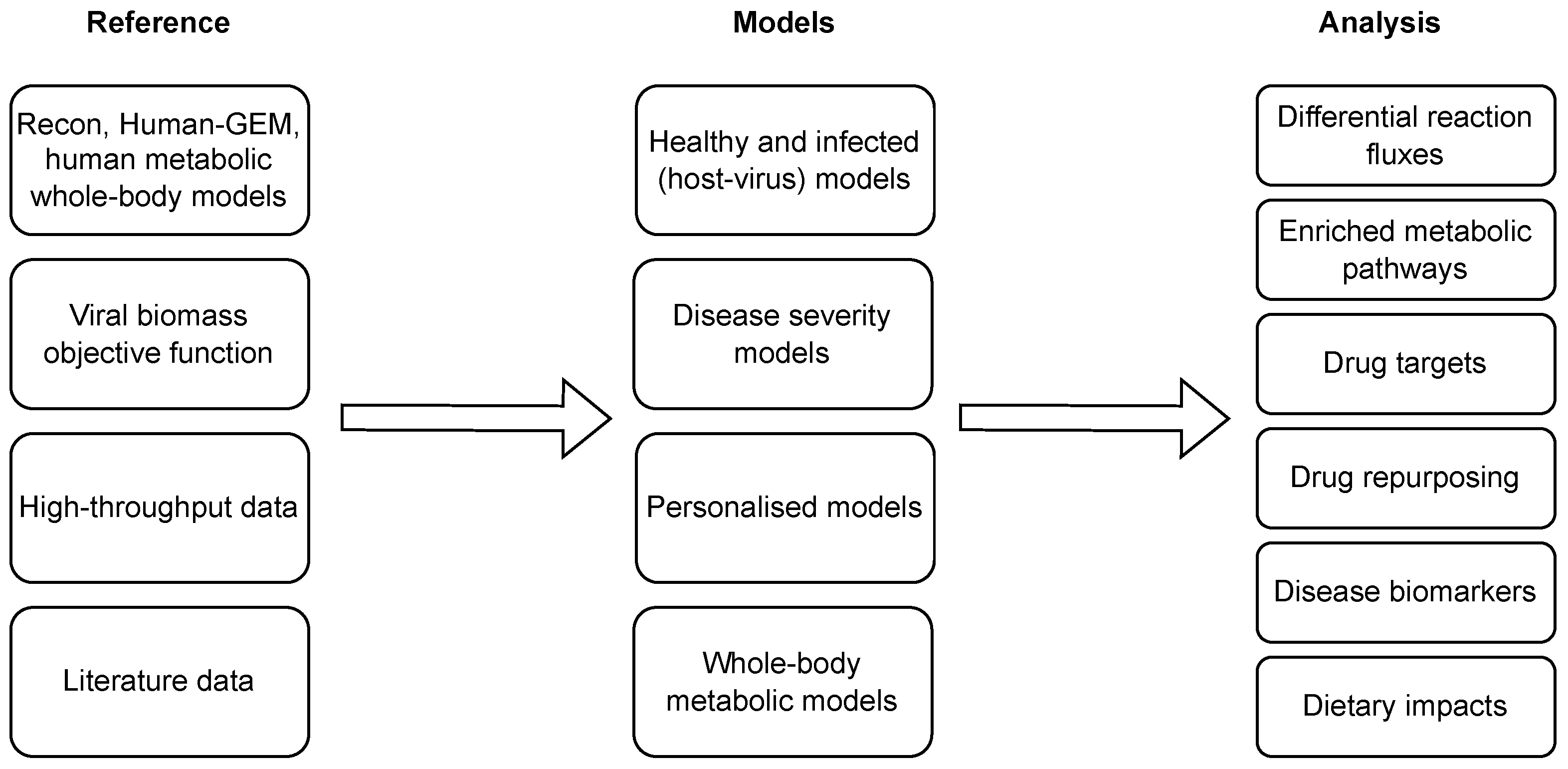
6. COVID-19 Applications of Context-Specific Genome-Scale Metabolic Modelling
7. Conclusions
Author Contributions
Funding
Conflicts of Interest
Abbreviations
| ATP | adenosine triphosphate |
| BOF | biomass objective function |
| CCLE | Cancer Cell Line Encyclopedia |
| COBRA | constraint-based reconstruction and analysis |
| CORDA | cost optimization reaction dependency assessment |
| COVID | coronavirus disease |
| EBI | European Bioinformatics Institute |
| EGA | European Genome-Phenome Archive |
| ENA | European Nucleotide Archive |
| FANTOM5 | Functional Annotation of the Mammalian Genome 5 |
| FASTCC | fast consistency checking |
| FBA | flux balance analysis |
| FVA | flux variability analysis |
| GEM | genome-scale metabolic model |
| GEO | Genome Expression Omnibus |
| GIM3E | gene inactivation moderated by metabolism, metabolomics and expression |
| GIMME | gene inactivity moderated by metabolism and expression |
| GIMMEp | gene inactivity moderated by metabolism and expression by proteome |
| GPR | gene-protein-reaction |
| GTEx | Genotype-Tissue Expression database |
| HPA | Human Protein Atlas |
| iMAT | integrative metabolic analysis tool |
| INIT | integrative network inference for tissues |
| LP | linear programming |
| MADE | metabolic adjustment by differential expression |
| MBA | model building algorithm |
| mCADRE | metabolic context-specificity assessed by deterministic reaction evaluation |
| METRADE | MEtabolic and TRanscriptomics ADaptation Estimator |
| MILP | mixed integer linear programming |
| MIQP | mixed integer quadratic programming |
| MTA | metabolic transformation algorithm |
| NCBI | National Center for Biotechnology Information |
| NGS | next-generation sequencing |
| NHBE | normal human bronchial epithelial |
| PCA | principal component analysis |
| PDC | Proteomic Data Commons |
| pFBA | parsimonious flux balance analysis |
| PRIME | personalized reconstructIon of metabolic models |
| QP | quadratic programming |
| RegrEx | regularized context-specific model extraction method |
| RIPTide | reaction inclusion by parsimony and transcript distribution |
| RMetD2 | relative metabolic differences version 2 |
| RMF | required metabolic function |
| RNA-seq | RNA sequencing |
| SARS-CoV-2 | Severe acute respiratory syndrome coronavirus 2 |
| scRNA-seq | single cell RNA sequencing |
| SRA | Sequence Read Archive |
| TCGA | The Cancer Genome Atlas |
| TIGER | toolbox for integrating genome-scale metabolism, expression, and regulation |
| tINIT | task-driven integrative network inference for tissues |
| TPM | transcripts per million |
| VBOF | viral biomass objective function |
| WBM | whole-body model |
References
- Zhang, C.; Hua, Q. Applications of genome-scale metabolic models in biotechnology and systems medicine. Front. Physiol. 2016, 6, 413. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Lewis, N.E.; Nagarajan, H.; Palsson, B.O. Constraining the metabolic genotype–phenotype relationship using a phylogeny of in silico methods. Nat. Rev. Microbiol. 2012, 10, 291–305. [Google Scholar] [CrossRef] [Green Version]
- O’brien, E.J.; Lerman, J.A.; Chang, R.L.; Hyduke, D.R.; Palsson, B.Ø. Genome-scale models of metabolism and gene expression extend and refine growth phenotype prediction. Mol. Syst. Biol. 2013, 9, 693. [Google Scholar] [CrossRef] [PubMed]
- Maarleveld, T.R.; Khandelwal, R.A.; Olivier, B.G.; Teusink, B.; Bruggeman, F.J. Basic concepts and principles of stoichiometric modeling of metabolic networks. Biotechnol. J. 2013, 8, 997–1008. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Nilsson, A.; Nielsen, J. Genome scale metabolic modeling of cancer. Metab. Eng. 2017, 43, 103–112. [Google Scholar] [CrossRef] [PubMed]
- Moolamalla, S.; Vinod, P. Genome-scale metabolic modelling predicts biomarkers and therapeutic targets for neuropsychiatric disorders. Comput. Biol. Med. 2020, 125, 103994. [Google Scholar] [CrossRef]
- Thiele, I.; Palsson, B.Ø. A protocol for generating a high-quality genome-scale metabolic reconstruction. Nat. Protoc. 2010, 5, 93–121. [Google Scholar] [CrossRef] [Green Version]
- Mendoza, S.N.; Olivier, B.G.; Molenaar, D.; Teusink, B. A systematic assessment of current genome-scale metabolic reconstruction tools. Genome Biol. 2019, 20, 1–20. [Google Scholar] [CrossRef] [Green Version]
- Heirendt, L.; Arreckx, S.; Pfau, T.; Mendoza, S.N.; Richelle, A.; Heinken, A.; Haraldsdóttir, H.S.; Wachowiak, J.; Keating, S.M.; Vlasov, V.; et al. Creation and analysis of biochemical constraint-based models using the COBRA Toolbox v. 3.0. Nat. Protoc. 2019, 14, 639–702. [Google Scholar] [CrossRef] [Green Version]
- Ebrahim, A.; Lerman, J.A.; Palsson, B.O.; Hyduke, D.R. COBRApy: Constraints-based reconstruction and analysis for python. BMC Syst. Biol. 2013, 7, 1–6. [Google Scholar] [CrossRef]
- Wang, H.; Marcišauskas, S.; Sánchez, B.J.; Domenzain, I.; Hermansson, D.; Agren, R.; Nielsen, J.; Kerkhoven, E.J. RAVEN 2.0: A versatile toolbox for metabolic network reconstruction and a case study on Streptomyces coelicolor. PLoS Comput. Biol. 2018, 14, e1006541. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Steffensen, J.L.; Dufault-Thompson, K.; Zhang, Y. PSAMM: A portable system for the analysis of metabolic models. PLoS Comput. Biol. 2016, 12, e1004732. [Google Scholar] [CrossRef]
- Karlsen, E.; Schulz, C.; Almaas, E. Automated generation of genome-scale metabolic draft reconstructions based on KEGG. BMC Bioinf. 2018, 19, 1–11. [Google Scholar] [CrossRef] [PubMed]
- Kostromins, A.; Stalidzans, E. Paint4Net: COBRA Toolbox extension for visualization of stoichiometric models of metabolism. Biosystems 2012, 109, 233–239. [Google Scholar] [CrossRef]
- Pan, S.; Reed, J.L. Advances in gap-filling genome-scale metabolic models and model-driven experiments lead to novel metabolic discoveries. Curr. Opin. Biotechnol. 2018, 51, 103–108. [Google Scholar] [CrossRef] [PubMed]
- Orth, J.D.; Thiele, I.; Palsson, B.Ø. What is flux balance analysis? Nat. Biotechnol. 2010, 28, 245–248. [Google Scholar] [CrossRef]
- Herrmann, H.A.; Dyson, B.C.; Vass, L.; Johnson, G.N.; Schwartz, J.M. Flux sampling is a powerful tool to study metabolism under changing environmental conditions. NPJ Syst. Biol. Appl. 2019, 5, 32. [Google Scholar] [CrossRef] [Green Version]
- Ye, C.; Wei, X.; Shi, T.; Sun, X.; Xu, N.; Gao, C.; Zou, W. Genome-scale metabolic network models: From first-generation to next-generation. Appl. Microbiol. Biotechnol. 2022, 106, 4907–4920. [Google Scholar] [CrossRef]
- Duarte, N.C.; Becker, S.A.; Jamshidi, N.; Thiele, I.; Mo, M.L.; Vo, T.D.; Srivas, R.; Palsson, B.Ø. Global reconstruction of the human metabolic network based on genomic and bibliomic data. Proc. Natl. Acad. Sci. USA 2007, 104, 1777–1782. [Google Scholar] [CrossRef] [Green Version]
- Swainston, N.; Smallbone, K.; Hefzi, H.; Dobson, P.D.; Brewer, J.; Hanscho, M.; Zielinski, D.C.; Ang, K.S.; Gardiner, N.J.; Gutierrez, J.M.; et al. Recon 2.2: From reconstruction to model of human metabolism. Metabolomics 2016, 12, 109. [Google Scholar] [CrossRef]
- Brunk, E.; Sahoo, S.; Zielinski, D.C.; Altunkaya, A.; Dräger, A.; Mih, N.; Gatto, F.; Nilsson, A.; Preciat Gonzalez, G.A.; Aurich, M.K.; et al. Recon3D enables a three-dimensional view of gene variation in human metabolism. Nat. Biotechnol. 2018, 36, 272–281. [Google Scholar] [CrossRef] [PubMed]
- Mardinoglu, A.; Agren, R.; Kampf, C.; Asplund, A.; Uhlen, M.; Nielsen, J. Genome-scale metabolic modelling of hepatocytes reveals serine deficiency in patients with non-alcoholic fatty liver disease. Nat. Commun. 2014, 5, 3083. [Google Scholar] [CrossRef] [Green Version]
- Robinson, J.L.; Kocabaş, P.; Wang, H.; Cholley, P.E.; Cook, D.; Nilsson, A.; Anton, M.; Ferreira, R.; Domenzain, I.; Billa, V.; et al. An atlas of human metabolism. Sci. Signal. 2020, 13, eaaz1482. [Google Scholar] [CrossRef] [PubMed]
- Bintener, T.; Pacheco, M.P.; Kishk, A.; Didier, J.; Sauter, T. Drug Target Prediction Using Context-Specific Metabolic Models Reconstructed from rFASTCORMICS. In Cancer Drug Resistance; Springer: Berlin/Heidelberg, Germany, 2022; pp. 221–240. [Google Scholar] [CrossRef]
- Tomi-Andrino, C.; Pandele, A.; Winzer, K.; King, J.; Rahman, R.; Kim, D.H. Metabolic modeling-based drug repurposing in Glioblastoma. Sci. Rep. 2022, 12, 11189. [Google Scholar] [CrossRef]
- Barata, T.; Vieira, V.; Rodrigues, R.; das Neves, R.P.; Rocha, M. Reconstruction of tissue-specific genome-scale metabolic models for human cancer stem cells. Comput. Biol. Med. 2022, 142, 105177. [Google Scholar] [CrossRef]
- Song, H.; Kim, T.Y.; Choi, B.K.; Choi, S.J.; Nielsen, L.K.; Chang, H.N.; Lee, S.Y. Development of chemically defined medium for Mannheimia succiniciproducens based on its genome sequence. Appl. Microbiol. Biotechnol. 2008, 79, 263–272. [Google Scholar] [CrossRef]
- Jiang, S.; Otero-Muras, I.; Banga, J.R.; Wang, Y.; Kaiser, M.; Krasnogor, N. OptDesign: Identifying Optimum Design Strategies in Strain Engineering for Biochemical Production. ACS Synth. Biol. 2022, 11, 1531–1541. [Google Scholar] [CrossRef]
- Lachance, J.C.; Matteau, D.; Brodeur, J.; Lloyd, C.J.; Mih, N.; King, Z.A.; Knight, T.F.; Feist, A.M.; Monk, J.M.; Palsson, B.O.; et al. Genome-scale metabolic modeling reveals key features of a minimal gene set. Mol. Syst. Biol. 2021, 17, e10099. [Google Scholar] [CrossRef]
- Loghmani, S.B.; Veith, N.; Sahle, S.; Bergmann, F.T.; Olivier, B.G.; Kummer, U. Inspecting the Solution Space of Genome-Scale Metabolic Models. Metabolites 2022, 12, 43. [Google Scholar] [CrossRef] [PubMed]
- Mahadevan, R.; Schilling, C.H. The effects of alternate optimal solutions in constraint-based genome-scale metabolic models. Metab. Eng. 2003, 5, 264–276. [Google Scholar] [CrossRef]
- García, M.M.; Pacheco, M.; Bintener, T.; Presta, L.; Sauter, T. Importance of the biomass formulation for cancer metabolic modeling and drug prediction. iScience 2021, 24, 103110. [Google Scholar] [CrossRef] [PubMed]
- Uhlén, M.; Fagerberg, L.; Hallström, B.M.; Lindskog, C.; Oksvold, P.; Mardinoglu, A.; Sivertsson, Å.; Kampf, C.; Sjöstedt, E.; Asplund, A.; et al. Tissue-based map of the human proteome. Science 2015, 347, 1260419. [Google Scholar] [CrossRef] [PubMed]
- Schultz, A.; Qutub, A.A. Reconstruction of Tissue-Specific Metabolic Networks Using CORDA. PLoS Comput. Biol. 2016, 12, 1–33. [Google Scholar] [CrossRef] [PubMed]
- Opdam, S.; Richelle, A.; Kellman, B.; Li, S.; Zielinski, D.C.; Lewis, N.E. A systematic evaluation of methods for tailoring genome-scale metabolic models. Cell Syst. 2017, 4, 318–329. [Google Scholar] [CrossRef] [Green Version]
- Åkesson, M.; Förster, J.; Nielsen, J. Integration of gene expression data into genome-scale metabolic models. Metab. Eng. 2004, 6, 285–293. [Google Scholar] [CrossRef] [PubMed]
- Di Filippo, M.; Damiani, C.; Pescini, D. GPRuler: Metabolic gene-protein-reaction rules automatic reconstruction. PLoS Comput. Biol. 2021, 17, e1009550. [Google Scholar] [CrossRef] [PubMed]
- Becker, S.A.; Palsson, B.O. Context-specific metabolic networks are consistent with experiments. PLoS Comput. Biol. 2008, 4, e1000082. [Google Scholar] [CrossRef] [Green Version]
- Grausa, K.; Mozga, I.; Pleiko, K.; Pentjuss, A. Integrative Gene Expression and Metabolic Analysis Tool IgemRNA. Biomolecules 2022, 12, 586. [Google Scholar] [CrossRef]
- Vlassis, N.; Pacheco, M.P.; Sauter, T. Fast Reconstruction of Compact Context-Specific Metabolic Network Models. PLoS Comput. Biol. 2014, 10, 1–10. [Google Scholar] [CrossRef]
- Pacheco, M.P.; John, E.; Kaoma, T.; Heinäniemi, M.; Nicot, N.; Vallar, L.; Bueb, J.L.; Sinkkonen, L.; Sauter, T. Integrated metabolic modelling reveals cell-type specific epigenetic control points of the macrophage metabolic network. BMC Genom. 2015, 16, 809. [Google Scholar] [CrossRef] [Green Version]
- Pacheco, M.P.; Bintener, T.; Ternes, D.; Kulms, D.; Haan, S.; Letellier, E.; Sauter, T. Identifying and targeting cancer-specific metabolism with network-based drug target prediction. EBioMedicine 2019, 43, 98–106. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Robaina Estévez, S.; Nikoloski, Z. Generalized framework for context-specific metabolic model extraction methods. Front. Plant Sci. 2014, 5, 491. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Bordbar, A.; Mo, M.L.; Nakayasu, E.S.; Schrimpe-Rutledge, A.C.; Kim, Y.M.; Metz, T.O.; Jones, M.B.; Frank, B.C.; Smith, R.D.; Peterson, S.N.; et al. Model-driven multi-omic data analysis elucidates metabolic immunomodulators of macrophage activation. Mol. Syst. Biol. 2012, 8, 558. [Google Scholar] [CrossRef]
- Schmidt, B.J.; Ebrahim, A.; Metz, T.O.; Adkins, J.N.; Palsson, B.Ø.; Hyduke, D.R. GIM3E: Condition-specific models of cellular metabolism developed from metabolomics and expression data. Bioinformatics 2013, 29, 2900–2908. [Google Scholar] [CrossRef] [Green Version]
- Jenior, M.L.; Moutinho, T.J., Jr.; Dougherty, B.V.; Papin, J.A. Transcriptome-guided parsimonious flux analysis improves predictions with metabolic networks in complex environments. PLoS Comput. Biol. 2020, 16, 1–26. [Google Scholar] [CrossRef] [Green Version]
- Zur, H.; Ruppin, E.; Shlomi, T. iMAT: An integrative metabolic analysis tool. Bioinformatics 2010, 26, 3140–3142. [Google Scholar] [CrossRef] [Green Version]
- Agren, R.; Bordel, S.; Mardinoglu, A.; Pornputtapong, N.; Nookaew, I.; Nielsen, J. Reconstruction of genome-scale active metabolic networks for 69 human cell types and 16 cancer types using INIT. PLoS Comput. Biol. 2012, 8, e1002518. [Google Scholar] [CrossRef]
- Agren, R.; Mardinoglu, A.; Asplund, A.; Kampf, C.; Uhlen, M.; Nielsen, J. Identification of anticancer drugs for hepatocellular carcinoma through personalized genome-scale metabolic modeling. Mol. Syst. Biol. 2014, 10, 721. [Google Scholar] [CrossRef]
- Lee, D.; Smallbone, K.; Dunn, W.B.; Murabito, E.; Winder, C.L.; Kell, D.B.; Mendes, P.; Swainston, N. Improving metabolic flux predictions using absolute gene expression data. BMC Syst. Biol. 2012, 6, 73. [Google Scholar] [CrossRef]
- Robaina Estévez, S.; Nikoloski, Z. Context-specific metabolic model extraction based on regularized least squares optimization. PLoS ONE 2015, 10, e0131875. [Google Scholar] [CrossRef]
- Jerby, L.; Shlomi, T.; Ruppin, E. Computational reconstruction of tissue-specific metabolic models: Application to human liver metabolism. Mol. Syst. Biol. 2010, 6, 401. [Google Scholar] [CrossRef] [PubMed]
- Wang, Y.; Eddy, J.A.; Price, N.D. Reconstruction of genome-scale metabolic models for 126 human tissues using mCADRE. BMC Syst. Biol. 2012, 6, 153. [Google Scholar] [CrossRef] [Green Version]
- Tefagh, M.; Boyd, S.P. SWIFTCORE: A tool for the context-specific reconstruction of genome-scale metabolic networks. BMC Bioinf. 2020, 21, 140. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Pacheco, M.P.; Ji, J.; Prohaska, T.; García, M.M.; Sauter, T. scFASTCORMICS: A Contextualization Algorithm to Reconstruct Metabolic Multi-Cell Population Models from Single-Cell RNAseq Data. Metabolites 2022, 12, 1211. [Google Scholar] [CrossRef]
- Jensen, P.A.; Papin, J.A. Functional integration of a metabolic network model and expression data without arbitrary thresholding. Bioinformatics 2011, 27, 541–547. [Google Scholar] [CrossRef] [Green Version]
- Zhang, C.; Lee, S.; Bidkhori, G.; Benfeitas, R.; Lovric, A.; Chen, S.; Uhlen, M.; Nielsen, J.; Mardinoglu, A. RMetD2: A tool for integration of relative transcriptomics data into Genome-scale metabolic models. BioRxiv 2019. [Google Scholar] [CrossRef]
- Ravi, S.; Gunawan, R. ΔFBA—Predicting metabolic flux alterations using genome-scale metabolic models and differential transcriptomic data. PLoS Comput. Biol. 2021, 17, 1–18. [Google Scholar] [CrossRef]
- Chan, S.H.; Cai, J.; Wang, L.; Simons-Senftle, M.N.; Maranas, C.D. Standardizing biomass reactions and ensuring complete mass balance in genome-scale metabolic models. Bioinformatics 2017, 33, 3603–3609. [Google Scholar] [CrossRef]
- Lewis, N.E.; Hixson, K.K.; Conrad, T.M.; Lerman, J.A.; Charusanti, P.; Polpitiya, A.D.; Adkins, J.N.; Schramm, G.; Purvine, S.O.; Lopez-Ferrer, D.; et al. Omic data from evolved E. coli are consistent with computed optimal growth from genome-scale models. Mol. Syst. Biol. 2010, 6, 390. [Google Scholar] [CrossRef]
- Lee, S.M.; Lee, G.; Kim, H.U. Machine learning-guided evaluation of extraction and simulation methods for cancer patient-specific metabolic models. Comput. Struct. Biotechnol. J. 2022, 20, 3041–3052. [Google Scholar] [CrossRef]
- Smith, A.B.; Jenior, M.L.; Keenan, O.; Hart, J.L.; Specker, J.; Abbas, A.; Rangel, P.C.; Di, C.; Green, J.; Bustin, K.A.; et al. Enterococci enhance Clostridioides difficile pathogenesis. Nature 2022, 611, 780–786. [Google Scholar] [CrossRef]
- Cho, J.S.; Gu, C.; Han, T.H.; Ryu, J.Y.; Lee, S.Y. Reconstruction of context-specific genome-scale metabolic models using multiomics data to study metabolic rewiring. Curr. Opin. Syst. Biol. 2019, 15, 1–11. [Google Scholar] [CrossRef]
- García Sánchez, C.E.; Torres Sáez, R.G. Comparison and analysis of objective functions in flux balance analysis. Biotechnol. Prog. 2014, 30, 985–991. [Google Scholar] [CrossRef]
- García-Dorival, I.; Cuesta-Geijo, M.Á.; Barrado-Gil, L.; Galindo, I.; Garaigorta, U.; Urquiza, J.; Del Puerto, A.; Campillo, N.E.; Martínez, A.; Gastaminza, P.; et al. Identification of Niemann-Pick C1 protein as a potential novel SARS-CoV-2 intracellular target. Antivir. Res. 2021, 194, 105167. [Google Scholar] [CrossRef]
- Bordbar, A.; Monk, J.M.; King, Z.A.; Palsson, B.O. Constraint-based models predict metabolic and associated cellular functions. Nat. Rev. Genet. 2014, 15, 107–120. [Google Scholar] [CrossRef]
- Shlomi, T.; Cabili, M.N.; Herrgård, M.J.; Palsson, B.Ø.; Ruppin, E. Network-based prediction of human tissue-specific metabolism. Nat. Biotechnol. 2008, 26, 1003–1010. [Google Scholar] [CrossRef]
- Ryu, J.Y.; Kim, H.U.; Lee, S.Y. Framework and resource for more than 11,000 gene-transcript-protein-reaction associations in human metabolism. Proc. Natl. Acad. Sci. USA 2017, 114, E9740–E9749. [Google Scholar] [CrossRef] [Green Version]
- Robaina-Estévez, S.; Daloso, D.M.; Zhang, Y.; Fernie, A.R.; Nikoloski, Z. Resolving the central metabolism of Arabidopsis guard cells. Sci. Rep. 2017, 7, 8307. [Google Scholar] [CrossRef] [Green Version]
- Gudmundsson, S.; Thiele, I. Computationally efficient flux variability analysis. BMC Bioinf. 2010, 11, 1–3. [Google Scholar] [CrossRef]
- Wu, W.H.; Li, F.Y.; Shu, Y.C.; Lai, J.M.; Chang, P.M.H.; Huang, C.Y.F.; Wang, F.S. Oncogene inference optimization using constraint-based modelling incorporated with protein expression in normal and tumour tissues. R. Soc. Open Sci. 2020, 7, 191241. [Google Scholar] [CrossRef] [Green Version]
- Jensen, P.A.; Lutz, K.A.; Papin, J.A. TIGER: Toolbox for integrating genome-scale metabolic models, expression data, and transcriptional regulatory networks. BMC Syst. Biol. 2011, 5, 1–12. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Angione, C.; Lió, P. Predictive analytics of environmental adaptability in multi-omic network models. Sci. Rep. 2015, 5, 15147. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Yaneske, E.; Zampieri, G.; Bertoldi, L.; Benvenuto, G.; Angione, C. Genome-scale metabolic modelling of SARS-CoV-2 in cancer cells reveals an increased shift to glycolytic energy production. FEBS Lett. 2021, 595, 2350–2365. [Google Scholar] [CrossRef] [PubMed]
- Barrett, T.; Wilhite, S.E.; Ledoux, P.; Evangelista, C.; Kim, I.F.; Tomashevsky, M.; Marshall, K.A.; Phillippy, K.H.; Sherman, P.M.; Holko, M.; et al. NCBI GEO: Archive for functional genomics data sets—update. Nucleic Acids Res. 2012, 41, D991–D995. [Google Scholar] [CrossRef] [Green Version]
- Edgar, R.; Domrachev, M.; Lash, A.E. Gene Expression Omnibus: NCBI gene expression and hybridization array data repository. Nucleic Acids Res. 2002, 30, 207–210. [Google Scholar] [CrossRef] [Green Version]
- Athar, A.; Füllgrabe, A.; George, N.; Iqbal, H.; Huerta, L.; Ali, A.; Snow, C.; Fonseca, N.A.; Petryszak, R.; Papatheodorou, I.; et al. ArrayExpress update–from bulk to single-cell expression data. Nucleic Acids Res. 2019, 47, D711–D715. [Google Scholar] [CrossRef]
- Papatheodorou, I.; Moreno, P.; Manning, J.; Fuentes, A.M.P.; George, N.; Fexova, S.; Fonseca, N.A.; Füllgrabe, A.; Green, M.; Huang, N.; et al. Expression Atlas update: From tissues to single cells. Nucleic Acids Res. 2020, 48, D77–D83. [Google Scholar] [CrossRef] [Green Version]
- Freeberg, M.A.; Fromont, L.A.; D’Altri, T.; Romero, A.F.; Ciges, J.I.; Jene, A.; Kerry, G.; Moldes, M.; Ariosa, R.; Bahena, S.; et al. The European genome-phenome archive in 2021. Nucleic Acids Res. 2022, 50, D980–D987. [Google Scholar] [CrossRef]
- Lizio, M.; Abugessaisa, I.; Noguchi, S.; Kondo, A.; Hasegawa, A.; Hon, C.C.; De Hoon, M.; Severin, J.; Oki, S.; Hayashizaki, Y.; et al. Update of the FANTOM web resource: Expansion to provide additional transcriptome atlases. Nucleic Acids Res. 2019, 47, D752–D758. [Google Scholar] [CrossRef]
- Weinstein, J.N.; Collisson, E.A.; Mills, G.B.; Shaw, K.R.; Ozenberger, B.A.; Ellrott, K.; Shmulevich, I.; Sander, C.; Stuart, J.M. The Cancer Genome Atlas Pan-Cancer analysis project. Nat. Genet. 2013, 45, 1113–1120. [Google Scholar] [CrossRef]
- Cerami, E.; Gao, J.; Dogrusoz, U.; Gross, B.E.; Sumer, S.O.; Aksoy, B.A.; Jacobsen, A.; Byrne, C.J.; Heuer, M.L.; Larsson, E.; et al. The cBio cancer genomics portal: An open platform for exploring multidimensional cancer genomics data. Cancer Discov. 2012, 2, 401–404. [Google Scholar] [CrossRef] [Green Version]
- Barretina, J.; Caponigro, G.; Stransky, N.; Venkatesan, K.; Margolin, A.A.; Kim, S.; Wilson, C.J.; Lehár, J.; Kryukov, G.V.; Sonkin, D.; et al. The Cancer Cell Line Encyclopedia enables predictive modelling of anticancer drug sensitivity. Nature 2012, 483, 603–607. [Google Scholar] [CrossRef] [Green Version]
- Deutsch, E.W.; Bandeira, N.; Sharma, V.; Perez-Riverol, Y.; Carver, J.J.; Kundu, D.J.; García-Seisdedos, D.; Jarnuczak, A.F.; Hewapathirana, S.; Pullman, B.S.; et al. The ProteomeXchange consortium in 2020: Enabling ‘big data’approaches in proteomics. Nucleic Acids Res. 2020, 48, D1145–D1152. [Google Scholar] [CrossRef] [Green Version]
- Haug, K.; Cochrane, K.; Nainala, V.C.; Williams, M.; Chang, J.; Jayaseelan, K.V.; O’Donovan, C. MetaboLights: A resource evolving in response to the needs of its scientific community. Nucleic Acids Res. 2020, 48, D440–D444. [Google Scholar] [CrossRef] [Green Version]
- Richelle, A.; Chiang, A.W.; Kuo, C.C.; Lewis, N.E. Increasing consensus of context-specific metabolic models by integrating data-inferred cell functions. PLoS Comput. Biol. 2019, 15, e1006867. [Google Scholar] [CrossRef] [Green Version]
- Pacheco, M.P.; Pfau, T.; Sauter, T. Benchmarking procedures for high-throughput context specific reconstruction algorithms. Front. Physiol. 2016, 6, 410. [Google Scholar] [CrossRef] [Green Version]
- Richelle, A.; Joshi, C.; Lewis, N.E. Assessing key decisions for transcriptomic data integration in biochemical networks. PLoS Comput. Biol. 2019, 15, e1007185. [Google Scholar] [CrossRef] [Green Version]
- Joshi, C.J.; Schinn, S.M.; Richelle, A.; Shamie, I.; O’Rourke, E.J.; Lewis, N.E. StanDep: Capturing transcriptomic variability improves context-specific metabolic models. PLoS Comput. Biol. 2020, 16, e1007764. [Google Scholar] [CrossRef]
- Ho, Y.C.; Pepyne, D.L. Simple explanation of the no-free-lunch theorem and its implications. J. Optim. Theory Appl. 2002, 115, 549–570. [Google Scholar] [CrossRef]
- Walakira, A.; Rozman, D.; Režen, T.; Mraz, M.; Moškon, M. Guided extraction of genome-scale metabolic models for the integration and analysis of omics data. Comput. Struct. Biotechnol. J. 2021, 8, 3521–3530. [Google Scholar] [CrossRef]
- Lorbek, G.; Perše, M.; Jeruc, J.; Juvan, P.; Gutierrez-Mariscal, F.M.; Lewinska, M.; Gebhardt, R.; Horvat, S.; Björkhem, I.; Rozman, D.; et al. Lessons from hepatocyte-specific Cyp51 knockout mice: Impaired cholesterol synthesis leads to oval cell-driven liver injury. Sci. Rep. 2015, 5, 1–11. [Google Scholar] [CrossRef] [Green Version]
- Correia, S.; Costa, B.; Rocha, M. Reconstruction of consensus tissue-specific metabolic models. bioRxiv 2018. [Google Scholar] [CrossRef]
- Hufsky, F.; Lamkiewicz, K.; Almeida, A.; Aouacheria, A.; Arighi, C.; Bateman, A.; Baumbach, J.; Beerenwinkel, N.; Brandt, C.; Cacciabue, M.; et al. Computational strategies to combat COVID-19: Useful tools to accelerate SARS-CoV-2 and coronavirus research. Brief. Bioinform. 2021, 22, 642–663. [Google Scholar] [CrossRef]
- Ostaszewski, M.; Mazein, A.; Gillespie, M.E.; Kuperstein, I.; Niarakis, A.; Hermjakob, H.; Pico, A.R.; Willighagen, E.L.; Evelo, C.T.; Hasenauer, J.; et al. COVID-19 Disease Map, building a computational repository of SARS-CoV-2 virus-host interaction mechanisms. Sci. Data 2020, 7, 136. [Google Scholar] [CrossRef]
- Ostaszewski, M.; Niarakis, A.; Mazein, A.; Kuperstein, I.; Phair, R.; Orta-Resendiz, A.; Singh, V.; Aghamiri, S.S.; Acencio, M.L.; Glaab, E.; et al. COVID-19 Disease Map, a computational knowledge repository of virus–host interaction mechanisms. Mol. Syst. Biol. 2021, 17, e10387. [Google Scholar] [CrossRef]
- Ng, Y.L.; Salim, C.K.; Chu, J.J.H. Drug repurposing for COVID-19: Approaches, challenges and promising candidates. Pharmacol. Ther. 2021, 228, 107930. [Google Scholar] [CrossRef]
- Renz, A.; Widerspick, L.; Dräger, A. FBA reveals guanylate kinase as a potential target for antiviral therapies against SARS-CoV-2. Bioinformatics 2020, 36, i813–i821. [Google Scholar] [CrossRef] [PubMed]
- Renz, A.; Widerspick, L.; Dräger, A. Genome-Scale Metabolic Model of Infection with SARS-CoV-2 Mutants Confirms Guanylate Kinase as Robust Potential Antiviral Target. Genes 2021, 12, 796. [Google Scholar] [CrossRef]
- Delattre, H.; Sasidharan, K.; Soyer, O.S. Inhibiting the reproduction of SARS-CoV-2 through perturbations in human lung cell metabolic network. Life Sci. Alliance 2021, 4, e202000869. [Google Scholar] [CrossRef]
- Santos-Beneit, F.; Raškevičius, V.; Skeberdis, V.A.; Bordel, S. A metabolic modeling approach reveals promising therapeutic targets and antiviral drugs to combat COVID-19. Sci. Rep. 2021, 11, 11982. [Google Scholar] [CrossRef]
- Cheng, K.; Martin-Sancho, L.; Pal, L.R.; Pu, Y.; Riva, L.; Yin, X.; Sinha, S.; Nair, N.U.; Chanda, S.K.; Ruppin, E. Genome-scale metabolic modeling reveals SARS-CoV-2-induced metabolic changes and antiviral targets. Mol. Syst. Biol. 2021, 17, e10260. [Google Scholar] [CrossRef]
- Kishk, A.; Pacheco, M.P.; Sauter, T. DCcov: Repositioning of drugs and drug combinations for SARS-CoV-2 infected lung through constraint-based modeling. Iscience 2021, 24, 103331. [Google Scholar] [CrossRef]
- Dillard, L.; Wase, N.; Ramakrishnan, G.; Park, J.; Sherman, N.; Carpenter, R.; Young, M.; Donlan, A.; Petri, W.; Papin, J. Leveraging metabolic modeling to identify functional metabolic alterations associated with COVID-19 disease severity. Metabolomics 2022, 18, 51. [Google Scholar] [CrossRef] [PubMed]
- Wang, F.S.; Chen, K.L.; Chu, S.W. Human/SARS-CoV-2 genome-scale metabolic modeling to discover potential antiviral targets for COVID-19. J. Taiwan Inst. Chem. Eng. 2022, 133, 104273. [Google Scholar] [CrossRef] [PubMed]
- Nanda, P.; Ghosh, A. Genome Scale-Differential Flux Analysis reveals deregulation of lung cell metabolism on SARS-CoV-2 infection. PLoS Comput. Biol. 2021, 17, e1008860. [Google Scholar] [CrossRef] [PubMed]
- Režen, T.; Martins, A.; Mraz, M.; Zimic, N.; Rozman, D.; Moškon, M. Integration of omics data to generate and analyse COVID-19 specific genome-scale metabolic models. Comput. Biol. Med. 2022, 145, 105428. [Google Scholar] [CrossRef]
- Ambikan, A.T.; Yang, H.; Krishnan, S.; Akusjärvi, S.S.; Gupta, S.; Lourda, M.; Sperk, M.; Arif, M.; Zhang, C.; Nordqvist, H.; et al. Multi-omics personalized network analyses highlight progressive disruption of central metabolism associated with COVID-19 severity. Cell Syst. 2022, 13, 665–681.e4. [Google Scholar] [CrossRef] [PubMed]
- Renz, A.; Hohner, M.; Breitenbach, M.; Josephs-Spaulding, J.; Dürrwald, J.; Best, L.; Jami, R.; Marinos, G.; Cabreiro, F.; Dräger, A.; et al. Metabolic Modeling Elucidates Phenformin and Atpenin A5 as Broad-Spectrum Antiviral Drugs. Preprints 2022, 1–31. [Google Scholar] [CrossRef]
- Thiele, I.; Fleming, R.M. Whole-body metabolic modelling predicts isoleucine dependency of SARS-CoV-2 replication. Comput. Struct. Biotechnol. J. 2022, 20, 4098–4109. [Google Scholar] [CrossRef] [PubMed]
- Aller, S.; Scott, A.; Sarkar-Tyson, M.; Soyer, O.S. Integrated human-virus metabolic stoichiometric modelling predicts host-based antiviral targets against Chikungunya, Dengue and Zika viruses. J. R. Soc. Interface 2018, 15, 20180125. [Google Scholar] [CrossRef] [Green Version]
- Bordbar, A.; Lewis, N.E.; Schellenberger, J.; Palsson, B.Ø.; Jamshidi, N. Insight into human alveolar macrophage and M. tuberculosis interactions via metabolic reconstructions. Mol. Syst. Biol. 2010, 6, 422. [Google Scholar] [CrossRef]
- Wishart, D.S.; Feunang, Y.D.; Guo, A.C.; Lo, E.J.; Marcu, A.; Grant, J.R.; Sajed, T.; Johnson, D.; Li, C.; Sayeeda, Z.; et al. DrugBank 5.0: A major update to the DrugBank database for 2018. Nucleic Acids Res. 2018, 46, D1074–D1082. [Google Scholar] [CrossRef]
- Jayk Bernal, A.; Gomes da Silva, M.M.; Musungaie, D.B.; Kovalchuk, E.; Gonzalez, A.; Delos Reyes, V.; Martín-Quirós, A.; Caraco, Y.; Williams-Diaz, A.; Brown, M.L.; et al. Molnupiravir for oral treatment of COVID-19 in nonhospitalized patients. N. Engl. J. Med. 2022, 386, 509–520. [Google Scholar] [CrossRef]
- Bordel, S. Constraint based modeling of metabolism allows finding metabolic cancer hallmarks and identifying personalized therapeutic windows. Oncotarget 2018, 9, 19716. [Google Scholar] [CrossRef] [Green Version]
- Yizhak, K.; Gaude, E.; Le Dévédec, S.; Waldman, Y.Y.; Stein, G.Y.; van de Water, B.; Frezza, C.; Ruppin, E. Phenotype-based cell-specific metabolic modeling reveals metabolic liabilities of cancer. Elife 2014, 3, e03641. [Google Scholar] [CrossRef]
- Mardinoglu, A.; Agren, R.; Kampf, C.; Asplund, A.; Nookaew, I.; Jacobson, P.; Walley, A.J.; Froguel, P.; Carlsson, L.M.; Uhlen, M.; et al. Integration of clinical data with a genome-scale metabolic model of the human adipocyte. Mol. Syst. Biol. 2013, 9, 649. [Google Scholar] [CrossRef]
- Blanco-Melo, D.; Nilsson-Payant, B.E.; Liu, W.C.; Uhl, S.; Hoagland, D.; Møller, R.; Jordan, T.X.; Oishi, K.; Panis, M.; Sachs, D.; et al. Imbalanced host response to SARS-CoV-2 drives development of COVID-19. Cell 2020, 181, 1036–1045. [Google Scholar] [CrossRef] [PubMed]
- Appelberg, S.; Gupta, S.; Svensson Akusjärvi, S.; Ambikan, A.T.; Mikaeloff, F.; Saccon, E.; Végvári, Á.; Benfeitas, R.; Sperk, M.; Ståhlberg, M.; et al. Dysregulation in Akt/mTOR/HIF-1 signaling identified by proteo-transcriptomics of SARS-CoV-2 infected cells. Emerg. Microbes Infect. 2020, 9, 1748–1760. [Google Scholar] [CrossRef] [PubMed]
- Keaty, T.C.; Jensen, P.A. Gapsplit: Efficient random sampling for non-convex constraint-based models. Bioinformatics 2020, 36, 2623–2625. [Google Scholar] [CrossRef] [PubMed]
- Donlan, A.N.; Sutherland, T.E.; Marie, C.; Preissner, S.; Bradley, B.T.; Carpenter, R.M.; Sturek, J.M.; Ma, J.Z.; Moreau, G.B.; Donowitz, J.R.; et al. IL-13 is a driver of COVID-19 severity. JCI Insight 2021, 6. [Google Scholar] [CrossRef]
- Gordon, D.E.; Jang, G.M.; Bouhaddou, M.; Xu, J.; Obernier, K.; White, K.M.; O’Meara, M.J.; Rezelj, V.V.; Guo, J.Z.; Swaney, D.L.; et al. A SARS-CoV-2 protein interaction map reveals targets for drug repurposing. Nature 2020, 583, 459–468. [Google Scholar] [CrossRef]
- Bouhaddou, M.; Memon, D.; Meyer, B.; White, K.M.; Rezelj, V.V.; Marrero, M.C.; Polacco, B.J.; Melnyk, J.E.; Ulferts, S.; Kaake, R.M.; et al. The global phosphorylation landscape of SARS-CoV-2 infection. Cell 2020, 182, 685–712. [Google Scholar] [CrossRef]
- Valcárcel, L.V.; Torrano, V.; Tobalina, L.; Carracedo, A.; Planes, F.J. rMTA: Robust metabolic transformation analysis. Bioinformatics 2019, 35, 4350–4355. [Google Scholar] [CrossRef]
- Wyler, E.; Mösbauer, K.; Franke, V.; Diag, A.; Gottula, L.T.; Arsiè, R.; Klironomos, F.; Koppstein, D.; Hönzke, K.; Ayoub, S.; et al. Transcriptomic profiling of SARS-CoV-2 infected human cell lines identifies HSP90 as target for COVID-19 therapy. iScience 2021, 24, 102151. [Google Scholar] [CrossRef]
- Thiele, I.; Sahoo, S.; Heinken, A.; Hertel, J.; Heirendt, L.; Aurich, M.K.; Fleming, R.M. Personalized whole-body models integrate metabolism, physiology, and the gut microbiome. Mol. Syst. Biol. 2020, 16, e8982. [Google Scholar] [CrossRef] [PubMed]
- Tabula Muris Consortium. A single-cell transcriptomic atlas characterizes ageing tissues in the mouse. Nature 2020, 583, 590–595. [Google Scholar] [CrossRef] [PubMed]
- Wang, Y.; Song, W.; Wang, J.; Wang, T.; Xiong, X.; Qi, Z.; Fu, W.; Yang, X.; Chen, Y.G. Single-cell transcriptome analysis reveals differential nutrient absorption functions in human intestine. J. Exp. Med. 2020, 217, e20191130. [Google Scholar] [CrossRef]
- Chua, R.L.; Lukassen, S.; Trump, S.; Hennig, B.P.; Wendisch, D.; Pott, F.; Debnath, O.; Thürmann, L.; Kurth, F.; Völker, M.T.; et al. COVID-19 severity correlates with airway epithelium–immune cell interactions identified by single-cell analysis. Nat. Biotechnol. 2020, 38, 970–979. [Google Scholar] [CrossRef]
- Noronha, A.; Modamio, J.; Jarosz, Y.; Guerard, E.; Sompairac, N.; Preciat, G.; Daníelsdóttir, A.D.; Krecke, M.; Merten, D.; Haraldsdóttir, H.S.; et al. The Virtual Metabolic Human database: Integrating human and gut microbiome metabolism with nutrition and disease. Nucleic Acids Res. 2019, 47, D614–D624. [Google Scholar] [CrossRef]
- Alqutami, F.; Senok, A.; Hachim, M. COVID-19 transcriptomic atlas: A comprehensive analysis of COVID-19 related transcriptomics datasets. Front. Genet. 2021, 12, 755222. [Google Scholar] [CrossRef]
- King, Z.A.; Lu, J.; Dräger, A.; Miller, P.; Federowicz, S.; Lerman, J.A.; Ebrahim, A.; Palsson, B.O.; Lewis, N.E. BiGG Models: A platform for integrating, standardizing and sharing genome-scale models. Nucleic Acids Res. 2016, 44, D515–D522. [Google Scholar] [CrossRef] [PubMed]
- Cruz, F.; Faria, J.P.; Rocha, M.; Rocha, I.; Dias, O. A review of methods for the reconstruction and analysis of integrated genome-scale models of metabolism and regulation. Biochem. Soc. Trans. 2020, 48, 1889–1903. [Google Scholar] [CrossRef] [PubMed]
- Chung, C.H.; Lin, D.W.; Eames, A.; Chandrasekaran, S. Next-generation genome-scale metabolic modeling through integration of regulatory mechanisms. Metabolites 2021, 11, 606. [Google Scholar] [CrossRef]
- Karr, J.R.; Sanghvi, J.C.; Macklin, D.N.; Gutschow, M.V.; Jacobs, J.M.; Bolival, B., Jr.; Assad-Garcia, N.; Glass, J.I.; Covert, M.W. A whole-cell computational model predicts phenotype from genotype. Cell 2012, 150, 389–401. [Google Scholar] [CrossRef] [Green Version]
- Ye, C.; Xu, N.; Gao, C.; Liu, G.; Xu, J.; Zhang, W.; Chen, X.; Nielsen, J.; Liu, L. Comprehensive understanding of Saccharomyces cerevisiae phenotypes with whole-cell model WM_S288C. Biotechnol. Bioeng. 2020, 117, 1562–1574. [Google Scholar] [CrossRef]
- Szigeti, B.; Roth, Y.D.; Sekar, J.A.; Goldberg, A.P.; Pochiraju, S.C.; Karr, J.R. A blueprint for human whole-cell modeling. Curr. Opin. Syst. Biol. 2018, 7, 8–15. [Google Scholar] [CrossRef] [Green Version]
- Hunter, P.; Coveney, P.V.; De Bono, B.; Diaz, V.; Fenner, J.; Frangi, A.F.; Harris, P.; Hose, R.; Kohl, P.; Lawford, P.; et al. A vision and strategy for the virtual physiological human in 2010 and beyond. Philos. Trans. Royal Soc. 2010, 368, 2595–2614. [Google Scholar] [CrossRef] [PubMed]
- Bernstein, D.B.; Sulheim, S.; Almaas, E.; Segrè, D. Addressing uncertainty in genome-scale metabolic model reconstruction and analysis. Genome Biol. 2021, 22, 64. [Google Scholar] [CrossRef]




{kind=link}
{kind=link}
{kind=link}
{kind=link}
| Family | Description |
|---|---|
| GIMME-like | Maximising the compliance with the experimental evidence while pertaining to a given RMF. |
| iMAT-like | Does not specify a RMF, matching of reactions states (active or inactive) with expression profiles (present or absent), employs MILP-based optimisation. |
| MBA-like | Defining core reactions and removing other reactions while pertaining to model consistency, support integration of different data types. |
| MADE-like | Employs differential gene expression data to identify flux differences between two or more conditions. |
| Algorithm | Reference | Family | Input Data | Comments |
|---|---|---|---|---|
| GIMME | Becker et al., 2008 [38] | GIMME-like | transcriptomics | Inactivate reactions below a threshold while maintaining RMF. |
| GIMMEp | Bordbar et al., 2012 [44] | GIMME-like | transcriptomics, proteomics | RMFs based on proteomics data. |
| GIM3E | Schmidt et al., 2013 [45] | GIMME-like | transcriptomics, metabolomics | No thresholding. |
| RIPTiDe | Jenior et al., 2020 [46] | GIMME-like | transcriptomics | Minimises the weighted flux values, no thresholding. |
| iMAT | Zur et al., 2010 [47] | iMAT-like | transcriptomics, proteomics | Matches reaction activities with expression profiles, no RMF. |
| INIT | Agren et al., 2012 [48] | iMAT-like | transcriptomics, proteomics, metabolomics (qualitative) | Reaction weights based on experimental evidence, integration of metabolomics data. |
| tINIT | Agren et al., 2014 [49] | iMAT-like | prior knowledge, transcriptomics, proteomics, metabolomics (qualitative) | Based on a set of required metabolic tasks. |
| Lee | Lee et al., 2012 [50] | iMAT-like | transcriptomics | Uses absolute expression data (RNA-seq). |
| RegrEx | Estevez et al., 2015 [51] | iMAT-like | transcriptomics | Uses absolute expression data (RNA-seq) and regularisation. |
| MBA | Jerby et al., 2010 [52] | MBA-like | prior knowledge, transcriptomics, proteomics, metabolomics, fluxomics | Removes non-core reactions and checks model consistency for core reactions. |
| mCADRE | Wang et al., 2012 [53] | MBA-like | transcriptomics, metabolomics | Different reaction scores to determine core reactions. |
| FASTCORE | Vlassis et al., 2014 [40] | MBA-like | a set of core reactions | Two LPs to find a minimal set of non-core reactions to activate all core reactions. |
| SWIFTCORE | Tefagh and Boyd, 2020 [54] | MBA-like | a set of core reactions | Enhanced runtime and network compactness in comparison to FASTCORE. |
| FASTCORMICS | Pires Pacheco at al., 2015 [41] | MBA-like | transcriptomics | FASTCORE workflow for microarray data. |
| rFASTCORMICS | Pires Pacheco at al., 2019 [42] | MBA-like | transcriptomics | FASTCORE workflow for RNA-seq data. |
| scFASTCORMICS | Pires Pacheco at al., 2022 [55] | MBA-like | transcriptomics | FASTCORE workflow for scRNA-seq data. |
| CORDA | Schultz and Qutub, 2016 [34] | MBA-like | a set of core reactions | Does not require to remove all non-core reactions. |
| MADE | Jensen and Papin, 2011 [56] | MADE-like | transcriptomics | Identifies reaction activities in a sequence of measurements. |
| RMetD2 | Zhang et al., 2019 [57] | MADE-like | transcriptomics | Sequentially pushes the constraints. |
| ΔFBA | Ravi et al., 2021 [58] | MADE-like | transcriptomics | Finds a consistent and minimal solution of flux differences between the conditions. |
| High-Throughput Data | Input Data | Algorithm | Data Repositories |
|---|---|---|---|
| Transcriptome | Gene expression value | GIMME-like | ArrayExpress |
| iMAT-like | cBioPortal | ||
| MBA-like | CCLE | ||
| PRIME | EGA | ||
| Differential gene expression value | ENA | ||
| Expression Atlas | |||
| FANTOM5 | |||
| MADE-like | GEO | ||
| METRADE | GTEx | ||
| HPA | |||
| SRA | |||
| TCGA | |||
| Proteome | Protein expression value | GIMME-like | cBioPortal |
| iMAT-like | CCLE | ||
| MBA-like | Expression Atlas | ||
| Differential protein expression value | HPA | ||
| PDC | |||
| METRADE | ProteomeXchange | ||
| TCGA | |||
| Metabolome | Metabolite concentration | GIMME-like | MetaboLights |
| iMAT-like | Metabolomics workbench | ||
| MBA-like |
| Reference | Reconstruction Algorithm(s) | Comments |
|---|---|---|
| Renz et al., 2020 [98] | none | Integration of VBOF into a human alveolar macrophage model. |
| Renz et al., 2021 [99] | none | A follow-up study on [98]. |
| Delatre et al., 2021 [100] | none | Integration of VBOF into a human lung cell model. |
| Yaneske et al., 2021 [74] | METRADE | A combination of manual curation with automated reconstruction using transcriptomics and proteomics data from Huh-7 cells. |
| Santos-Beneit et al., 2021 [101] | pyTARG (for a healthy lung model) | Manual curation of a healthy lung model with literature data. |
| Cheng et al., 2021 [102] | iMAT | An integration of data from 12 datasets, validation of identified targets with additional experiment. |
| Kishk et al., 2021 [103] | rFASTCORMICS | An integration of data from two RNA-seq studies on lung cells. |
| Dillard et al., 2022 [104] | RIPTide | Combining GEMs with machine learning analysis on plasma metabolomes of non-acute and severe COVID-19 patients. |
| Wang et al., 2022 [105] | none | Extension and integration of VBOF into Recon3D. |
| Nanda and Ghosh, 2021 [106] | tINIT | An integration of NHBE and lung biopsy RNA-seq data into HumanGEM. |
| Režen et al., 2022 [107] | GIMME, iMAT, INIT, tINIT | An integration of different cell lines and patient samples data following the protocol proposed in [91]. |
| Ambikan et al., 2022 [108] | tINIT | A reconstruction of personalised and group-specific models with integration of RNA-seq data (blood), constraining exchange reactions with metabolomics data (plasma). |
| Renz et al., 2022 [109] | FASTCORE | A computational pipeline for identification of broad-spectrum antiviral drugs using scRNA-seq data. |
| Thiele and Fleming, 2022 [110] | none | An integration of VBOF and other virus-specific reactions into metabolic sex-specific WBM. |
Disclaimer/Publisher’s Note: The statements, opinions and data contained in all publications are solely those of the individual author(s) and contributor(s) and not of MDPI and/or the editor(s). MDPI and/or the editor(s) disclaim responsibility for any injury to people or property resulting from any ideas, methods, instructions or products referred to in the content. |
© 2023 by the authors. Licensee MDPI, Basel, Switzerland. This article is an open access article distributed under the terms and conditions of the Creative Commons Attribution (CC BY) license (https://creativecommons.org/licenses/by/4.0/).
Share and Cite
Moškon, M.; Režen, T. Context-Specific Genome-Scale Metabolic Modelling and Its Application to the Analysis of COVID-19 Metabolic Signatures. Metabolites 2023, 13, 126. https://doi.org/10.3390/metabo13010126
Moškon M, Režen T. Context-Specific Genome-Scale Metabolic Modelling and Its Application to the Analysis of COVID-19 Metabolic Signatures. Metabolites. 2023; 13(1):126. https://doi.org/10.3390/metabo13010126
Chicago/Turabian StyleMoškon, Miha, and Tadeja Režen. 2023. "Context-Specific Genome-Scale Metabolic Modelling and Its Application to the Analysis of COVID-19 Metabolic Signatures" Metabolites 13, no. 1: 126. https://doi.org/10.3390/metabo13010126
APA StyleMoškon, M., & Režen, T. (2023). Context-Specific Genome-Scale Metabolic Modelling and Its Application to the Analysis of COVID-19 Metabolic Signatures. Metabolites, 13(1), 126. https://doi.org/10.3390/metabo13010126