
Experimental Basis Sets of Quantification of Brain 1H-Magnetic Resonance Spectroscopy at 3.0 T

Abstract
:1. Introduction
2. Materials and Methods
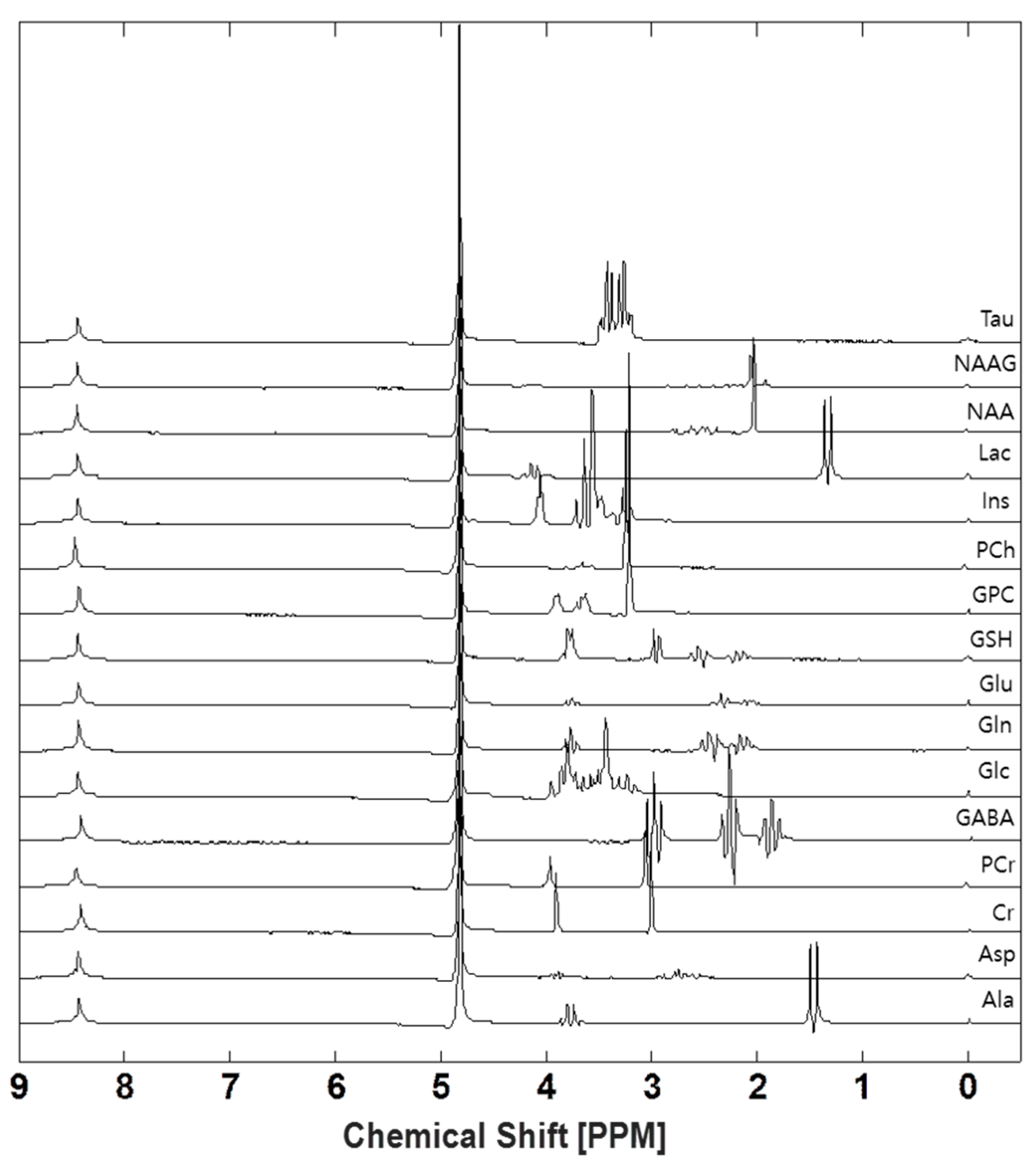
2.1. Generating Experimental Basis Set
2.2. In Vivo MRI and 1H-MRS Study
2.3. In Vitro 1H-MRS Phantom Study
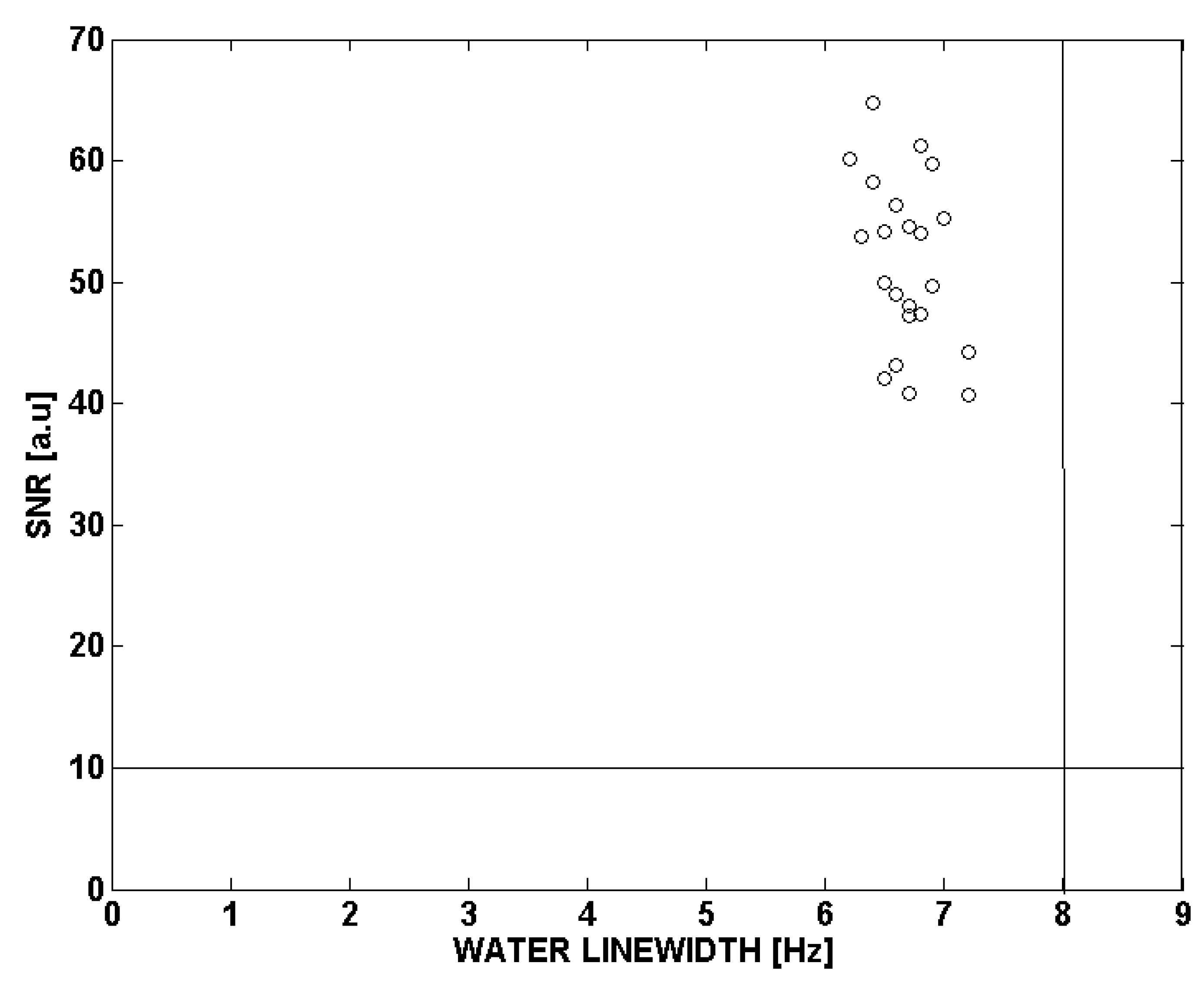
2.4. Quality Control of 1H-MRS Spectra
2.5. LCModel Spectral Analysis
2.6. Quantification of Metabolite Concentrations and CSF Correction
2.7. Statistical Analysis
3. Results
3.1. Generating Experimental Basis Set
3.2. Quality Control of 1H-MRS Spectra
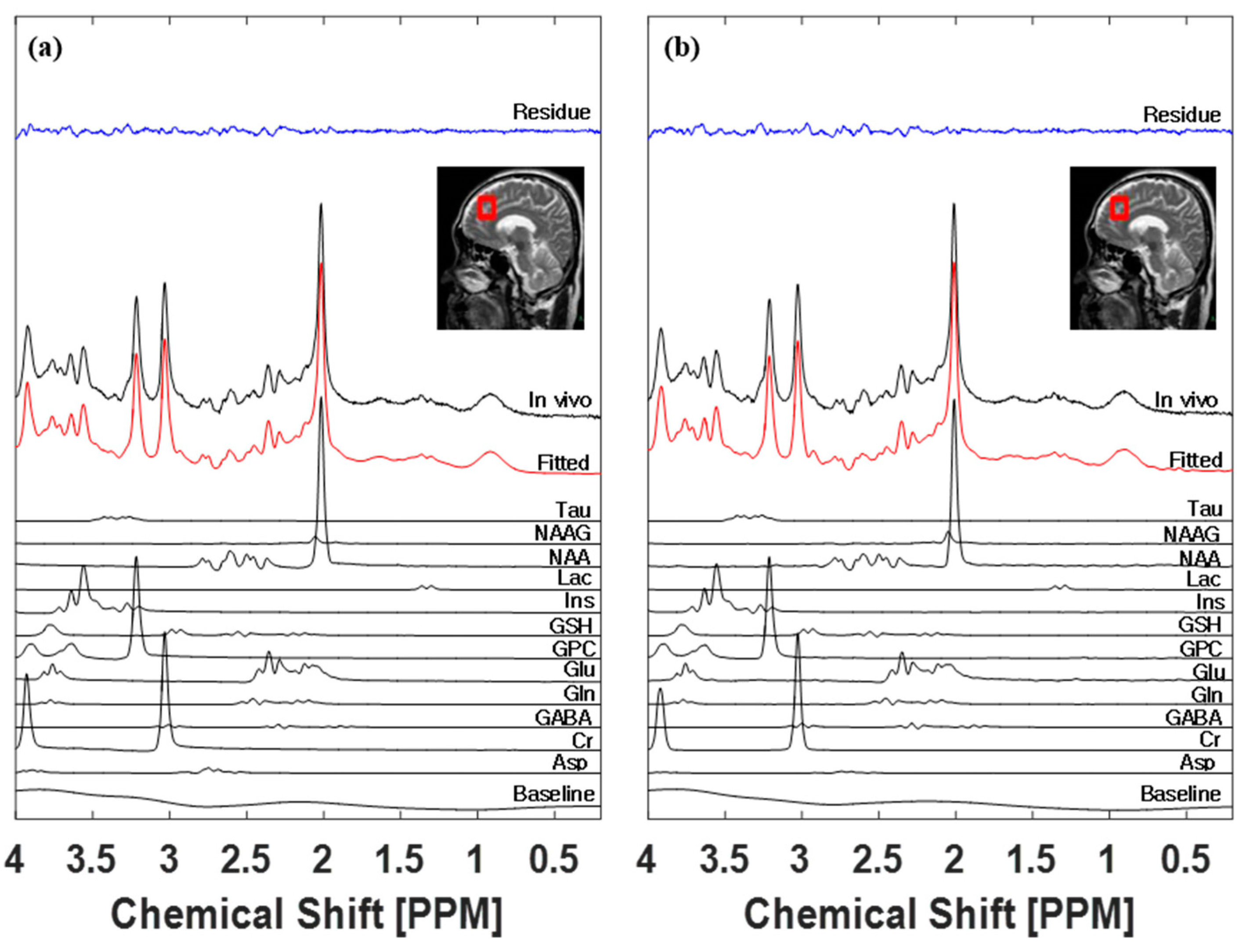
3.3. LCModel Spectral Fitting Analysis
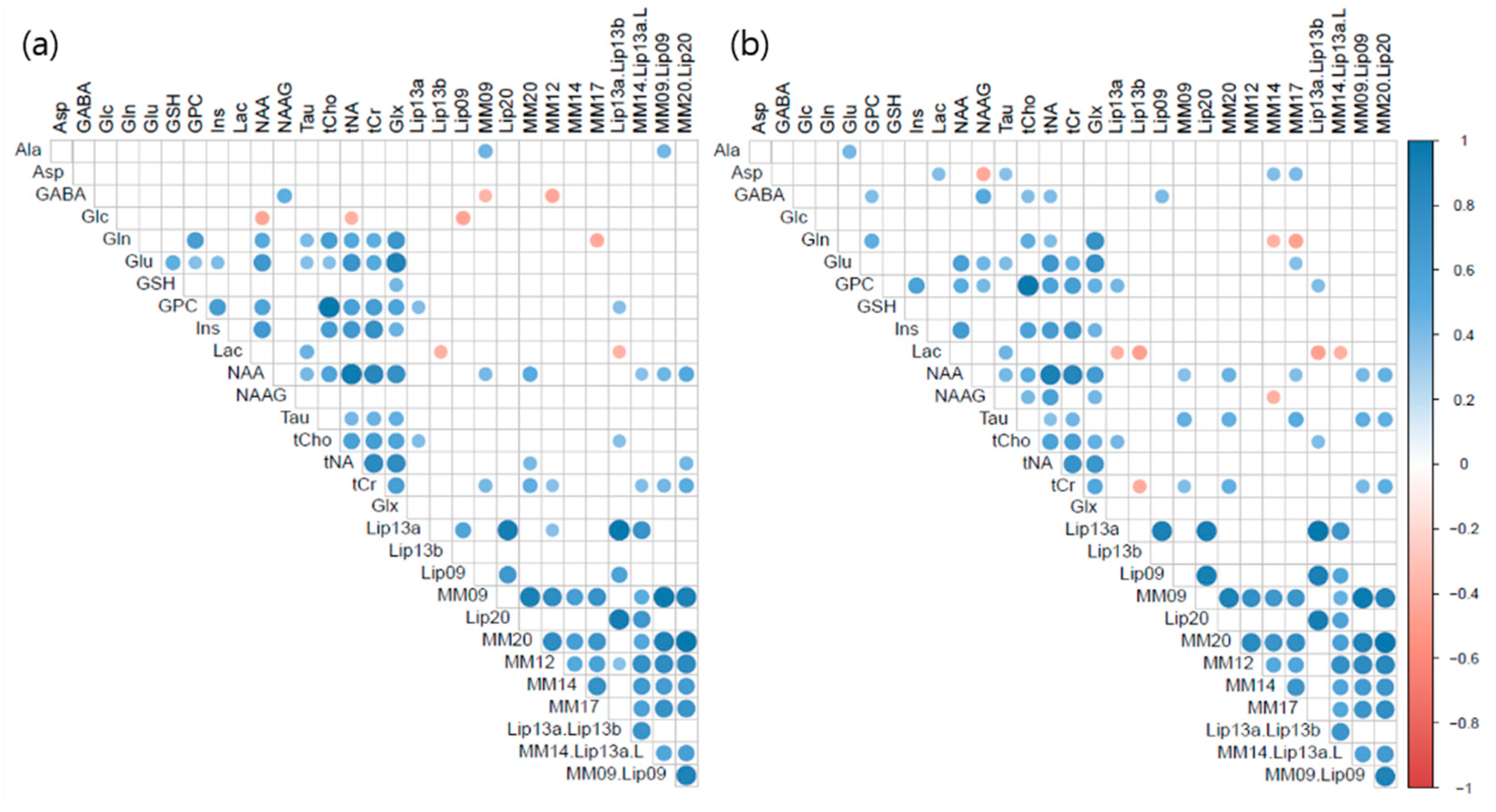
3.4. Correlation Matrices
3.5. In Vitro 1H-MRS Phantom Study
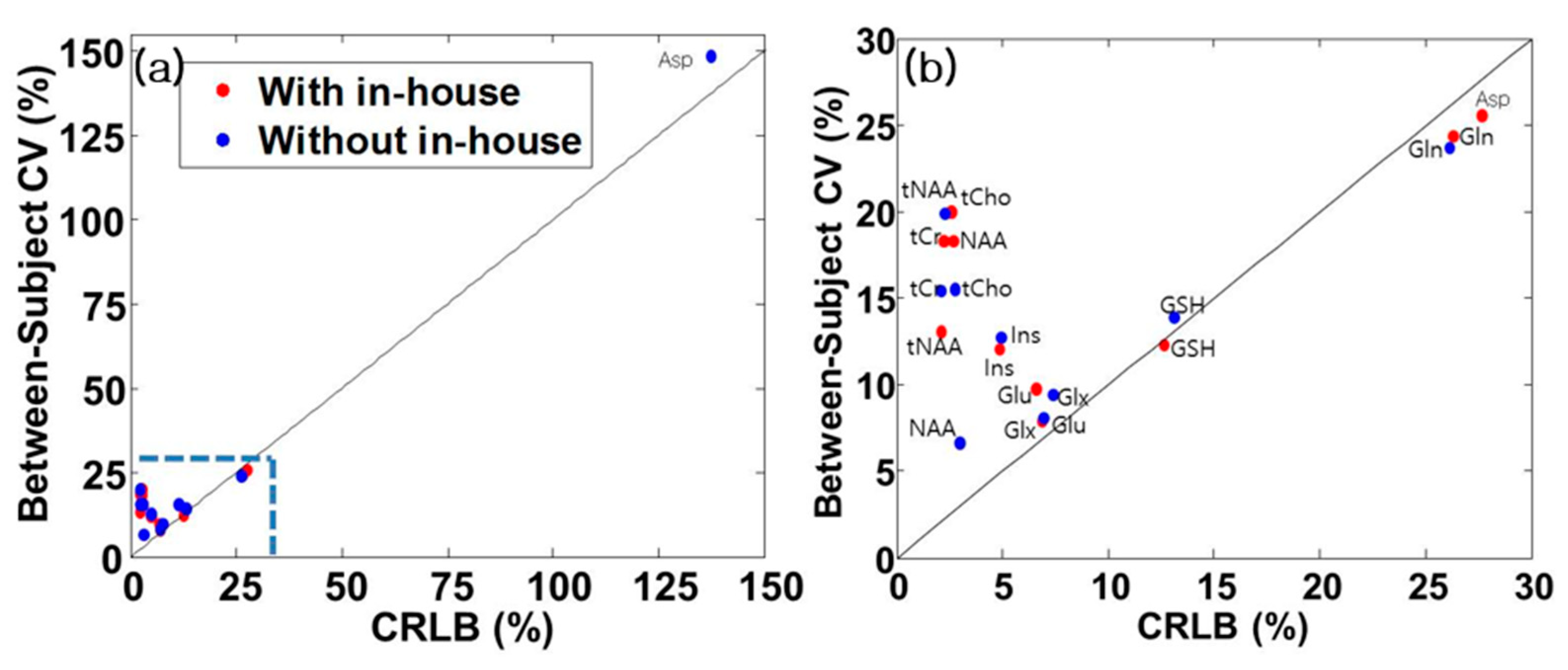
3.6. Coefficient of Variation
4. Discussion
5. Conclusions
Funding
Institutional Review Board Statement
Informed Consent Statement
Data Availability Statement
Acknowledgments
Conflicts of Interest
References
- Brambilla, P.; Stanley, J.A.; Nicoletti, M.A.; Sassi, R.B.; Mallinger, A.G.; Frank, E.; Kupfer, D.; Keshavan, M.S.; Soares, J.C. 1H magnetic resonance spectroscopy investigation of the dorsolateral prefrontal cortex in bipolar disorder patients. J. Affect. Disord. 2005, 86, 61–67. [Google Scholar] [CrossRef] [PubMed]
- Brambilla, P.; Stanley, J.A.; Sassi, R.B.; Nicoletti, M.A.; Mallinger, A.G.; Keshavan, M.S.; Soares, J.C. 1H MRS Study of Dorsolateral Prefrontal Cortex in Healthy Individuals before and after Lithium Administration. Neuropsychopharmacology 2004, 26, 1918–1924. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Chawla, S.; Wang, S.; Moore, P.; Woo, J.H.; Elman, L.; McCluskey, L.F.; Melhem, E.R.; Grossman, M.; Poptani, H. Quantitative proton magnetic resonance spectroscopy detects abnormalities in dorsolateral prefrontal cortex and motor cortex of patients with frontotemporal lobar degeneration. J. Neurol. 2010, 257, 114–121. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Yue, Q.; Liu, M.; Nie, X.; Wu, Q.; Li, J.; Zhang, W.; Huang, X.; Gong, Q. Quantitative 3.0T MR Spectroscopy Reveals Decreased Creatine Concentration in the Dorsolateral Prefrontal Cortex of Patients with Social Anxiety Disorder. PLoS ONE 2012, 7, e48105. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Horder, J.; Lavender, T.; Mendez, M.A.; O’gorman, R.; Daly, E.; Craig, M.C.; Lythgoe, D.J.; Barker, G.J.; Murphy, D.G. Reduced subcortical glutamate/glutamine in adults with autism spectrum disorders: A [1H] MRS study. Transl. Psychiatry 2013, 3, e279. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Murphy, P.S.; Viviers, L.; Abson, C.; Rowland, I.J.; Brada, M.; Leach, M.O.; Dzik-Jurasz, A.S.K. Monitoring temozolomide treatment of low-grade glioma with proton magnetic resonance spectroscopy. Br. J. Cancer 2004, 90, 781–786. [Google Scholar] [CrossRef] [PubMed]
- Govindaraju, V.; Young, K.; Maudsley, A.A. Proton NMR chemical shifts and coupling constants for brain metabolites. NMR Biomed 2000, 13, 129–153. [Google Scholar] [CrossRef]
- Provencher, S.W. Automatic quantitation of localized in vivo 1H spectra with LCModel. NMR Biomed 2001, 14, 260–264. [Google Scholar] [CrossRef] [PubMed]
- Poullet, J.B.; Sima, D.M.; Simonetti, A.W.; Neuter, B.D.; Vanhamme, L.; Lemmerling, P.; Huffel, S.V. An automated quantitation of short echo time MRS spectra in an open source software environment: AQSES. NMR Biomed 2007, 20, 493–504. [Google Scholar] [CrossRef]
- Wilson, M.; Reynolds, G.; Kauppinen, R.A.; Arvanitis, T.N.; Peet, A.C. A Constrained Least-Squares Approach to the Automated Quantitation of In Vivo 1H Magnetic Resonance Spectroscopy Data. Magn. Reson. Med. 2011, 65, 1–12. [Google Scholar] [CrossRef]
- Kanowski, M.; Kaufmann, J.; Braun, J.; Bernarding, J.; Tempelmann, C. Quantitation of Simulated Short Echo Time 1H Human Brain Spectra by LCModel and AMARES. Magn. Reson. Med. 2004, 51, 904–912. [Google Scholar] [CrossRef] [PubMed]
- Mosconi, E.; Sima, D.M.; Osorio Garcia, M.I.; Fontanella, M.; Fiorini, S.; Van Huffel, S.; Marzola, P. Different quantification algorithms may lead to different results: A comparison using proton MRS lipid signals. NMR Biomed. 2014, 27, 431–443. [Google Scholar] [CrossRef] [PubMed]
- Ratiney, H.; Sdika, M.; Coenradie, Y.; Cavassila, S.; Ormondt, D.; Graveron-Demilly, D. Time-domain semi-parametric estimation based on a metabolite basis set. NMR Biomed 2005, 18, 1–13. [Google Scholar] [CrossRef] [PubMed]
- Provencher, S.W. LCModel & LCMgui User’s Manual. 2014. Available online: http://s-provencher.com/lcm-manual (accessed on 1 November 2013).
- Cudalbu, C.; Cavassila, S.; Rabeson, H.; Ormondt, D.V.; Graveron-Demilly, D. Influence of measured and simulated basis sets on metabolite concentration estimates. NMR Biomed. 2008, 21, 627–636. [Google Scholar] [CrossRef]
- Wilson, M.; Davies, N.P.; Sun, Y.; Natarajan, K.; Arvanitis, T.N.; Kauppinen, R.A.; Peet, A.C. A comparison between simulated and experimental basis sets for assessing short-TE in vivo 1H MRS data at 1.5 T. NMR Biomed. 2010, 23, 1117–1126. [Google Scholar]
- Cudalbu, C.; Mlynarik, V.; Xin, L.; Gruetter, R. Quantification of in vivo short echo-time proton magnetic resonance spectra at 14.1 T using two different approaches of modeling the macromolecule spectrum. Meas. Sci. Technol. 2009, 20, 104034. [Google Scholar] [CrossRef] [Green Version]
- Bottomley, P.A. Spatial Localization in NMR Spectroscopy in vivo. Ann. N. Y. Acad. Sci. 1987, 508, 333–348. [Google Scholar] [CrossRef]
- Tkac, I.; Starcuk, Z.; Choi, I.Y.; Gruetter, R. In Vivo 1H NMR Spectroscopy of Rat Brain at 1 ms Echo Time. Magn. Reson. Med. 1999, 41, 649–656. [Google Scholar] [CrossRef]
- Van Der Graaf, M.; Julià-Sapé, M.; Howe, F.A.; Ziegler, A.; Majós, C.; Moreno-Torres, A.; Rijpkema, M.; Acosta, D.; Opstad, K.S.; van Der Meulen, Y.M.; et al. MRS quality assessment in a multicenter study on MRS-based classification of brain tumours. NMR Biomed. 2008, 21, 148–158. [Google Scholar] [CrossRef]
- Provencher, S.W. Estimation of Metabolite Concentrations from Localized in Vivo Proton NMR Spectra. Magn. Reson. Med. 1993, 30, 672–679. [Google Scholar] [CrossRef]
- Provencher, S.W. Constrained regularization method for inverting data represented by linear algebraic or integral equations. Comput. Phys. Commun. 1982, 27, 213–227. [Google Scholar] [CrossRef]
- Mandal, P.K. In vivo proton magnetic resonance spectroscopic signal processing for the absolute quantitation of brain metabolites. Eur. J. Radiol. 2012, 81, e653–e664. [Google Scholar] [CrossRef] [PubMed]
- Ashburner, J.; Barnes, G.; Chen, C.; Daunizeau, J.; Flandin, G.; Friston, K.; Kiebel, S.; Kilner, J.; Litvak, V.; Moran, R.; et al. SPM12 Manual; Wellcome Trust Centre for Neuroimaging: London, UK, 2014. [Google Scholar]
- Chen, C.; Hall, E. MRS_MRI_libs. Available online: https://github.com/chenkonturek/MRS_MRI_libs (accessed on 12 June 2017).
- Sujji, G.E. MRI Brain Image Segmentation based on Thresholding. Int. J. Adv. Comput. Res. 2013, 3, 2277–7970. [Google Scholar]
- Helms, G. The principles of quantification applied to in vivo proton MR spectroscopy. Eur. J. Radiol. 2008, 67, 218–229. [Google Scholar] [CrossRef] [PubMed]
- Deelchand, D.K.; Adanyeguh, I.M.; Emir, U.E.; Nguyen, T.M.; Valabregue, R.; Henry, P.G.; Mochel, F.; Öz, G. Two-Site Reproducibility of Cerebellar and Brainstem Neurochemical Profiles with Short-Echo, Single-Voxel MRS at 3T. Magn. Reson. Med. 2014, 73, 18–25. [Google Scholar] [CrossRef] [Green Version]
- Kaiser, L.G.; Young, K.; Matson, G.B. Numerical Simulations of localized high field 1H MR Spectroscopy. J. Magn. Reson. 2008, 195, 67–75. [Google Scholar] [CrossRef] [Green Version]
- Deelchand, D.K.; Iltis, I.; Henry, P. Improved Quantification Precision of Human Brain Short Echo-Time 1H Magnetic Resonance Spectroscopy at High Magnetic Field: A Simulation Study. Magn. Reson. Med. 2014, 72, 20–25. [Google Scholar] [CrossRef] [Green Version]
- Marjanska, M.; Deelchand, D.K.; Kreis, R. Results and interpretation of a fitting challenge for magnetic resonance spectroscopy set up by the MRS study group of ISMRM. Magn. Reson. Med. 2022, 87, 11–32. [Google Scholar] [CrossRef]
- Schaller, B.; Xin, L.; Cudalbu, C.; Gruetter, R. Quantification of the neurochemical profile using simulated macromolecule resonances at 3T. NMR Biomed. 2013, 26, 593–599. [Google Scholar] [CrossRef] [Green Version]






{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
| CRLB [mM] | ||||||
|---|---|---|---|---|---|---|
| Metabolite and Macromolecule | In-House | Simulated | In-House—Simulated | |||
| Mean | SE | Mean | SE | CI | p | |
| Ala | 0.08 | 0.02 | 0.10 | 0.02 | [−0.06, 0.02] | 0.25 |
| Asp | 0.33 | 0.004 | 0.42 | 0.02 | [−0.13, −0.04] | *** <0.001 |
| GABA | 0.22 | 0.004 | 0.29 | 0.004 | [−0.07, −0.06] | *** <0.001 |
| Glc | 0.05 | 0.02 | 0.07 | 0.02 | [−0.05, 0.01] | 0.24 |
| Gln | 0.37 | 0.008 | 0.43 | 0.007 | [−0.07, −0.04] | *** <0.001 |
| Glu | 0.47 | 0.006 | 0.40 | 0.005 | [−0.004, 0.02] | 0.17 |
| GSH | 0.12 | 0.002 | 0.16 | 0.002 | [−0.044, −0.038] | *** <0.001 |
| Ins | 0.14 | 0.002 | 0.19 | 0.003 | [−0.05, −0.04] | *** <0.001 |
| Lac | 0.18 | 0.003 | 0.22 | 0.006 | [−0.05, −0.04] | *** <0.001 |
| NAA | 0.13 | 0.006 | 0.20 | 0.004 | [−0.05, −0.02] | *** <0.001 |
| NAAG | 0.12 | 0.006 | 0.18 | 0.005 | [−0.08, −0.04] | *** <0.001 |
| Tau | 0.20 | 0.003 | 0.24 | 0.004 | [−0.044, −0.035] | *** <0.001 |
| tCho | 0.02 | 0.0006 | 0.04 | 0.001 | [−0.03, −0.02] | *** <0.001 |
| tNA | 0.14 | 0.004 | 0.16 | 0.005 | [−0.04, −0.02] | *** <0.001 |
| tCr | 0.11 | 0.003 | 0.01 | 0.002 | [0.004, 0.01] | ** <0.01 |
| Glx | 0.53 | 0.01 | 0.56 | 0.008 | [−0.05, −0.01] | *** <0.001 |
| Lip13a | 1.14 | 0.09 | 1.14 | 1.12 | [−0.20, 0.18] | 0.95 |
| Lip13b | 0.05 | 0.03 | 0.02 | 0.01 | [−0.01, 0.08] | 0.14 |
| Lip09 | 0.32 | 0.06 | 0.41 | 0.05 | [−0.20, 0.01] | 0.09 |
| MM09 | 0.74 | 0.02 | 0.74 | 0.02 | [−0.03, 0.03] | 0.99 |
| Lip20 | 0.27 | 0.03 | 0.24 | 0.03 | [0.0001, 0.06] | * <0.05 |
| MM20 | 1.16 | 0.03 | 1.10 | 0.03 | [0.04, 0.09] | *** <0.001 |
| MM12 | 0.54 | 0.02 | 0.57 | 0.02 | [−0.05, −0.01] | ** <0.01 |
| MM14 | 1.07 | 0.03 | 1.07 | 0.03 | [−0.02, 0.03] | 0.56 |
| MM17 | 0.72 | 0.02 | 0.67 | 0.02 | [0.02, 0.09] | ** <0.01 |
| Lip13a + Lip13b | 1.11 | 0.09 | 1.14 | 0.11 | [−0.18, 0.13] | 0.70 |
| MM14 + Lip13a + Lip13b + MM12 | 1.47 | 0.05 | 1.42 | 0.05 | [−0.02, 0.11] | 0.14 |
| MM09 + Lip09 | 0.71 | 0.02 | 0.07 | 0.02 | [−0.01, 0.03] | 0.35 |
| MM20 + Lip20 | 1.15 | 0.03 | 1.10 | 0.03 | [0.03, 0.08] | *** <0.001 |
| Concentration [mM] | ||||||
|---|---|---|---|---|---|---|
| Metabolite and Macromolecule | In-House | Simulated | In-House—Simulated | |||
| Mean | SE | Mean | SE | CI | p | |
| Ala | 0.06 | 0.02 | 0.05 | 0.02 | [−0.03, 0.04] | 0.58 |
| Asp | 1.24 | 0.07 | 0.68 | 0.09 | [0.46, 0.67] | *** <0.001 |
| GABA | 0.47 | 0.03 | 0.89 | 0.04 | [−0.48, −0.37] | *** <0.001 |
| Glc | 0.04 | 0.02 | 0.06 | 0.03 | [−0.05, 0.003] | 0.078 |
| Gln | 1.46 | 0.07 | 1.70 | 0.09 | [−0.32, −0.17] | *** <0.001 |
| Glu | 6.26 | 0.11 | 5.84 | 0.09 | [0.34, 0.51] | *** <0.001 |
| GSH | 0.94 | 0.03 | 1.20 | 0.03 | [−0.29, −0.23] | *** <0.001 |
| Ins | 2.96 | 0.06 | 3.78 | 0.08 | [−0.88, −0.78] | *** <0.001 |
| Lac | 0.33 | 0.03 | 0.42 | 0.03 | [−0.12, −0.06] | *** <0.001 |
| NAA | 6.24 | 0.11 | 6.62 | 0.10 | [−0.43, −0.32] | *** <0.001 |
| NAAG | 0.24 | 0.03 | 0.58 | 0.05 | [−0.42, −0.26] | *** <0.001 |
| Tau | 0.52 | 0.04 | 0.81 | 0.05 | [−0.33, −0.25] | *** <0.001 |
| tCho | 0.76 | 0.11 | 1.56 | 0.03 | [−0.83, −0.77] | *** <0.001 |
| tNA | 6.49 | 0.03 | 7.21 | 0.13 | [−0.76, −0.67] | *** <0.001 |
| tCr | 5.00 | 0.10 | 4.71 | 0.09 | [0.26, 0.33] | *** <0.001 |
| Glx | 7.73 | 0.15 | 7.55 | 0.14 | [0.05, 0.31] | ** 0.0086 |
| Lip13a | 1.19 | 0.17 | 1.20 | 0.20 | [−0.12, 0.08] | 0.72 |
| Lip13b | 0.02 | 0.02 | 0.03 | 0.03 | [−0.04, 0.02] | 0.78 |
| Lip09 | 0.10 | 0.02 | 0.19 | 0.03 | [−0.13, −0.05] | *** <0.001 |
| MM09 | 5.08 | 0.14 | 5.03 | 0.15 | [0.004, 0.10] | * 0.034 |
| Lip20 | 0.15 | 0.02 | 0.11 | 0.02 | [0.02, 0.05] | *** <0.001 |
| MM20 | 10.03 | 0.28 | 9.41 | 0.26 | [0.48, 0.77] | *** <0.001 |
| MM12 | 1.81 | 0.07 | 1.72 | 0.07 | [0.07, 0.12] | *** <0.001 |
| MM14 | 4.20 | 0.14 | 4.12 | 0.15 | [−0.009, 0.16] | 0.078 |
| MM17 | 2.63 | 0.11 | 2.30 | 0.10 | [0.28, 0.38] | *** <0.001 |
| Lip13a + Lip13b | 1.21 | 0.17 | 1.23 | 0.20 | [−0.11, 0.07] | 0.65 |
| MM14 + Lip13a + Lip13b + MM12 | 7.22 | 0.27 | 7.07 | 0.28 | [0.02, 0.28] | * 0.023 |
| MM09 + Lip09 | 5.18 | 0.13 | 5.22 | 0.14 | [−0.10, 0.01] | 0.13 |
| MM20 + Lip20 | 10.18 | 0.28 | 9.52 | 0.26 | [0.51, 0.80] | *** <0.001 |
| Metabolite | Phantom Concentration | In-House | Simulated | ||
|---|---|---|---|---|---|
| Concentration | CRLB | Concentration | CRLB | ||
| Ins | 7.5 | 6.10 | 0.30 (5) | 7.89 | 0.37 (5) |
| Lac | 5 | 3.37 | 0.30 (9) | 4.33 | 0.39 (9) |
| Cho | 3 | 2.45 | 0.07 (3) | 3.54 | 0.11 (3) |
| NAA | 12.5 | 11.58 | 0.23 (2) | 13.33 | 0.27 (2) |
| Cr | 10 | 9.85 | 0.30 (3) | 10.37 | 0.21 (2) |
| Glu | 12.5 | 11.85 | 0.71 (6) | 11.76 | 0.71 (6) |
Disclaimer/Publisher’s Note: The statements, opinions and data contained in all publications are solely those of the individual author(s) and contributor(s) and not of MDPI and/or the editor(s). MDPI and/or the editor(s) disclaim responsibility for any injury to people or property resulting from any ideas, methods, instructions or products referred to in the content. |
© 2023 by the author. Licensee MDPI, Basel, Switzerland. This article is an open access article distributed under the terms and conditions of the Creative Commons Attribution (CC BY) license (https://creativecommons.org/licenses/by/4.0/).
Share and Cite
Baek, H.-M. Experimental Basis Sets of Quantification of Brain 1H-Magnetic Resonance Spectroscopy at 3.0 T. Metabolites 2023, 13, 368. https://doi.org/10.3390/metabo13030368
Baek H-M. Experimental Basis Sets of Quantification of Brain 1H-Magnetic Resonance Spectroscopy at 3.0 T. Metabolites. 2023; 13(3):368. https://doi.org/10.3390/metabo13030368
Chicago/Turabian StyleBaek, Hyeon-Man. 2023. "Experimental Basis Sets of Quantification of Brain 1H-Magnetic Resonance Spectroscopy at 3.0 T" Metabolites 13, no. 3: 368. https://doi.org/10.3390/metabo13030368
APA StyleBaek, H. -M. (2023). Experimental Basis Sets of Quantification of Brain 1H-Magnetic Resonance Spectroscopy at 3.0 T. Metabolites, 13(3), 368. https://doi.org/10.3390/metabo13030368