
Obesity-Induced Brain Neuroinflammatory and Mitochondrial Changes



{kind=link}
{kind=link}
Abstract
:1. Introduction
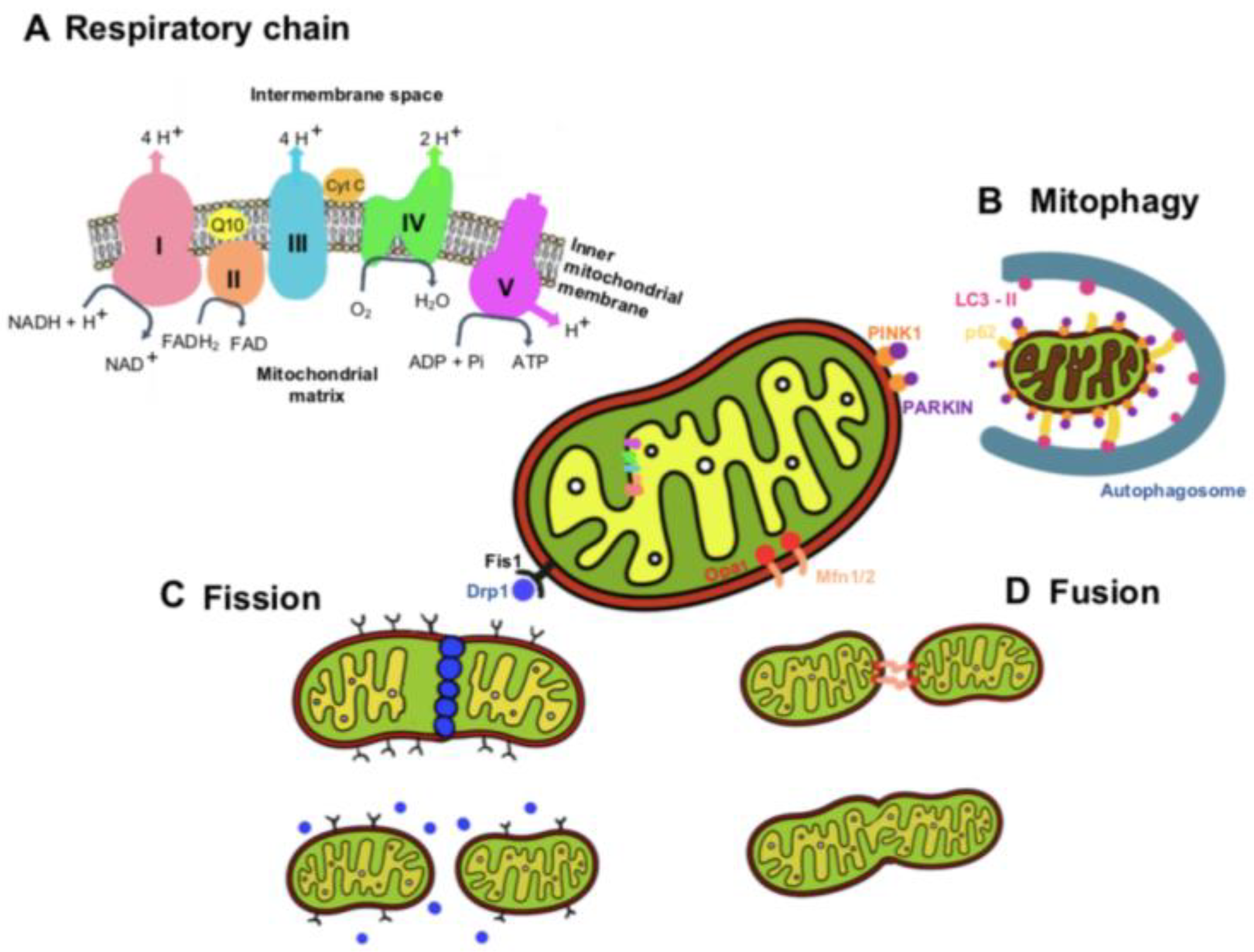
2. Mitochondria Functions and Dynamics
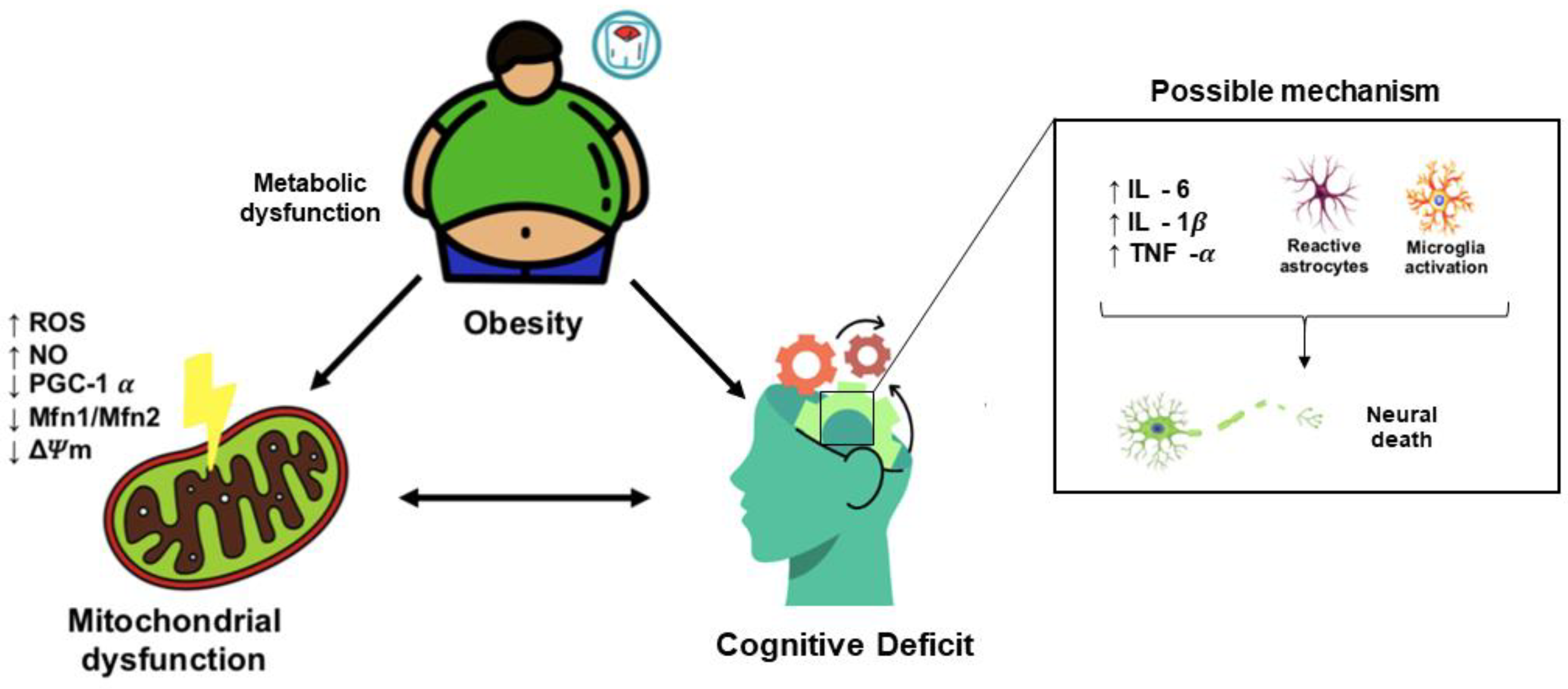
3. Obesity Induces Cognitive Decline
3.1. Obesity-Induced Cognitive Decline: Role of Neuroinflammation
3.1.1. Hypothalamic Neuroinflammation
3.1.2. Hippocampal Neuroinflammation
3.2. Obesity-Induced Cognitive Decline: Role of Mitochondrial Dysfunction
4. Conclusions
Author Contributions
Funding
Conflicts of Interest
References
- Blüher, M. Obesity: Global epidemiology and pathogenesis. Nat. Rev. Endocrinol. 2019, 15, 288–298. [Google Scholar] [CrossRef]
- Collaborators, G.B.D.O.; Afshin, A.; Forouzanfar, M.H.; Reitsma, M.B.; Sur, P.; Estep, K.; Lee, A.; Marczak, L.; Mokdad, A.H.; Moradi-Lakeh, M.; et al. Health Effects of Overweight and Obesity in 195 Countries over 25 Years. N. Engl. J. Med. 2017, 377, 13–27. [Google Scholar] [CrossRef]
- Morys, F.; Dadar, M.; Dagher, A. Association Between Midlife Obesity and Its Metabolic Consequences, Cerebrovascular Disease, and Cognitive Decline. J. Clin. Endocrinol. Metab. 2021, 106, e4260–e4274. [Google Scholar] [CrossRef]
- De Lorenzo, A.; Gratteri, S.; Gualtieri, P. Why primary obesity is a disease? J. Transl. Med. 2019, 17, 169. [Google Scholar] [CrossRef] [Green Version]
- Pratchayasakul, W.; Kerdphoo, S.; Petsophonsakul, P.; Pongchaidecha, A.; Chattipakorn, N.; Chattipakorn, S. Effects of high-fat diet on insulin receptor function in rat hippocampus and the level of neuronal corticosterone. Life Sci. 2011, 88, 619–627. [Google Scholar] [CrossRef]
- Pipatpiboon, N.; Pratchayasakul, W.; Chattipakorn, N.; Chattipakorn, S.C. PPARγ agonist improves neuronal insulin receptor function in hippocampus and brain mitochondria function in rats with insulin resistance induced by long term high-fat diets. Endocrinology 2012, 153, 329–338. [Google Scholar] [CrossRef]
- Abbasnejad, Z.; Nasseri, B.; Zardooz, H.; Ghasemi, R. Time-course study of high fat diet induced alterations in spatial memory, hippocampal JNK, P38, ERK and Akt activity. Metab. Brain Dis. 2019, 34, 659–673. [Google Scholar] [CrossRef]
- Gómez-Apo, E.; Mondragón-Maya, A.; Ferrari-Díaz, M.; Silva-Pereyra, J. Structural Brain Changes Associated with Overweight and Obesity. J. Obes. 2021, 2021, 6613385. [Google Scholar] [CrossRef]
- Gaspar, J.M.; Baptista, F.I.; Macedo, M.P.; Ambrósio, A.F. Inside the Diabetic Brain: Role of Different Players Involved in Cognitive Decline. ACS Chem. Neurosci. 2016, 7, 131–142. [Google Scholar] [CrossRef]
- Gispen, W.H.; Biessels, G.J. Cognition and synaptic plasticity in diabetes mellitus. Trends Neurosci. 2000, 23, 542–549. [Google Scholar] [CrossRef] [PubMed]
- Fitzpatrick, A.L.; Kuller, L.H.; Lopez, O.L.; Diehr, P.; O’Meara, E.S.; Longstreth, W.T., Jr.; Luchsinger, J.A. Midlife and late-life obesity and the risk of dementia: Cardiovascular health study. Arch. Neurol. 2009, 66, 336–342. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Hughes, T.F.; Borenstein, A.R.; Schofield, E.; Wu, Y.; Larson, E.B. Association between late-life body mass index and dementia: The Kame Project. Neurology 2009, 72, 1741–1746. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Whitmer, R.A.; Gunderson, E.P.; Quesenberry, C.P., Jr.; Zhou, J.; Yaffe, K. Body mass index in midlife and risk of Alzheimer disease and vascular dementia. Curr. Alzheimer Res. 2007, 4, 103–109. [Google Scholar] [CrossRef] [PubMed]
- Biessels, G.J.; Staekenborg, S.; Brunner, E.; Brayne, C.; Scheltens, P. Risk of dementia in diabetes mellitus: A systematic review. Lancet. Neurol. 2006, 5, 64–74. [Google Scholar] [CrossRef] [PubMed]
- Lu, F.P.; Lin, K.P.; Kuo, H.K. Diabetes and the risk of multi-system aging phenotypes: A systematic review and meta-analysis. PLoS ONE 2009, 4, e4144. [Google Scholar] [CrossRef] [Green Version]
- Marseglia, A.; Darin-Mattsson, A.; Skoog, J.; Rydén, L.; Hadarsson-Bodin, T.; Kern, S.; Rydberg Sterner, T.; Shang, Y.; Zettergren, A.; Westman, E.; et al. Metabolic Syndrome Is Associated With Poor Cognition: A Population-Based Study of 70-Year-Old Adults Without Dementia. J. Gerontol. Ser. A 2021, 76, 2275–2283. [Google Scholar] [CrossRef]
- Giacomello, M.; Pyakurel, A.; Glytsou, C.; Scorrano, L. The cell biology of mitochondrial membrane dynamics. Nat. Rev. Mol. Cell Biol. 2020, 21, 204–224. [Google Scholar] [CrossRef]
- Bras, M.; Queenan, B.; Susin, S.A. Programmed cell death via mitochondria: Different modes of dying. Biochemistry 2005, 70, 231–239. [Google Scholar] [CrossRef]
- Mitchell, P. Coupling of Phosphorylation to Electron and Hydrogen Transfer by a Chemi-Osmotic type of Mechanism. Nature 1961, 191, 144–148. [Google Scholar] [CrossRef]
- Hatefi, Y. The Mitochondrial Electron Transport and Oxidative Phosphorylation System. Annu. Rev. Biochem. 1985, 54, 1015–1069. [Google Scholar] [CrossRef]
- Zhao, R.Z.; Jiang, S.; Zhang, L.; Yu, Z.B. Mitochondrial electron transport chain, ROS generation and uncoupling. Int. J. Mol. Med. 2019, 44, 3–15. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Nolfi-Donegan, D.; Braganza, A.; Shiva, S. Mitochondrial electron transport chain: Oxidative phosphorylation, oxidant production, and methods of measurement. Redox Biol. 2020, 37, 101674. [Google Scholar] [CrossRef] [PubMed]
- Chan, D.C. Mitochondrial Dynamics and Its Involvement in Disease. Annu. Rev. Pathol. Mech. Dis. 2020, 15, 235–259. [Google Scholar] [CrossRef] [Green Version]
- Galizzi, G.; Di Carlo, M. Insulin and Its Key Role for Mitochondrial Function/Dysfunction and Quality Control: A Shared Link between Dysmetabolism and Neurodegeneration. Biology 2022, 11, 943. [Google Scholar] [CrossRef] [PubMed]
- Cardoso, S.; López, I.P.; Piñeiro-Hermida, S.; Pichel, J.G.; Moreira, P.I. IGF1R Deficiency Modulates Brain Signaling Pathways and Disturbs Mitochondria and Redox Homeostasis. Biomedicines 2021, 9, 158. [Google Scholar] [CrossRef]
- Ribeiro, M.; Rosenstock, T.R.; Oliveira, A.M.; Oliveira, C.R.; Rego, A.C. Insulin and IGF-1 improve mitochondrial function in a PI-3K/Akt-dependent manner and reduce mitochondrial generation of reactive oxygen species in Huntington’s disease knock-in striatal cells. Free. Radic. Biol. Med. 2014, 74, 129–144. [Google Scholar] [CrossRef]
- Cheng, Y.; Buchan, M.; Vitanova, K.; Aitken, L.; Gunn-Moore, F.J.; Ramsay, R.R.; Doherty, G. Neuroprotective actions of leptin facilitated through balancing mitochondrial morphology and improving mitochondrial function. J. Neurochem. 2020, 155, 191–206. [Google Scholar] [CrossRef] [Green Version]
- Jong, C.J.; Yeung, J.; Tseung, E.; Karmazyn, M. Leptin-induced cardiomyocyte hypertrophy is associated with enhanced mitochondrial fission. Mol. Cell. Biochem. 2019, 454, 33–44. [Google Scholar] [CrossRef]
- Yang, F.; Li, B.; Yang, Y.; Huang, M.; Liu, X.; Zhang, Y.; Liu, H.; Zhang, L.; Pan, Y.; Tian, S.; et al. Leptin enhances glycolysis via OPA1-mediated mitochondrial fusion to promote mesenchymal stem cell survival. Int. J. Mol. Med. 2019, 44, 301–312. [Google Scholar] [CrossRef]
- Wauman, J.; Tavernier, J. The intracellular domain of the leptin receptor prevents mitochondrial depolarization and mitophagy. Biochim. Biophys. Acta. Mol. Cell Res. 2018, 1865, 1312–1325. [Google Scholar] [CrossRef]
- Otera, H.; Wang, C.; Cleland, M.M.; Setoguchi, K.; Yokota, S.; Youle, R.J.; Mihara, K. Mff is an essential factor for mitochondrial recruitment of Drp1 during mitochondrial fission in mammalian cells. J. Cell Biol. 2010, 191, 1141–1158. [Google Scholar] [CrossRef] [Green Version]
- Losón, O.C.; Song, Z.; Chen, H.; Chan, D.C. Fis1, Mff, MiD49, and MiD51 mediate Drp1 recruitment in mitochondrial fission. Mol. Biol. Cell 2013, 24, 659–667. [Google Scholar] [CrossRef]
- Chen, H.; Detmer, S.A.; Ewald, A.J.; Griffin, E.E.; Fraser, S.E.; Chan, D.C. Mitofusins Mfn1 and Mfn2 coordinately regulate mitochondrial fusion and are essential for embryonic development. J. Cell Biol. 2003, 160, 189–200. [Google Scholar] [CrossRef]
- Koshiba, T.; Detmer, S.A.; Kaiser, J.T.; Chen, H.; McCaffery, J.M.; Chan, D.C. Structural basis of mitochondrial tethering by mitofusin complexes. Science 2004, 305, 858–862. [Google Scholar] [CrossRef] [Green Version]
- Qi, Y.; Yan, L.; Yu, C.; Guo, X. Structures of human mitofusin 1 provide insight into mitochondrial tethering. J. Cell Biol. 2016, 215, 621–629. [Google Scholar] [CrossRef] [Green Version]
- Song, Z.; Ghochani, M.; McCaffery, J.M.; Frey, T.G.; Chan, D.C. Mitofusins and OPA1 mediate sequential steps in mitochondrial membrane fusion. Mol. Biol. Cell 2009, 20, 3525–3532. [Google Scholar] [CrossRef]
- Ward, M.A.; Carlsson, C.M.; Trivedi, M.A.; Sager, M.A.; Johnson, S.C. The effect of body mass index on global brain volume in middle-aged adults: A cross sectional study. BMC Neurol. 2005, 5, 23. [Google Scholar] [CrossRef] [Green Version]
- Haltia, L.T.; Viljanen, A.; Parkkola, R.; Kemppainen, N.; Rinne, J.O.; Nuutila, P.; Kaasinen, V. Brain white matter expansion in human obesity and the recovering effect of dieting. J. Clin. Endocrinol. Metab. 2007, 92, 3278–3284. [Google Scholar] [CrossRef] [Green Version]
- Gazdzinski, S.; Kornak, J.; Weiner, M.W.; Meyerhoff, D.J. Body mass index and magnetic resonance markers of brain integrity in adults. Ann. Neurol. 2008, 63, 652–657. [Google Scholar] [CrossRef] [Green Version]
- Chen, R.; Cai, G.; Xu, S.; Sun, Q.; Luo, J.; Wang, Y.; Li, M.; Lin, H.; Liu, J. Body mass index related to executive function and hippocampal subregion volume in subjective cognitive decline. Front. Aging Neurosci. 2022, 14, 905035. [Google Scholar] [CrossRef] [PubMed]
- Kharabian Masouleh, S.; Arélin, K.; Horstmann, A.; Lampe, L.; Kipping, J.A.; Luck, T.; Riedel-Heller, S.G.; Schroeter, M.L.; Stumvoll, M.; Villringer, A.; et al. Higher body mass index in older adults is associated with lower gray matter volume: Implications for memory performance. Neurobiol. Aging 2016, 40, 1–10. [Google Scholar] [CrossRef] [PubMed]
- Gustafson, D.; Rothenberg, E.; Blennow, K.; Steen, B.; Skoog, I. An 18-year follow-up of overweight and risk of Alzheimer disease. Arch. Intern. Med. 2003, 163, 1524–1528. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Dahl, A.; Hassing, L.B.; Fransson, E.; Berg, S.; Gatz, M.; Reynolds, C.A.; Pedersen, N.L. Being overweight in midlife is associated with lower cognitive ability and steeper cognitive decline in late life. J. Gerontol. Ser. A Biol. Sci. Med. Sci. 2010, 65, 57–62. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Gunstad, J.; Lhotsky, A.; Wendell, C.R.; Ferrucci, L.; Zonderman, A.B. Longitudinal examination of obesity and cognitive function: Results from the Baltimore longitudinal study of aging. Neuroepidemiology 2010, 34, 222–229. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Gunstad, J.; Paul, R.H.; Cohen, R.A.; Tate, D.F.; Spitznagel, M.B.; Gordon, E. Elevated body mass index is associated with executive dysfunction in otherwise healthy adults. Compr. Psychiatry 2007, 48, 57–61. [Google Scholar] [CrossRef] [PubMed]
- Quaye, E.; Galecki, A.T.; Tilton, N.; Whitney, R.; Briceño, E.M.; Elkind, M.S.V. Association of Obesity with Cognitive Decline in Black and White Americans. Neurology 2022. [Google Scholar] [CrossRef]
- Baym, C.L.; Khan, N.A.; Monti, J.M.; Raine, L.B.; Drollette, E.S.; Moore, R.D.; Scudder, M.R.; Kramer, A.F.; Hillman, C.H.; Cohen, N.J. Dietary lipids are differentially associated with hippocampal-dependent relational memory in prepubescent children. Am. J. Clin. Nutr. 2014, 99, 1026–1032. [Google Scholar] [CrossRef] [Green Version]
- Francis, H.M.; Stevenson, R.J. Higher reported saturated fat and refined sugar intake is associated with reduced hippocampal-dependent memory and sensitivity to interoceptive signals. Behav. Neurosci. 2011, 125, 943–955. [Google Scholar] [CrossRef]
- Attuquayefio, T.; Stevenson, R.J.; Oaten, M.J.; Francis, H.M. A four-day Western-style dietary intervention causes reductions in hippocampal-dependent learning and memory and interoceptive sensitivity. PLoS ONE 2017, 12, e0172645. [Google Scholar] [CrossRef] [Green Version]
- Wolf, P.A.; Beiser, A.; Elias, M.F.; Au, R.; Vasan, R.S.; Seshadri, S. Relation of obesity to cognitive function: Importance of central obesity and synergistic influence of concomitant hypertension. The Framingham Heart Study. Curr. Alzheimer Res. 2007, 4, 111–116. [Google Scholar] [CrossRef]
- Snyder, L.L.; Foland-Ross, L.C.; Cato, A.; Reiss, A.L.; Shah, C.; Hossain, J.; Elmufti, H.; Nelly, M. Impact of dysglycemia and obesity on the brain in adolescents with and without type 2 diabetes: A pilot study. Pediatr. Diabetes 2022, 23, 1674–1686. [Google Scholar] [CrossRef] [PubMed]
- Cserjési, R.; Molnár, D.; Luminet, O.; Lénárd, L. Is there any relationship between obesity and mental flexibility in children? Appetite 2007, 49, 675–678. [Google Scholar] [CrossRef] [PubMed]
- Liang, J.; Matheson, B.E.; Kaye, W.H.; Boutelle, K.N. Neurocognitive correlates of obesity and obesity-related behaviors in children and adolescents. Int. J. Obes. 2014, 38, 494–506. [Google Scholar] [CrossRef] [Green Version]
- Beeri, M.S.; Tirosh, A.; Lin, H.M.; Golan, S.; Boccara, E.; Sano, M.; Zhu, C.W. Stability in BMI over time is associated with a better cognitive trajectory in older adults. Alzheimer’s Dement. 2022, 18, 2131–2139. [Google Scholar] [CrossRef] [PubMed]
- Li, C.M.; Song, J.R.; Zhao, J.; Wang, C.F.; Zhang, C.S.; Wang, H.D.; Zhang, Q.; Liu, D.F.; Ma, Z.Y.; Yuan, J.H.; et al. The effects of bariatric surgery on cognition in patients with obesity: A systematic review and meta-analysis. Surg. Obes. Relat. Dis. 2022, 18, 1323–1338. [Google Scholar] [CrossRef]
- Dardano, A.; Aghakhanyan, G.; Moretto, C.; Ciccarone, A.; Bellini, R.; Ceccarini, G.; Sancho Bornez, V.; Santini, F.; Volterrani, D.; Del Prato, S.; et al. Brain effect of bariatric surgery in people with obesity. Int. J. Obes. 2022, 46, 1671–1677. [Google Scholar] [CrossRef]
- de Paula, G.C.; Brunetta, H.S.; Engel, D.F.; Gaspar, J.M.; Velloso, L.A.; Engblom, D.; de Oliveira, J.; de Bem, A.F. Hippocampal Function Is Impaired by a Short-Term High-Fat Diet in Mice: Increased Blood-Brain Barrier Permeability and Neuroinflammation as Triggering Events. Front. Neurosci. 2021, 15, 734158. [Google Scholar] [CrossRef]
- Hao, S.; Dey, A.; Yu, X.; Stranahan, A.M. Dietary obesity reversibly induces synaptic stripping by microglia and impairs hippocampal plasticity. Brain Behav. Immun. 2016, 51, 230–239. [Google Scholar] [CrossRef] [Green Version]
- Hsu, T.M.; Konanur, V.R.; Taing, L.; Usui, R.; Kayser, B.D.; Goran, M.I.; Kanoski, S.E. Effects of sucrose and high fructose corn syrup consumption on spatial memory function and hippocampal neuroinflammation in adolescent rats. Hippocampus 2015, 25, 227–239. [Google Scholar] [CrossRef]
- Stranahan, A.M.; Arumugam, T.V.; Cutler, R.G.; Lee, K.; Egan, J.M.; Mattson, M.P. Diabetes impairs hippocampal function through glucocorticoid-mediated effects on new and mature neurons. Nat. Neurosci. 2008, 11, 309–317. [Google Scholar] [CrossRef]
- Boitard, C.; Cavaroc, A.; Sauvant, J.; Aubert, A.; Castanon, N.; Layé, S.; Ferreira, G. Impairment of hippocampal-dependent memory induced by juvenile high-fat diet intake is associated with enhanced hippocampal inflammation in rats. Brain Behav. Immun. 2014, 40, 9–17. [Google Scholar] [CrossRef] [PubMed]
- Lizarbe, B.; Soares, A.F.; Larsson, S.; Duarte, J.M.N. Neurochemical Modifications in the Hippocampus, Cortex and Hypothalamus of Mice Exposed to Long-Term High-Fat Diet. Front. Neurosci. 2018, 12, 985. [Google Scholar] [CrossRef] [PubMed]
- Howe, A.M.; Burke, S.; O’Reilly, M.E.; McGillicuddy, F.C.; Costello, D.A. Palmitic Acid and Oleic Acid Differently Modulate TLR2-Mediated Inflammatory Responses in Microglia and Macrophages. Mol. Neurobiol. 2022, 59, 2348–2362. [Google Scholar] [CrossRef]
- Valdearcos, M.; Robblee, M.M.; Benjamin, D.I.; Nomura, D.K.; Xu, A.W.; Koliwad, S.K. Microglia dictate the impact of saturated fat consumption on hypothalamic inflammation and neuronal function. Cell Rep. 2014, 9, 2124–2138. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Carraro, R.S.; Souza, G.F.; Solon, C.; Razolli, D.S.; Chausse, B.; Barbizan, R.; Victorio, S.C.; Velloso, L.A. Hypothalamic mitochondrial abnormalities occur downstream of inflammation in diet-induced obesity. Mol. Cell. Endocrinol. 2017, 460, 238–245. [Google Scholar] [CrossRef]
- Dalvi, P.S.; Chalmers, J.A.; Luo, V.; Han, D.Y.; Wellhauser, L.; Liu, Y.; Tran, D.Q.; Castel, J.; Luquet, S.; Wheeler, M.B.; et al. High fat induces acute and chronic inflammation in the hypothalamus: Effect of high-fat diet, palmitate and TNF-alpha on appetite-regulating NPY neurons. Int. J. Obes. 2017, 41, 149–158. [Google Scholar] [CrossRef] [PubMed]
- Gaspar, J.M.; Castilho, Á.; Baptista, F.I.; Liberal, J.; Ambrósio, A.F. Long-term exposure to high glucose induces changes in the content and distribution of some exocytotic proteins in cultured hippocampal neurons. Neuroscience 2010, 171, 981–992. [Google Scholar] [CrossRef] [PubMed]
- Beilharz, J.E.; Maniam, J.; Morris, M.J. Short exposure to a diet rich in both fat and sugar or sugar alone impairs place, but not object recognition memory in rats. Brain Behav. Immun. 2014, 37, 134–141. [Google Scholar] [CrossRef]
- Xu, H.; Barnes, G.T.; Yang, Q.; Tan, G.; Yang, D.; Chou, C.J.; Sole, J.; Nichols, A.; Ross, J.S.; Tartaglia, L.A.; et al. Chronic inflammation in fat plays a crucial role in the development of obesity-related insulin resistance. J. Clin. Investig. 2003, 112, 1821–1830. [Google Scholar] [CrossRef]
- Hotamisligil, G.S.; Shargill, N.S.; Spiegelman, B.M. Adipose expression of tumor necrosis factor-alpha: Direct role in obesity-linked insulin resistance. Science 1993, 259, 87–91. [Google Scholar] [CrossRef]
- Hotamisligil, G.S.; Arner, P.; Caro, J.F.; Atkinson, R.L.; Spiegelman, B.M. Increased adipose tissue expression of tumor necrosis factor-alpha in human obesity and insulin resistance. J. Clin. Investig. 1995, 95, 2409–2415. [Google Scholar] [CrossRef] [PubMed]
- Fain, J.N.; Madan, A.K.; Hiler, M.L.; Cheema, P.; Bahouth, S.W. Comparison of the release of adipokines by adipose tissue, adipose tissue matrix, and adipocytes from visceral and subcutaneous abdominal adipose tissues of obese humans. Endocrinology 2004, 145, 2273–2282. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Thaler, J.P.; Yi, C.X.; Schur, E.A.; Guyenet, S.J.; Hwang, B.H.; Dietrich, M.O.; Zhao, X.; Sarruf, D.A.; Izgur, V.; Maravilla, K.R.; et al. Obesity is associated with hypothalamic injury in rodents and humans. J. Clin. Investig. 2012, 122, 153–162. [Google Scholar] [CrossRef] [Green Version]
- De Souza, C.T.; Araujo, E.P.; Bordin, S.; Ashimine, R.; Zollner, R.L.; Boschero, A.C.; Saad, M.J.; Velloso, L.A. Consumption of a fat-rich diet activates a proinflammatory response and induces insulin resistance in the hypothalamus. Endocrinology 2005, 146, 4192–4199. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Yu, G.; Cao, F.; Hou, T.; Cheng, Y.; Jia, B.; Yu, L.; Chen, W.; Xu, Y.; Chen, M.; Wang, Y. Astrocyte reactivation in medial prefrontal cortex contributes to obesity-promoted depressive-like behaviors. J. Neuroinflammation 2022, 19, 166. [Google Scholar] [CrossRef]
- Salas-Venegas, V.; Flores-Torres, R.P.; Rodríguez-Cortés, Y.M.; Rodríguez-Retana, D.; Ramírez-Carreto, R.J.; Concepción-Carrillo, L.E.; Pérez-Flores, L.J.; Alarcón-Aguilar, A.; López-Díazguerrero, N.E.; Gómez-González, B.; et al. The Obese Brain: Mechanisms of Systemic and Local Inflammation, and Interventions to Reverse the Cognitive Deficit. Front. Integr. Neurosci. 2022, 16, 798995. [Google Scholar] [CrossRef] [PubMed]
- Guo, D.-H.; Yamamoto, M.; Hernandez, C.M.; Khodadadi, H.; Baban, B.; Stranahan, A.M. Visceral adipose NLRP3 impairs cognition in obesity via IL-1R1 on CX3CR1+ cells. J. Clin. Investig. 2020, 130, 1961–1976. [Google Scholar] [CrossRef] [Green Version]
- Buckman, L.B.; Hasty, A.H.; Flaherty, D.K.; Buckman, C.T.; Thompson, M.M.; Matlock, B.K.; Weller, K.; Ellacott, K.L. Obesity induced by a high-fat diet is associated with increased immune cell entry into the central nervous system. Brain Behav. Immun. 2014, 35, 33–42. [Google Scholar] [CrossRef] [Green Version]
- Zhang, X.; Zhang, G.; Zhang, H.; Karin, M.; Bai, H.; Cai, D. Hypothalamic IKKbeta/NF-kappaB and ER stress link overnutrition to energy imbalance and obesity. Cell 2008, 135, 61–73. [Google Scholar] [CrossRef] [Green Version]
- Everard, A.; Lazarevic, V.; Derrien, M.; Girard, M.; Muccioli, G.G.; Neyrinck, A.M.; Possemiers, S.; Van Holle, A.; François, P.; de Vos, W.M.; et al. Responses of gut microbiota and glucose and lipid metabolism to prebiotics in genetic obese and diet-induced leptin-resistant mice. Diabetes 2011, 60, 2775–2786. [Google Scholar] [CrossRef]
- Bruce-Keller, A.J.; Salbaum, J.M.; Luo, M.; Blanchard, E.; Taylor, C.M.; Welsh, D.A.; Berthoud, H.R. Obese-type gut microbiota induce neurobehavioral changes in the absence of obesity. Biol. Psychiatry 2015, 77, 607–615. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Timper, K.; Bruning, J.C. Hypothalamic circuits regulating appetite and energy homeostasis: Pathways to obesity. Dis. Model. Mech. 2017, 10, 679–689. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Velloso, L.A.; Schwartz, M.W. Altered hypothalamic function in diet-induced obesity. Int. J. Obes. 2011, 35, 1455–1465. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Bergen, H.T.; Mizuno, T.; Taylor, J.; Mobbs, C.V. Resistance to diet-induced obesity is associated with increased proopiomelanocortin mRNA and decreased neuropeptide Y mRNA in the hypothalamus. Brain Res. 1999, 851, 198–203. [Google Scholar] [CrossRef]
- Briggs, D.I.; Lemus, M.B.; Kua, E.; Andrews, Z.B. Diet-induced obesity attenuates fasting-induced hyperphagia. J. Neuroendocrinol. 2011, 23, 620–626. [Google Scholar] [CrossRef]
- Kleinridders, A.; Schenten, D.; Konner, A.C.; Belgardt, B.F.; Mauer, J.; Okamura, T.; Wunderlich, F.T.; Medzhitov, R.; Bruning, J.C. MyD88 signaling in the CNS is required for development of fatty acid-induced leptin resistance and diet-induced obesity. Cell Metab. 2009, 10, 249–259. [Google Scholar] [CrossRef] [Green Version]
- Milanski, M.; Degasperi, G.; Coope, A.; Morari, J.; Denis, R.; Cintra, D.E.; Tsukumo, D.M.; Anhe, G.; Amaral, M.E.; Takahashi, H.K.; et al. Saturated fatty acids produce an inflammatory response predominantly through the activation of TLR4 signaling in hypothalamus: Implications for the pathogenesis of obesity. J. Neurosci. 2009, 29, 359–370. [Google Scholar] [CrossRef] [Green Version]
- Berkseth, K.E.; Guyenet, S.J.; Melhorn, S.J.; Lee, D.; Thaler, J.P.; Schur, E.A.; Schwartz, M.W. Hypothalamic gliosis associated with high-fat diet feeding is reversible in mice: A combined immunohistochemical and magnetic resonance imaging study. Endocrinology 2014, 155, 2858–2867. [Google Scholar] [CrossRef] [Green Version]
- Gupta, S.; Knight, A.G.; Gupta, S.; Keller, J.N.; Bruce-Keller, A.J. Saturated long-chain fatty acids activate inflammatory signaling in astrocytes. J. Neurochem. 2012, 120, 1060–1071. [Google Scholar] [CrossRef] [Green Version]
- Douglass, J.D.; Dorfman, M.D.; Fasnacht, R.; Shaffer, L.D.; Thaler, J.P. Astrocyte IKKβ/NF-κB signaling is required for diet-induced obesity and hypothalamic inflammation. Mol. Metab. 2017, 6, 366–373. [Google Scholar] [CrossRef]
- Jin, S.; Kim, K.K.; Park, B.S.; Kim, D.H.; Jeong, B.; Kang, D.; Lee, T.H.; Park, J.W.; Kim, J.G.; Lee, B.J. Function of astrocyte MyD88 in high-fat-diet-induced hypothalamic inflammation. J. Neuroinflammation 2020, 17, 195. [Google Scholar] [CrossRef] [PubMed]
- Mendes, N.F.; Gaspar, J.M.; Lima-Junior, J.C.; Donato, J., Jr.; Velloso, L.A.; Araujo, E.P. TGF-beta1 down-regulation in the mediobasal hypothalamus attenuates hypothalamic inflammation and protects against diet-induced obesity. Metabolism 2018, 85, 171–182. [Google Scholar] [CrossRef] [PubMed]
- Gaspar, J.M.; Mendes, N.F.; Correa-da-Silva, F.; Lima-Junior, J.C.; Gaspar, R.C.; Ropelle, E.R.; Araujo, E.P.; Carvalho, H.M.; Velloso, L.A. Downregulation of HIF complex in the hypothalamus exacerbates diet-induced obesity. Brain Behav. Immun. 2018, 73, 550–561. [Google Scholar] [CrossRef] [PubMed]
- Valdearcos, M.; Douglass, J.D.; Robblee, M.M.; Dorfman, M.D.; Stifler, D.R.; Bennett, M.L.; Gerritse, I.; Fasnacht, R.; Barres, B.A.; Thaler, J.P.; et al. Microglial Inflammatory Signaling Orchestrates the Hypothalamic Immune Response to Dietary Excess and Mediates Obesity Susceptibility. Cell Metab. 2017, 26, 185–197.e183. [Google Scholar] [CrossRef] [Green Version]
- Sobesky, J.L.; Barrientos, R.M.; De May, H.S.; Thompson, B.M.; Weber, M.D.; Watkins, L.R.; Maier, S.F. High-fat diet consumption disrupts memory and primes elevations in hippocampal IL-1β, an effect that can be prevented with dietary reversal or IL-1 receptor antagonism. Brain Behav. Immun. 2014, 42, 22–32. [Google Scholar] [CrossRef] [Green Version]
- Gannon, O.J.; Robison, L.S.; Salinero, A.E.; Abi-Ghanem, C.; Mansour, F.M.; Kelly, R.D.; Tyagi, A.; Brawley, R.R.; Ogg, J.D.; Zuloaga, K.L. High-fat diet exacerbates cognitive decline in mouse models of Alzheimer’s disease and mixed dementia in a sex-dependent manner. J. Neuroinflammation 2022, 19, 110. [Google Scholar] [CrossRef] [PubMed]
- Jones, N.S.; Watson, K.Q.; Rebeck, G.W. High-fat diet increases gliosis and immediate early gene expression in APOE3 mice, but not APOE4 mice. J. Neuroinflammation 2021, 18, 214. [Google Scholar] [CrossRef]
- Cavaliere, G.; Trinchese, G.; Penna, E.; Cimmino, F.; Pirozzi, C.; Lama, A.; Annunziata, C.; Catapano, A.; Mattace Raso, G.; Meli, R.; et al. High-Fat Diet Induces Neuroinflammation and Mitochondrial Impairment in Mice Cerebral Cortex and Synaptic Fraction. Front. Cell. Neurosci. 2019, 13, 509. [Google Scholar] [CrossRef]
- Lorena, F.B.; Nascimento, B.P.P.D.; Camargo, E.L.R.A.; Bernardi, M.M.; Fukushima, A.R.; Panizza, J.D.N.; Nogueira, P.D.B.; Brandão, M.E.S.; Ribeiro, M.O. Long-term obesity is associated with depression and neuroinflammation. Arch. Endocrinol. Metab. 2021, 65, 537–548. [Google Scholar] [CrossRef]
- Antunes, M.M.; Godoy, G.; Masi, L.N.; Curi, R.; Bazotte, R.B. Prefrontal Cortex and Hippocampus Inflammation in Mice Fed High-Carbohydrate or High-Fat Diets. J. Med. Food 2022, 25, 110–113. [Google Scholar] [CrossRef]
- Vega-Torres, J.D.; Ontiveros-Angel, P.; Stuffle, E.C.; Solak, S.; Terrones, E.; Tyner, E.; Oropeza, M.; Dela Peña, I.; Obenaus, A.; Ford, B.D.; et al. Short-term exposure to an obesogenic diet during adolescence elicits anxiety-related behavior and neuroinflammation: Modulatory effects of exogenous neuregulin-1. Transl. Psychiatry 2022, 12, 83. [Google Scholar] [CrossRef] [PubMed]
- Erion, J.R.; Wosiski-Kuhn, M.; Dey, A.; Hao, S.; Davis, C.L.; Pollock, N.K.; Stranahan, A.M. Obesity elicits interleukin 1-mediated deficits in hippocampal synaptic plasticity. J. Neurosci. 2014, 34, 2618–2631. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Kang, E.B.; Koo, J.H.; Jang, Y.C.; Yang, C.H.; Lee, Y.; Cosio-Lima, L.M.; Cho, J.Y. Neuroprotective Effects of Endurance Exercise Against High-Fat Diet-Induced Hippocampal Neuroinflammation. J. Neuroendocrinol. 2016, 28. [Google Scholar] [CrossRef]
- Wardzinski, E.K.; Kistenmacher, A.; Melchert, U.H.; Jauch-Chara, K.; Oltmanns, K.M. Impaired brain energy gain upon a glucose load in obesity. Metabolism 2018, 85, 90–96. [Google Scholar] [CrossRef]
- Davidson, T.L.; Hargrave, S.L.; Swithers, S.E.; Sample, C.H.; Fu, X.; Kinzig, K.P.; Zheng, W. Inter-relationships among diet, obesity and hippocampal-dependent cognitive function. Neuroscience 2013, 253, 110–122. [Google Scholar] [CrossRef] [PubMed]
- Kratz, M.; Baars, T.; Guyenet, S. The relationship between high-fat dairy consumption and obesity, cardiovascular, and metabolic disease. Eur. J. Nutr. 2013, 52, 1–24. [Google Scholar] [CrossRef]
- Yan, J.; Zhang, H.; Yin, Y.; Li, J.; Tang, Y.; Purkayastha, S.; Li, L.; Cai, D. Obesity- and aging-induced excess of central transforming growth factor-beta potentiates diabetic development via an RNA stress response. Nat. Med. 2014, 20, 1001–1008. [Google Scholar] [CrossRef] [Green Version]
- Ma, W.; Yuan, L.; Yu, H.; Xi, Y.; Xiao, R. Mitochondrial dysfunction and oxidative damage in the brain of diet-induced obese rats but not in diet-resistant rats. Life Sci. 2014, 110, 53–60. [Google Scholar] [CrossRef]
- Galizzi, G.; Palumbo, L.; Amato, A.; Conigliaro, A.; Nuzzo, D.; Terzo, S.; Caruana, L.; Picone, P.; Alessandro, R.; Mulè, F.; et al. Altered insulin pathway compromises mitochondrial function and quality control both in in vitro and in vivo model systems. Mitochondrion 2021, 60, 178–188. [Google Scholar] [CrossRef]
- Osorio-Paz, I.; Ramírez-Pérez, G.; Hernández-Ramírez, J.E.; Uribe-Carvajal, S.; Salceda, R. Mitochondrial activity in different regions of the brain at the onset of streptozotocin-induced diabetes in rats. Mol. Biol. Rep. 2018, 45, 871–879. [Google Scholar] [CrossRef]
- Chandrasekaran, K.; Choi, J.; Arvas, M.I.; Salimian, M.; Singh, S.; Xu, S.; Gullapalli, R.P.; Kristian, T.; Russell, J.W. Nicotinamide Mononucleotide Administration Prevents Experimental Diabetes-Induced Cognitive Impairment and Loss of Hippocampal Neurons. Int. J. Mol. Sci. 2020, 21, 3756. [Google Scholar] [CrossRef] [PubMed]
- Tyagi, A.; Nguyen, C.U.; Chong, T.; Michel, C.R.; Fritz, K.S.; Reisdorph, N.; Knaub, L.; Reusch, J.E.B.; Pugazhenthi, S. SIRT3 deficiency-induced mitochondrial dysfunction and inflammasome formation in the brain. Sci. Rep. 2018, 8, 17547. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Raza, H.; John, A.; Howarth, F.C. Increased oxidative stress and mitochondrial dysfunction in zucker diabetic rat liver and brain. Cell. Physiol. Biochem. 2015, 35, 1241–1251. [Google Scholar] [CrossRef] [PubMed]
- Chunchai, T.; Thunapong, W.; Yasom, S.; Wanchai, K.; Eaimworawuthikul, S.; Metzler, G.; Lungkaphin, A.; Pongchaidecha, A.; Sirilun, S.; Chaiyasut, C.; et al. Decreased microglial activation through gut-brain axis by prebiotics, probiotics, or synbiotics effectively restored cognitive function in obese-insulin resistant rats. J. Neuroinflammation 2018, 15, 11. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Pintana, H.; Tanajak, P.; Pratchayasakul, W.; Sa-Nguanmoo, P.; Chunchai, T.; Satjaritanun, P.; Leelarphat, L.; Chattipakorn, N.; Chattipakorn, S.C. Energy restriction combined with dipeptidyl peptidase-4 inhibitor exerts neuroprotection in obese male rats. Br. J. Nutr. 2016, 116, 1700–1708. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Pintana, H.; Apaijai, N.; Chattipakorn, N.; Chattipakorn, S.C. DPP-4 inhibitors improve cognition and brain mitochondrial function of insulin-resistant rats. J. Endocrinol. 2013, 218, 1–11. [Google Scholar] [CrossRef] [Green Version]
- Sa-Nguanmoo, P.; Tanajak, P.; Kerdphoo, S.; Jaiwongkam, T.; Wang, X.; Liang, G.; Li, X.; Jiang, C.; Pratchayasakul, W.; Chattipakorn, N.; et al. FGF21 and DPP-4 inhibitor equally prevents cognitive decline in obese rats. BioMed. Pharmacother. 2018, 97, 1663–1672. [Google Scholar] [CrossRef]
- Chunchai, T.; Samniang, B.; Sripetchwandee, J.; Pintana, H.; Pongkan, W.; Kumfu, S.; Shinlapawittayatorn, K.; KenKnight, B.H.; Chattipakorn, N.; Chattipakorn, S.C. Vagus Nerve Stimulation Exerts the Neuroprotective Effects in Obese-Insulin Resistant Rats, Leading to the Improvement of Cognitive Function. Sci. Rep. 2016, 6, 26866. [Google Scholar] [CrossRef] [Green Version]
- Park, H.S.; Cho, H.S.; Kim, T.W. Physical exercise promotes memory capability by enhancing hippocampal mitochondrial functions and inhibiting apoptosis in obesity-induced insulin resistance by high fat diet. Metab. Brain Dis. 2018, 33, 283–292. [Google Scholar] [CrossRef]
- Siino, V.; Jensen, P.; James, P.; Vasto, S.; Amato, A.; Mulè, F.; Accardi, G.; Larsen, M.R. Obesogenic Diets Cause Alterations on Proteins and Theirs Post-Translational Modifications in Mouse Brains. Nutr. Metab. Insights 2021, 14, 11786388211012405. [Google Scholar] [CrossRef]
- Wohua, Z.; Weiming, X. Glutaredoxin 2 (GRX2) deficiency exacerbates high fat diet (HFD)-induced insulin resistance, inflammation and mitochondrial dysfunction in brain injury: A mechanism involving GSK-3β. Biomed. Pharmacother. 2019, 118, 108940. [Google Scholar] [CrossRef] [PubMed]
- Nasrallah, C.M.; Horvath, T.L. Mitochondrial dynamics in the central regulation of metabolism. Nat. Rev. Endocrinol. 2014, 10, 650–658. [Google Scholar] [CrossRef] [PubMed]
- Gómez-Valadés, A.G.; Pozo, M.; Varela, L.; Boudjadja, M.B.; Ramírez, S.; Chivite, I.; Eyre, E.; Haddad-Tóvolli, R.; Obri, A.; Milà-Guasch, M.; et al. Mitochondrial cristae-remodeling protein OPA1 in POMC neurons couples Ca(2+) homeostasis with adipose tissue lipolysis. Cell Metab. 2021, 33, 1820–1835.e1829. [Google Scholar] [CrossRef] [PubMed]
- Thaler, J.P.; Guyenet, S.J.; Dorfman, M.D.; Wisse, B.E.; Schwartz, M.W. Hypothalamic inflammation: Marker or mechanism of obesity pathogenesis? Diabetes 2013, 62, 2629–2634. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Schneeberger, M.; Dietrich, M.O.; Sebastian, D.; Imbernon, M.; Castano, C.; Garcia, A.; Esteban, Y.; Gonzalez-Franquesa, A.; Rodriguez, I.C.; Bortolozzi, A.; et al. Mitofusin 2 in POMC neurons connects ER stress with leptin resistance and energy imbalance. Cell 2013, 155, 172–187. [Google Scholar] [CrossRef] [Green Version]
- Dietrich, M.O.; Liu, Z.W.; Horvath, T.L. Mitochondrial dynamics controlled by mitofusins regulate Agrp neuronal activity and diet-induced obesity. Cell 2013, 155, 188–199. [Google Scholar] [CrossRef] [Green Version]
- Diaz, B.; Fuentes-Mera, L.; Tovar, A.; Montiel, T.; Massieu, L.; Martínez-Rodríguez, H.G.; Camacho, A. Saturated lipids decrease mitofusin 2 leading to endoplasmic reticulum stress activation and insulin resistance in hypothalamic cells. Brain Res. 2015, 1627, 80–89. [Google Scholar] [CrossRef]
- Colombani, A.L.; Carneiro, L.; Benani, A.; Galinier, A.; Jaillard, T.; Duparc, T.; Offer, G.; Lorsignol, A.; Magnan, C.; Casteilla, L.; et al. Enhanced hypothalamic glucose sensing in obesity: Alteration of redox signaling. Diabetes 2009, 58, 2189–2197. [Google Scholar] [CrossRef] [Green Version]
- Terrien, J.; Seugnet, I.; Seffou, B.; Herrero, M.J.; Bowers, J.; Chamas, L.; Decherf, S.; Duvernois-Berthet, E.; Djediat, C.; Ducos, B.; et al. Reduced central and peripheral inflammatory responses and increased mitochondrial activity contribute to diet-induced obesity resistance in WSB/EiJ mice. Sci. Rep. 2019, 9, 19696. [Google Scholar] [CrossRef] [Green Version]
- Jayashankar, V.; Selwan, E.; Hancock, S.E.; Verlande, A.; Goodson, M.O.; Eckenstein, K.H.; Milinkeviciute, G.; Hoover, B.M.; Chen, B.; Fleischman, A.G.; et al. Drug-like sphingolipid SH-BC-893 opposes ceramide-induced mitochondrial fission and corrects diet-induced obesity. EMBO Mol. Med. 2021, 13, e13086. [Google Scholar] [CrossRef]
- Tenório, M.C.D.S.; Graciliano, N.G.; Moura, F.A.; Oliveira, A.C.M.D.; Goulart, M.O.F. N-Acetylcysteine (NAC): Impacts on Human Health. Antioxidants 2021, 10, 967. [Google Scholar] [CrossRef] [PubMed]
- Keshk, W.A.; Ibrahim, M.A.; Shalaby, S.M.; Zalat, Z.A.; Elseady, W.S. Redox status, inflammation, necroptosis and inflammasome as indispensable contributors to high fat diet (HFD)-induced neurodegeneration; Effect of N-acetylcysteine (NAC). Arch. Biochem. Biophys. 2020, 680, 108227. [Google Scholar] [CrossRef] [PubMed]
- Garcia-Serrano, A.M.; Vieira, J.P.P.; Fleischhart, V.; Duarte, J.M.N. Taurine and N-acetylcysteine treatments prevent memory impairment and metabolite profile alterations in the hippocampus of high-fat diet-fed female mice. Nutr. Neurosci. 2022, 1–13. [Google Scholar] [CrossRef] [PubMed]
- Kumar, P.; Liu, C.; Hsu, J.W.; Chacko, S.; Minard, C.; Jahoor, F.; Sekhar, R.V. Glycine and N-acetylcysteine (GlyNAC) supplementation in older adults improves glutathione deficiency, oxidative stress, mitochondrial dysfunction, inflammation, insulin resistance, endothelial dysfunction, genotoxicity, muscle strength, and cognition: Results of a pilot clinical trial. Clin. Transl. Med. 2021, 11, e372. [Google Scholar] [PubMed]


Disclaimer/Publisher’s Note: The statements, opinions and data contained in all publications are solely those of the individual author(s) and contributor(s) and not of MDPI and/or the editor(s). MDPI and/or the editor(s) disclaim responsibility for any injury to people or property resulting from any ideas, methods, instructions or products referred to in the content. |
© 2023 by the authors. Licensee MDPI, Basel, Switzerland. This article is an open access article distributed under the terms and conditions of the Creative Commons Attribution (CC BY) license (https://creativecommons.org/licenses/by/4.0/).
Share and Cite
Schmitt, L.O.; Gaspar, J.M. Obesity-Induced Brain Neuroinflammatory and Mitochondrial Changes. Metabolites 2023, 13, 86. https://doi.org/10.3390/metabo13010086
Schmitt LO, Gaspar JM. Obesity-Induced Brain Neuroinflammatory and Mitochondrial Changes. Metabolites. 2023; 13(1):86. https://doi.org/10.3390/metabo13010086
Chicago/Turabian StyleSchmitt, Luisa O., and Joana M. Gaspar. 2023. "Obesity-Induced Brain Neuroinflammatory and Mitochondrial Changes" Metabolites 13, no. 1: 86. https://doi.org/10.3390/metabo13010086
APA StyleSchmitt, L. O., & Gaspar, J. M. (2023). Obesity-Induced Brain Neuroinflammatory and Mitochondrial Changes. Metabolites, 13(1), 86. https://doi.org/10.3390/metabo13010086