
Neurodegeneration, Mitochondria, and Antibiotics

 , ,
, , 
Abstract
:1. Introduction
2. Iron Accumulation and Lipid Peroxidation in Neurodegenerative Diseases
3. Neuroinflammation
4. Alzheimer’s Disease
5. Parkinson’s Disease
6. Huntington’s Disease
7. Amyotrophic Lateral Sclerosis
8. Prion Diseases
9. Primary Mitochondrial Diseases
10. Cerebral Ischemia
11. Neuropsychiatric Diseases: Schizophrenia and Depression
12. Conclusions
Author Contributions
Funding
Acknowledgments
Conflicts of Interest
Abbreviations
| AD | Alzheimer’s disease |
| ALS | Amyotrophic lateral sclerosis |
| Aβ | β-amyloid |
| BBB | Blood–brain barrier |
| CI | Cerebral ischemia |
| CNS | Central nervous system |
| DAMP | Danger-associated molecular pattern |
| FQ | Fluoroquinolone |
| HD | Huntington’s disease |
| HTT | Huntingtin |
| IL | Interleukin |
| KGDH | Ketoglutarate dehydrogenase |
| LB | Lewis bodies |
| LPS | Lipopolysaccharide |
| MELAS | Mitochondrial encephalopathy, lactic acidosis, and stroke-like episodes |
| mtDNA | Mitochondrial DNA |
| MVD | Mitochondria-derived vesicles |
| TOR | Main target of rapamycin |
| NBIA | Neurodegeneration with brain iron accumulation |
| NLRP3 | NLR family pyrin domain containing 3 |
| NFT | Neurofibrillary tangles |
| PD | Parkinson’s disease |
| PDH | Pyruvate dehydrogenase |
| PrP | Prion protein |
| PrPC | Cellular prion protein |
| ROS | Reactive oxygen species |
| SNpc | Substantia nigra pars compacta |
| TLR | Toll-like receptor |
| TNF-α | Tumor necrosis factor-α |
| UPRmt | Mitochondrial unfolded protein response |
References
- Beach, T.G. A Review of Biomarkers for Neurodegenerative Disease: Will They Swing Us Across the Valley? Neurol. Ther. 2017, 6 (Suppl. S1), 5–13. [Google Scholar] [CrossRef] [Green Version]
- Evidente, V.G.; Adler, C.H.; Sabbagh, M.N.; Connor, D.J.; Hentz, J.G.; Caviness, J.N.; Sue, L.I.; Beach, T.G. Neuropathological findings of PSP in the elderly without clinical PSP: Possible incidental PSP? Park. Relat. Disord. 2011, 17, 365–371. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Frigerio, R.; Fujishiro, H.; Ahn, T.B.; Josephs, K.A.; Maraganore, D.M.; DelleDonne, A.; Parisi, J.E.; Klos, K.J.; Boeve, B.F.; Dickson, D.W.; et al. Incidental Lewy body disease: Do some cases represent a preclinical stage of dementia with Lewy bodies? Neurobiol. Aging 2011, 32, 857–863. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Lin, M.T.; Beal, M.F. Mitochondrial dysfunction and oxidative stress in neurodegenerative diseases. Nature 2006, 443, 787–795. [Google Scholar] [CrossRef]
- Burchell, V.S.; Gandhi, S.; Deas, E.; Wood, N.; Abramov, A.; Plun-Favreau, H. Targeting mitochondrial dysfunction in neurodegenerative disease: Part I. Expert Opin. Ther. Targets 2010, 14, 369–385. [Google Scholar] [CrossRef] [PubMed]
- Ferris, S.H.; de Leon, M.J.; Wolf, A.P.; Farkas, T.; Christman, D.R.; Reisberg, B.; Fowler, J.S.; MacGregor, R.; Goldman, A.; George, A.E.; et al. Positron Emission Tomography in the Study of Aging and Senile Dementia. Neurobiol. Aging 1981, 1, 127–131. [Google Scholar] [CrossRef] [PubMed]
- Tohgi, H.; Yonezawa, H.; Takahashi, S.; Sato, N.; Kato, E.; Kudo, M.; Hatano, K.; Sasaki, T. Cerebral blood flow and oxygen metabolism in senile dementia of Alzheimer’s type and vascular dementia with deep white matter changes. Neuroradiology 1998, 40, 131–137. [Google Scholar] [CrossRef]
- Parker, W.D., Jr.; Parks, J.; Filley, C.M. Electron transport chain defects in Alzheimer’s disease brain. Neurology 1994, 44, 1090. [Google Scholar] [CrossRef]
- Kerr, J.S.; Adriaanse, B.A.; Greig, N.H.; Mattson, M.P.; Cader, M.Z.; Bohr, V.A.; Fang, E.F. Mitophagy and Alzheimer’s Disease: Cellular and Molecular Mechanisms. Trends Neurosci. 2017, 40, 151–166. [Google Scholar] [CrossRef] [Green Version]
- Fang, E.F.; Hou, Y.; Palikaras, K.; Adriaanse, B.A.; Kerr, J.S.; Yang, B.; Lautrup, S.; Hasan-Olive, M.M.; Caponio, D.; Dan, X.; et al. Mitophagy inhibits amyloid-β and tau pathology and reverses cognitive deficits in models of Alzheimer’s disease. Nat. Neurosci. 2019, 22, 401–412. [Google Scholar] [CrossRef] [PubMed]
- Schapira, A.H.; Cooper, J.M.; Dexter, D.; Clark, J.B.; Jenner, P.; Marsden, C.D. Mitochondrial Complex I Deficiency in Parkinson’s Disease. Lancet 1989, 333, 1269. [Google Scholar] [CrossRef] [PubMed]
- Schapira, A.H.; Mann, V.M.; Cooper, J.M.; Dexter, D.; Daniel, S.E.; Jenner, P.; Clark, J.B.; Marsden, C.D. Anatomic and Disease Specificity of NADH CoQ1 Reductase (Complex I) Deficiency in Parkinson’s Disease. J. Neurochem. 1990, 55, 2142–2145. [Google Scholar] [CrossRef] [PubMed]
- Davis, G.C.; Williams, A.C.; Markey, S.P.; Ebert, M.H.; Caine, E.D.; Reichert, C.M.; Kopin, I.J. Chronic parkinsonism secondary to intravenous injection of meperidine analogues. Psychiatry Res. 1979, 1, 249–254. [Google Scholar] [CrossRef]
- Heikkila, R.E.; Hess, A.; Duvoisin, R.C. Dopaminergic Neurotoxicity of 1-Methyl-4-Phenyl-1,2,5,6-Tetrahydropyridine in Mice. Science 1984, 224, 1451–1453. [Google Scholar] [CrossRef]
- Beal, M.F. Experimental models of Parkinson’s disease. Nat. Rev. Neurosci. 2001, 2, 325–332. [Google Scholar] [CrossRef]
- Betarbet, R.; Sherer, T.B.; MacKenzie, G.; Garcia-Osuna, M.; Panov, A.V.; Greenamyre, J.T. Chronic systemic pesticide exposure reproduces features of Parkinson’s disease. Nat. Neurosci. 2000, 3, 1301–1306. [Google Scholar] [CrossRef] [Green Version]
- Bender, A.; Krishnan, K.J.; Morris, C.M.; Taylor, G.A.; Reeve, A.K.; Perry, R.H.; Jaros, E.; Hersheson, J.S.; Betts, J.; Klopstock, T.; et al. High levels of mitochondrial DNA deletions in substantia nigra neurons in aging and Parkinson disease. Nat. Genet. 2006, 38, 515–517. [Google Scholar] [CrossRef] [PubMed]
- Dölle, C.; Flønes, I.; Nido, G.S.; Miletic, H.; Osuagwu, N.; Kristoffersen, S.; Lilleng, P.K.; Larsen, J.P.; Tysnes, O.-B.; Haugarvoll, K.; et al. Defective mitochondrial DNA homeostasis in the substantia nigra in Parkinson disease. Nat. Commun. 2016, 7, 13548. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Pyle, A.; Brennan, R.; Kurzawa-Akanbi, M.; Yarnall, A.; Thouin, A.; Mollenhauer, B.; Burn, D.; Chinnery, P.F.; Hudson, G. Reduced cerebrospinal fluid mitochondrial DNA is a biomarker for early-stage Parkinson’s disease. Ann. Neurol. 2015, 78, 1000–1004. [Google Scholar] [CrossRef] [PubMed]
- Kitada, T.; Asakawa, S.; Hattori, N.; Matsumine, H.; Yamamura, Y.; Minoshima, S.; Yokochi, M.; Mizuno, Y.; Shimizu, N. Mutations in the parkin gene cause autosomal recessive juvenile parkinsonism. Nature 1998, 392, 605–608. [Google Scholar] [CrossRef]
- Valente, E.M.; Abou-Sleiman, P.M.; Caputo, V.; Muqit, M.M.; Harvey, K.; Gispert, S.; Ali, Z.; Del Turco, D.; Bentivoglio, A.R.; Healy, D.G.; et al. Hereditary Early-Onset Parkinson’s Disease Caused by Mutations in PINK1. Science 2004, 304, 1158. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Álvarez-Córdoba, M.; Khoury, A.F.; Villanueva-Paz, M.; Gómez-Navarro, C.; Villalón-García, I.; Suárez-Rivero, J.M.; Povea-Cabello, S.; de la Mata, M.; Cotán, D.; Talaverón-Rey, M.; et al. Pantothenate Rescues Iron Accumulation in Pantothenate Kinase-Associated Neurodegeneration Depending on the Type of Mutation. Mol. Neurobiol. 2018, 56, 3638–3656. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Álvarez-Córdoba, M.; Talaverón-Rey, M.; Villalón-García, I.; Povea-Cabello, S.; Suárez-Rivero, J.M.; Suárez-Carrillo, A.; Munuera-Cabeza, M.; Salas, J.J.; Sánchez-Alcázar, J.A. Down regulation of the expression of mitochondrial phosphopantetheinyl-proteins in pantothenate kinase-associated neurodegeneration: Pathophysiological consequences and therapeutic perspectives. Orphanet J. Rare Dis. 2021, 16, 201. [Google Scholar] [CrossRef]
- Lezi, E.; Swerdlow, R.H. Mitochondria in neurodegeneration. Adv. Exp. Med. Biol. 2012, 942, 269–286. [Google Scholar]
- Villalón-García, I.; Álvarez-Córdoba, M.; Povea-Cabello, S.; Talaverón-Rey, M.; Villanueva-Paz, M.; Luzón-Hidalgo, R.; Suárez-Rivero, J.M.; Suárez-Carrillo, A.; Munuera-Cabeza, M.; Salas, J.J.; et al. Vitamin E prevents lipid peroxidation and iron accumulation in PLA2G6-Associated Neurodegeneration. Neurobiol. Dis. 2022, 165, 105649. [Google Scholar] [CrossRef] [PubMed]
- Sánchez-Alcázar, J.; Villalón-García, I.; Povea-Cabello, S.; Álvarez-Córdoba, M.; Talaverón-Rey, M.; Suárez-Rivero, J.; Suárez-Carrillo, A.; Munuera-Cabeza, M.; Reche-López, D.; Cilleros-Holgado, P.; et al. Vicious cycle of lipid peroxidation and iron accumulation in neurodegeneration. Neural Regen. Res. 2023, 18, 1196. [Google Scholar] [CrossRef]
- Chaturvedi, R.K.; Beal, M.F. Mitochondrial Approaches for Neuroprotection. Ann. N. Y. Acad. Sci. 2008, 1147, 395–412. [Google Scholar] [CrossRef] [PubMed]
- Bender, A.; Koch, W.; Elstner, M.; Schombacher, Y.; Bender, J.; Moeschl, M.; Gekeler, F.; Muller-Myhsok, B.; Gasser, T.; Tatsch, K.; et al. Creatine supplementation in Parkinson disease: A placebo-controlled randomized pilot trial. Neurology 2006, 67, 1262–1264. [Google Scholar] [CrossRef]
- Zhu, L.; Zhou, Q.; He, L.; Chen, L. Mitochondrial unfolded protein response: An emerging pathway in human diseases. Free. Radic. Biol. Med. 2020, 163, 125–134. [Google Scholar] [CrossRef]
- Yi, H.-S.; Chang, J.Y.; Shong, M. The mitochondrial unfolded protein response and mitohormesis: A perspective on metabolic diseases. J. Mol. Endocrinol. 2018, 61, R91–R105. [Google Scholar] [CrossRef] [Green Version]
- Wang, S.; Gao, K.; Liu, Y. UPR mt coordinates immunity to maintain mitochondrial homeostasis and animal fitness. Mitochondrion 2018, 41, 9–13. [Google Scholar] [CrossRef] [PubMed]
- Anderson, N.S.; Haynes, C.M. Folding the Mitochondrial UPR into the Integrated Stress Response. Trends Cell Biol. 2020, 30, 428–439. [Google Scholar] [CrossRef]
- Cabezas, R.; El-Bachá, R.S.; González, J.; Barreto, G.E. Mitochondrial functions in astrocytes: Neuroprotective implications from oxidative damage by rotenone. Neurosci. Res. 2012, 74, 80–90. [Google Scholar] [CrossRef]
- Pellegrino, M.W.; Nargund, A.M.; Kirienko, N.V.; Gillis, R.; Fiorese, C.J.; Haynes, C.M. Mitochondrial UPR-regulated innate immunity provides resistance to pathogen infection. Nature 2014, 516, 414–417. [Google Scholar] [CrossRef] [Green Version]
- Yoneda, T.; Benedetti, C.; Urano, F.; Clark, S.G.; Harding, H.; Ron, D. Compartment-specific perturbation of protein handling activates genes encoding mitochondrial chaperones. J. Cell Sci. 2004, 117, 4055–4066. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Runkel, E.D.; Liu, S.; Baumeister, R.; Schulze, E. Surveillance-Activated Defenses Block the ROS–Induced Mitochondrial Unfolded Protein Response. PLoS Genet. 2013, 9, e1003346. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Kohanski, M.A.; Dwyer, D.J.; Collins, J.J. How antibiotics kill bacteria: From targets to networks. Nat. Rev. Microbiol. 2010, 8, 423–435. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Suárez-Rivero, J.M.; Pastor-Maldonado, C.J.; Povea-Cabello, S.; Álvarez-Córdoba, M.; Villalón-García, I.; Talaverón-Rey, M.; Suárez-Carrillo, A.; Munuera-Cabeza, M.; Sánchez-Alcázar, J.A. Mitochondria and Antibiotics: For Good or for Evil? Biomolecules 2021, 11, 1050. [Google Scholar] [CrossRef]
- Kalghatgi, S.; Spina, C.S.; Costello, J.C.; Liesa, M.; Morones-Ramirez, J.R.; Slomovic, S.; Molina, A.; Shirihai, O.S.; Collins, J.J. Bactericidal Antibiotics Induce Mitochondrial Dysfunction and Oxidative Damage in Mammalian Cells. Sci. Transl. Med. 2013, 5, 192ra85. [Google Scholar] [CrossRef] [Green Version]
- Wang, X.; Ryu, D.; Houtkooper, R.H.; Auwerx, J. Antibiotic use and abuse: A threat to mitochondria and chloroplasts with impact on research, health, and environment. Bioessays 2015, 37, 1045–1053. [Google Scholar] [CrossRef]
- Dethlefsen, L.; Relman, D.A. Incomplete recovery and individualized responses of the human distal gut microbiota to repeated antibiotic perturbation. Proc. Natl. Acad. Sci. USA 2011, 108 (Suppl. S1), 4554–4561. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Giedraitienė, A.; Vitkauskienė, A.; Naginienė, R.; Pavilonis, A. Antibiotic Resistance Mechanisms of Clinically Important Bacteria. Medicina 2011, 47, 19–46. [Google Scholar] [CrossRef]
- Cryan, J.F.; O’Riordan, K.J.; Cowan, C.S.; Sandhu, K.V.; Bastiaanssen, T.F.; Boehme, M.; Codagnone, M.G.; Cussotto, S.; Fulling, C.; Golubeva, A.V.; et al. The Microbiota-Gut-Brain Axis. Physiol. Rev. 2019, 99, 1877–2013. [Google Scholar] [CrossRef]
- Intili, G.; Paladino, L.; Rappa, F.; Alberti, G.; Plicato, A.; Calabrò, F.; Fucarino, A.; Cappello, F.; Bucchieri, F.; Tomasello, G.; et al. From Dysbiosis to Neurodegenerative Diseases through Different Communication Pathways: An Overview. Biology 2023, 12, 195. [Google Scholar] [CrossRef]
- Liu, H.; Su, W.; Li, S.; Du, W.; Ma, X.; Jin, Y.; Li, K.; Chen, H. Eradication of Helicobacter pylori infection might improve clinical status of patients with Parkinson’s disease, especially on bradykinesia. Clin. Neurol. Neurosurg. 2017, 160, 101–104. [Google Scholar] [CrossRef]
- Hegelmaier, T.; Lebbing, M.; Duscha, A.; Tomaske, L.; Tönges, L.; Holm, J.B.; Nielsen, H.B.; Gatermann, S.G.; Przuntek, H.; Haghikia, A. Interventional Influence of the Intestinal Microbiome through Dietary Intervention and Bowel Cleansing Might Improve Motor Symptoms in Parkinson’s Disease. Cells 2020, 9, 376. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Nelson, M.L.; Levy, S.B. The history of the tetracyclines. Ann. N. Y. Acad. Sci. 2011, 1241, 17–32. [Google Scholar] [CrossRef] [PubMed]
- Kriz, J.; Nguyen, M.D.; Julien, J.-P. Minocycline Slows Disease Progression in a Mouse Model of Amyotrophic Lateral Sclerosis. Neurobiol. Dis. 2002, 10, 268–278. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- González-Lizárraga, F.; Socías, S.B.; Ávila, C.L.; Torres-Bugeau, C.M.; Barbosa, L.R.S.; Binolfi, A.; Sepúlveda-Díaz, J.E.; Del-Bel, E.; Fernandez, C.O.; Papy-Garcia, D.; et al. Repurposing doxycycline for synucleinopathies: Remodelling of α-synuclein oligomers towards non-toxic parallel beta-sheet structured species. Sci. Rep. 2017, 7, srep41755. [Google Scholar] [CrossRef] [Green Version]
- Amaral, L.D.; dos Santos, N.A.G.; Sisti, F.M.; Del Bel, E.; dos Santos, A.C. The antibiotic doxycycline mimics the NGF signaling in PC12 cells: A relevant mechanism for neuroprotection. Chem. Interact. 2021, 341, 109454. [Google Scholar] [CrossRef]
- Tremblay, P.; Meiner, Z.; Galou, M.; Heinrich, C.; Petromilli, C.; Lisse, T.; Cayetano, J.; Torchia, M.; Mobley, W.; Bujard, H.; et al. Doxycycline control of prion protein transgene expression modulates prion disease in mice. Proc. Natl. Acad. Sci. USA 1998, 95, 12580–12585. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Tagliavini, F.; Forloni, G.; Colombo, L.; Rossi, G.; Girola, L.; Canciani, B.; Angeretti, N.; Giampaolo, L.; Peressini, E.; Awan, T.; et al. Tetracycline affects abnormal properties of synthetic PrP peptides and PrP(Sc) in vitro. J. Mol. Biol. 2000, 300, 1309–1322. [Google Scholar] [CrossRef] [PubMed]
- Stoilova, T.; Colombo, L.; Forloni, G.; Tagliavini, F.; Salmona, M. A New Face for Old Antibiotics: Tetracyclines in Treatment of Amyloidoses. J. Med. Chem. 2013, 56, 5987–6006. [Google Scholar] [CrossRef] [PubMed]
- Chen, M.; Ona, V.O.; Li, M.; Ferrante, R.J.; Fink, K.B.; Zhu, S.; Bian, J.; Guo, L.; Farrell, L.A.; Hersch, S.M.; et al. Minocycline inhibits caspase-1 and caspase-3 expression and delays mortality in a transgenic mouse model of Huntington disease. Nat. Med. 2000, 6, 797–801. [Google Scholar] [CrossRef] [PubMed]
- Ruan, L.; Zhou, C.; Jin, E.; Kucharavy, A.; Zhang, Y.; Wen, Z.; Florens, L.; Li, R. Cytosolic proteostasis through importing of misfolded proteins into mitochondria. Nature 2017, 543, 443–446. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Zhao, F.; Hua, Y.; He, Y.; Keep, R.; Xi, G. Minocycline-Induced Attenuation of Iron Overload and Brain Injury after Experimental Intracerebral Hemorrhage. Stroke 2011, 42, 3587–3593. [Google Scholar] [CrossRef] [Green Version]
- Minocycline in Patients with Huntington’s Disease. Available online: https://ClinicalTrials.gov/show/NCT00029874 (accessed on 4 March 2023).
- Tetracycline-Derivatives for Treatment of Cerebral Arteriovenous Malformations and Aneurysms. Available online: https://ClinicalTrials.gov/show/NCT00243893 (accessed on 4 March 2023).
- Efficacy of Doxycycline on Metakaryote Cell Death in Patients with Resectable Pancreatic Cancer. Available online: https://ClinicalTrials.gov/show/NCT02775695 (accessed on 4 March 2023).
- The Effect of Tetracycline in Degradation and Permeability of Collagen Membrane. Available online: https://ClinicalTrials.gov/show/NCT02057926 (accessed on 4 March 2023).
- A Pilot Study of Inflammatory Markers in Alzheimer’s Disease. Available online: https://ClinicalTrials.gov/show/NCT00715858 (accessed on 4 March 2023).
- Effect of Topical Tetracycline and Dexamethasone on Periodontal and Pulpal Regeneration of Replanted Avulsed Teeth. Available online: https://ClinicalTrials.gov/show/NCT03611920 (accessed on 4 March 2023).
- Shapiro, L.E.; Knowles, S.R.; Shear, N.H. Comparative Safety of Tetracycline, Minocycline, and Doxycycline. Arch. Dermatol. 1997, 133, 1224–1230. [Google Scholar] [CrossRef]
- Ezelarab, H.; Abbas, S.; Hassan, H.A.; Abuo-Rahma, G.E.-D.A. Recent updates of fluoroquinolones as antibacterial agents. Arch. der Pharm. 2018, 351, e1800141. [Google Scholar] [CrossRef]
- Tauber, S.C.; Nau, R. Immunomodulatory properties of antibiotics. Curr. Mol. Pharmacol. 2008, 1, 68–79. [Google Scholar]
- Shalit, I. Immunological aspects of new quinolones. Eur. J. Clin. Microbiol. Infect. Dis. 1991, 10, 262–266. [Google Scholar] [CrossRef]
- Yoshimura, T.; Kurita, C.; Usami, E.; Nakao, T.; Watanabe, S.; Kobayashi, J.; Yamazaki, F.; Nagai, H. Immunomodulatory Action of Levofloxacin on Cytokine Production by Human Peripheral Blood Mononuclear Cells. Chemotherapy 1996, 42, 459–464. [Google Scholar] [CrossRef] [PubMed]
- S9809, Ciprofloxacin Compared with Cephalexin in Treating Patients with Bladder Cancer. Available online: https://ClinicalTrials.gov/show/NCT00003824 (accessed on 4 March 2023).
- Efficacy and Safety of Oral OPS-2071 in Participants with Crohn’s Disease Showing Symptoms of Active Inflammation. Available online: https://ClinicalTrials.gov/show/NCT03850509 (accessed on 4 March 2023).
- Combination Antibiotic Therapy Compared to Monotherapy in the Treatment of Acute COPD. Available online: https://ClinicalTrials.gov/show/NCT04879030 (accessed on 4 March 2023).
- Alves, C.; Mendes, D.; Marques, F.B. Fluoroquinolones and the risk of tendon injury: A systematic review and meta-analysis. Eur. J. Clin. Pharmacol. 2019, 75, 1431–1443. [Google Scholar] [CrossRef]
- Dai, X.-C.; Yang, X.-X.; Ma, L.; Tang, G.-M.; Pan, Y.-Y.; Hu, H.-L. Relationship between fluoroquinolones and the risk of aortic diseases: A meta-analysis of observational studies. BMC Cardiovasc. Disord. 2020, 20, 49. [Google Scholar] [CrossRef] [Green Version]
- Owens, R.C., Jr.; Ambrose, P.G. Antimicrobial safety: Focus on fluoroquinolones. Clin. Infect. Dis. 2005, 41 (Suppl. S2), S144–S157. [Google Scholar] [CrossRef] [PubMed]
- Kaur, K.; Fayad, R.; Saxena, A.; Frizzell, N.; Chanda, A.; Das, S.; Chatterjee, S.; Hegde, S.; Baliga, M.S.; Ponemone, V.; et al. Fluoroquinolone-related neuropsychiatric and mitochondrial toxicity: A collaborative investigation by scientists and members of a social network. J. Community Support. Oncol. 2016, 14, 54–65. [Google Scholar] [CrossRef] [PubMed]
- Akahane, K.; Tsutomi, Y.; Kimura, Y.; Kitano, Y. Levofloxacin, an Optical Isomer of Ofloxacin, Has Attenuated Epileptogenic Activity in Mice and Inhibitory Potency in GABA Receptor Binding. Chemotherapy 1994, 40, 412–417. [Google Scholar] [CrossRef]
- Grill, M.F.; Maganti, R.K. Neurotoxic effects associated with antibiotic use: Management considerations. Br. J. Clin. Pharmacol. 2011, 72, 381–393. [Google Scholar] [CrossRef] [Green Version]
- Park-Wyllie, L.Y.; Juurlink, D.N.; Kopp, A.; Shah, B.R.; Stukel, T.A.; Stumpo, C.; Dresser, L.; Low, D.E.; Mamdani, M.M. Outpatient Gatifloxacin Therapy and Dysglycemia in Older Adults. N. Engl. J. Med. 2006, 354, 1352–1361. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Abulfathi, A.A.; Decloedt, E.H.; Svensson, E.M.; Diacon, A.H.; Donald, P.; Reuter, H. Clinical Pharmacokinetics and Pharmacodynamics of Rifampicin in Human Tuberculosis. Clin. Pharmacokinet. 2019, 58, 1103–1129. [Google Scholar] [CrossRef] [PubMed]
- Liang, Y.; Jing, X.; Zeng, Z.; Bi, W.; Chen, Y.; Wu, X.; Yang, L.; Liu, J.; Xiao, S.; Liu, S.; et al. Rifampicin attenuates rotenone-induced inflammation via suppressing NLRP3 inflammasome activation in microglia. Brain Res. 2015, 1622, 43–50. [Google Scholar] [CrossRef]
- Wu, X.; Liang, Y.; Jing, X.; Lin, D.; Chen, Y.; Zhou, T.; Peng, S.; Zheng, D.; Zeng, Z.; Lei, M.; et al. Rifampicin Prevents SH-SY5Y Cells from Rotenone-Induced Apoptosis via the PI3K/Akt/GSK-3beta/CREB Signaling Pathway. Neurochem. Res. 2018, 43, 886–893. [Google Scholar] [CrossRef]
- Liang, Y.; Zheng, D.; Peng, S.; Lin, D.; Jing, X.; Zeng, Z.; Chen, Y.; Huang, K.; Xie, Y.; Zhou, T.; et al. Rifampicin attenuates rotenone-treated microglia inflammation via improving lysosomal function. Toxicol. Vitr. 2019, 63, 104690. [Google Scholar] [CrossRef] [PubMed]
- Investigation of Rifampin to Reduce Pedal Amputations for Osteomyelitis in Diabetics. Available online: https://ClinicalTrials.gov/show/NCT03012529 (accessed on 4 March 2023).
- Pregnane X Receptor (PXR) Agonist Rifampicin Effects on Glucose, Lipid and Hormone Homeostasis. Available online: https://ClinicalTrials.gov/show/NCT00985270 (accessed on 4 March 2023).
- Girling, D.J.; Hitze, K.L. Adverse reactions to rifampicin. Bull. World Health Organ. 1979, 57, 45–49. [Google Scholar] [PubMed]
- Li, J.; Kim, S.G.; Blenis, J. Rapamycin: One drug, many effects. Cell Metab. 2014, 19, 373–379. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Moors, T.E.; Hoozemans, J.J.M.; Ingrassia, A.; Beccari, T.; Parnetti, L.; Chartier-Harlin, M.-C.; van de Berg, W.D.J. Therapeutic potential of autophagy-enhancing agents in Parkinson’s disease. Mol. Neurodegener. 2017, 12, 11. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Pupyshev, A.B.; Tikhonova, M.A.; Akopyan, A.A.; Tenditnik, M.V.; Dubrovina, N.I.; Korolenko, T.A. Therapeutic activation of autophagy by combined treatment with rapamycin and trehalose in a mouse MPTP-induced model of Parkinson’s disease. Pharmacol. Biochem. Behav. 2019, 177, 1–11. [Google Scholar] [CrossRef] [PubMed]
- Ding, L.; Nan, W.; Zhu, X.; Li, X.; Zhou, L.; Chen, H.; Yu, L.; Khan, F.U.; Zhong, H.; Shi, X. Rapamycin and FK506 derivative TH2849 could ameliorate neurodegenerative diseases through autophagy with low immunosuppressive effect. CNS Neurosci. Ther. 2018, 25, 452–464. [Google Scholar] [CrossRef] [Green Version]
- Zhang, G.; Yin, L.; Luo, Z.; Chen, X.; He, Y.; Yu, X.; Wang, M.; Tian, F.; Luo, H. Effects and potential mechanisms of rapamycin on MPTP-induced acute Parkinson’s disease in mice. Ann. Palliat. Med. 2021, 10, 2889–2897. [Google Scholar] [CrossRef]
- Socias, S.B.; González-Lizárraga, F.; Avila, C.L.; Vera, C.; Acuña, L.; Sepulveda-Diaz, J.E.; Del-Bel, E.; Raisman-Vozari, R.; Chehin, R.N. Exploiting the therapeutic potential of ready-to-use drugs: Repurposing antibiotics against amyloid aggregation in neurodegenerative diseases. Prog. Neurobiol. 2018, 162, 17–36. [Google Scholar] [CrossRef] [Green Version]
- Reglodi, D.; Renaud, J.; Tamas, A.; Tizabi, Y.; Socías, S.B.; Del-Bel, E.; Raisman-Vozari, R. Novel tactics for neuroprotection in Parkinson’s disease: Role of antibiotics, polyphenols and neuropeptides. Prog. Neurobiol. 2017, 155, 120–148. [Google Scholar] [CrossRef]
- Schmukler, E.; Solomon, S.; Simonovitch, S.; Goldshmit, Y.; Wolfson, E.; Michaelson, D.M.; Pinkas-Kramarski, R. Altered mitochondrial dynamics and function in APOE4-expressing astrocytes. Cell Death Dis. 2020, 11, 578. [Google Scholar] [CrossRef]
- Rapamycin—Effects on Alzheimer’s and Cognitive Health. Available online: https://ClinicalTrials.gov/show/NCT04629495 (accessed on 4 March 2023).
- Rapamycin Treatment for ALS. Available online: https://ClinicalTrials.gov/show/NCT03359538 (accessed on 4 March 2023).
- Impacts of Mechanistic Target of Rapamycin (mTOR) Inhibition on Aged Human Muscle (Rapamune). Available online: https://ClinicalTrials.gov/show/NCT05414292 (accessed on 4 March 2023).
- Temsirolimus in Myelodysplastic Syndrome (MDS). Available online: https://ClinicalTrials.gov/show/NCT01111448 (accessed on 4 March 2023).
- Effect of the Inhibition of the Mammalian Target of Rapamycin on Metabolism and Exercise. Available online: https://ClinicalTrials.gov/show/NCT01561404 (accessed on 4 March 2023).
- Nguyen, L.S.; Vautier, M.; Allenbach, Y.; Zahr, N.; Benveniste, O.; Funck-Brentano, C.; Salem, J.E. Sirolimus and mTOR Inhibitors: A Review of Side Effects and Specific Management in Solid Organ Trans-plantation. Drug Saf. 2019, 42, 813–825. [Google Scholar] [CrossRef]
- Ceftriaxone. LiverTox: Clinical and Research Information on Drug-Induced Liver Injury; National Institute of Diabetes and Digestive and Kidney Diseases (NIDDK): Bethesda, MD, USA, 2012.
- Ruzza, P.; Siligardi, G.; Hussain, R.; Marchiani, A.; Islami, M.; Bubacco, L.; Delogu, G.; Fabbri, D.; Dettori, M.A.; Sechi, M.; et al. Ceftriaxone Blocks the Polymerization of α-Synuclein and Exerts Neuroprotective Effects in Vitro. ACS Chem. Neurosci. 2013, 5, 30–38. [Google Scholar] [CrossRef] [Green Version]
- Ho, Y.-J.; Weng, J.-C.; Lin, C.-L.; Shen, M.-S.; Li, H.-H.; Liao, W.-C.; Tsai, N.-M.; Hung, C.-S.; Lai, T.-J.; Lee, I.-Y. Ceftriaxone Treatment for Neuronal Deficits: A Histological and MEMRI Study in a Rat Model of Dementia with Lewy Bodies. Behav. Neurol. 2018, 2018, 4618716. [Google Scholar] [CrossRef]
- Chotibut, T.; Meadows, S.; Kasanga, E.A.; McInnis, T.; Cantu, M.A.; Bishop, C.; Salvatore, M.F. Ceftriaxone reduces L-dopa-induced dyskinesia severity in 6-hydroxydopamine Parkinson’s disease model. Mov. Disord. 2017, 32, 1547–1556. [Google Scholar] [CrossRef] [PubMed]
- Sun, C.; Armstrong, M. Treatment of Parkinson’s Disease with Cognitive Impairment: Current Approaches and Future Directions. Behav. Sci. 2021, 11, 54. [Google Scholar] [CrossRef] [PubMed]
- Zhou, X.; Lu, J.; Wei, K.; Wei, J.; Tian, P.; Yue, M.; Wang, Y.; Hong, D.; Li, F.; Wang, B.; et al. Neuroprotective Effect of Ceftriaxone on MPTP-Induced Parkinson’s Disease Mouse Model by Regulating Inflammation and Intestinal Microbiota. Oxid. Med. Cell Longev. 2021, 2021, 9424582. [Google Scholar] [CrossRef]
- Zhang, Y.; Zhang, X.; Qu, S. Ceftriaxone Protects Astrocytes from MPP(+) via Suppression of NF-kappaB/JNK/c-Jun Signaling. Mol. Neurobiol. 2015, 52, 78–92. [Google Scholar] [CrossRef] [PubMed]
- Hsu, C.Y.; Hung, C.S.; Chang, H.M.; Liao, W.C.; Ho, S.C.; Ho, Y.J. Ceftriaxone prevents and reverses behavioral and neuronal deficits in an MPTP-induced animal model of Parkin-son’s disease dementia. Neuropharmacology 2015, 91, 43–56. [Google Scholar] [CrossRef] [PubMed]
- Hsieh, M.-H.; Meng, W.-Y.; Liao, W.-C.; Weng, J.-C.; Li, H.-H.; Su, H.-L.; Lin, C.-L.; Hung, C.-S.; Ho, Y.-J. Ceftriaxone reverses deficits of behavior and neurogenesis in an MPTP-induced rat model of Parkinson’s disease dementia. Brain Res. Bull. 2017, 132, 129–138. [Google Scholar] [CrossRef]
- Clinical Trial Ceftriaxone in Subjects with ALS. Available online: https://ClinicalTrials.gov/show/NCT00349622 (accessed on 4 March 2023).
- Compassionate Use of Ceftriaxone in Patients with Amyotrophic Lateral Sclerosis (ALS). Available online: https://ClinicalTrials.gov/show/NCT00718393 (accessed on 4 March 2023).
- A Glutamate Transporter GLT1, in the Treatment of Bipolar Disorder. Available online: https://ClinicalTrials.gov/show/NCT00512616 (accessed on 4 March 2023).
- A Placebo-controlled Efficacy Study of IV Ceftriaxone for Refractory Psychosis. Available online: https://ClinicalTrials.gov/show/NCT00591318 (accessed on 4 March 2023).
- Shalviri, G.; Yousefian, S.; Gholami, K. Adverse events induced by ceftriaxone: A 10-year review of reported cases to Iranian Pharmacovigilance Centre. J. Clin. Pharm. Ther. 2011, 37, 448–451. [Google Scholar] [CrossRef]
- Gorska, M.; Popowska, U.; Sielicka-Dudzin, A.; Kuban-Jankowska, A.; Sawczuk, W.; Knap, N.; Cicero, G.; Wozniak, F. Geldanamycin and its derivatives as Hsp90 inhibitors. Front. Biosci. 2012, 17, 2269–2277. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Paul, S.; Mahanta, S. Association of heat-shock proteins in various neurodegenerative disorders: Is it a master key to open the therapeutic door? Mol. Cell Biochem. 2014, 386, 45–61. [Google Scholar] [CrossRef]
- Shimomura, A.; Yamamoto, N.; Kondo, S.; Fujiwara, Y.; Suzuki, S.; Yanagitani, N.; Horiike, A.; Kitazono, S.; Ohyanagi, F.; Doi, T.; et al. First-in-Human Phase I Study of an Oral HSP90 Inhibitor, TAS-116, in Patients with Advanced Solid Tumors. Mol. Cancer Ther. 2019, 18, 531–540. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Hamill, R.J. Amphotericin B Formulations: A Comparative Review of Efficacy and Toxicity. Drugs 2013, 73, 919–934. [Google Scholar] [CrossRef] [PubMed]
- Mangé, A.; Nishida, N.; Milhavet, O.; McMahon, H.E.M.; Casanova, D.; Lehmann, S. Amphotericin B Inhibits the Generation of the Scrapie Isoform of the Prion Protein in Infected Cultures. J. Virol. 2000, 74, 3135–3140. [Google Scholar] [CrossRef] [Green Version]
- Laniado-Laborín, R.; Cabrales-Vargas, M.N. Amphotericin B: Side effects and toxicity. Rev. Iberoam. Micol. 2009, 26, 223–227. [Google Scholar] [CrossRef]
- Stockwell, B.R.; Angeli, J.P.F.; Bayir, H.; Bush, A.I.; Conrad, M.; Dixon, S.J.; Fulda, S.; Gascón, S.; Hatzios, S.K.; Kagan, V.E.; et al. Ferroptosis: A Regulated Cell Death Nexus Linking Metabolism, Redox Biology, and Disease. Cell 2017, 171, 273–285. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Ke, Y.; Qian, Z.M. Brain iron metabolism: Neurobiology and neurochemistry. Prog. Neurobiol. 2007, 83, 149–173. [Google Scholar] [CrossRef]
- Li, C.; Wu, Z.; Xue, H.; Gao, Q.; Zhang, Y.; Wang, C.; Zhao, P. Ferroptosis contributes to hypoxic-ischemic brain injury in neonatal rats: Role of the SIRT1/Nrf2/GPx4 signaling pathway. CNS Neurosci. Ther. 2022, 28, 2268–2280. [Google Scholar] [CrossRef] [PubMed]
- Corti, O.; Blomgren, K.; Poletti, A.; Beart, P.M. Autophagy in neurodegeneration: New insights underpinning therapy for neurological diseases. J. Neurochem. 2020, 154, 354–371. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Grenier, D.; Huot, M.P.; Mayrand, D. Iron-chelating activity of tetracyclines and its impact on the susceptibility of Actinobacillus actinomycetemcomitans to these antibiotics. Antimicrob. Agents Chemother. 2000, 44, 763–766. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Zhao, F.; Xi, G.; Liu, W.; Keep, R.F.; Hua, Y. Minocycline Attenuates Iron-Induced Brain Injury. Acta Neurochir. Suppl. 2016, 121, 361–365. [Google Scholar] [CrossRef] [PubMed]
- Leyden, J.J. Absorption of minocycline hydrochloride and tetracycline hydrochloride: Effect of food, milk, and iron. J. Am. Acad. Dermatol. 1985, 12, 308–312. [Google Scholar] [CrossRef] [PubMed]
- Geria, A.N.; Tajirian, A.L.; Kihiczak, G.; Schwartz, R.A. Minocycline-induced skin pigmentation: An update. Acta Dermatovenerol. Croat. 2009, 17, 123–126. [Google Scholar]
- Chen-Roetling, J.; Chen, L.; Regan, R.F. Minocycline attenuates iron neurotoxicity in cortical cell cultures. Biochem. Biophys. Res. Commun. 2009, 386, 322–326. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Regan, R.F.; Chen, M.; Li, Z.; Zhang, X.; Benvenisti-Zarom, L.; Chen-Roetling, J. Neurons lacking iron regulatory protein-2 are highly resistant to the toxicity of hemoglobin. Neurobiol. Dis. 2008, 31, 242–249. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Barza, M.; Brown, R.B.; Shanks, C.; Gamble, C.; Weinstein, L. Relation between Lipophilicity and Pharmacological Behavior of Minocycline, Doxycycline, Tetracycline, and Oxytetracycline in Dogs. Antimicrob. Agents Chemother. 1975, 8, 713–720. [Google Scholar] [CrossRef] [Green Version]
- Leng, F.; Edison, P. Neuroinflammation and microglial activation in Alzheimer disease: Where do we go from here? Nat. Rev. Neurol. 2021, 17, 157–172. [Google Scholar] [CrossRef]
- Kolmychkova, K.I.; Zhelankin, A.V.; Karagodin, V.P.; Orekhov, A.N. Mitochondria and inflammation. Zhurnal Patol. Fiziol. Eksperimentalnaia Ter. 2016, 60, 114–121. [Google Scholar]
- Banoth, B.; Cassel, S.L. Mitochondria in innate immune signaling. Transl. Res. 2018, 202, 52–68. [Google Scholar] [CrossRef] [PubMed]
- Acuña, L.; Hamadat, S.; Corbalán, N.S.; González-Lizárraga, F.; dos-Santos-Pereira, M.; Rocca, J.; Sepúlveda Díaz, J.; Del-Bel, E.; Papy-García, D.; Chehín, R.N.; et al. Rifampicin and Its Derivative Rifampicin Quinone Reduce Microglial Inflammatory Responses and Neuro-degeneration Induced In Vitro by alpha-Synuclein Fibrillary Aggregates. Cells 2019, 8, 776. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Cankaya, S.; Cankaya, B.; Kilic, U.; Kilic, E.; Yulug, B. The therapeutic role of minocycline in Parkinson’s disease. Drugs Context 2019, 8, 212553. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Zusso, M.; Lunardi, V.; Franceschini, D.; Pagetta, A.; Lo, R.; Stifani, S.; Frigo, A.C.; Giusti, P.; Moro, S. Ciprofloxacin and levofloxacin attenuate microglia inflammatory response via TLR4/NF-kB pathway. J. Neuroinflammation 2019, 16, 148. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Khanna, D.; Kalra, S. Minocycline and Doxycycline: More Than Antibiotics. Curr. Mol. Pharmacol. 2021, 14, 1046–1065. [Google Scholar] [CrossRef]
- Karachitos, A.; Solis Garcia del Pozo, J.; WJ de Groot, P.; Kmita, H.; Jordán, J. Minocycline mediated mitochondrial cytoprotection: Premises for therapy of cerebrovascular and neuro-degenerative diseases. Curr. Drug Targets 2013, 14, 47–55. [Google Scholar] [CrossRef]
- Kim, H.-S.; Suh, Y.-H. Minocycline and neurodegenerative diseases. Behav. Brain Res. 2009, 196, 168–179. [Google Scholar] [CrossRef] [PubMed]
- Li, C.; Yuan, K.; Schluesener, H. Impact of minocycline on neurodegenerative diseases in rodents: A meta-analysis. Rev. Neurosci. 2013, 24, 553–562. [Google Scholar] [CrossRef]
- Romero-Miguel, D.; Lamanna-Rama, N.; Casquero-Veiga, M.; Gómez-Rangel, V.; Desco, M.; Soto-Montenegro, M.L. Minocycline in neurodegenerative and psychiatric diseases: An update. Eur. J. Neurol. 2020, 28, 1056–1081. [Google Scholar] [CrossRef]
- Brogden, R.N.; Speight, T.M.; Avery, G.S. Minocycline: A review of its antibacterial and pharmacokinetic properties and therapeutic use. Drugs 1975, 9, 251–291. [Google Scholar] [CrossRef]
- Paldino, E.; Balducci, C.; La Vitola, P.; Artioli, L.; D’Angelo, V.; Giampà, C.; Artuso, V.; Forloni, G.; Fusco, F.R. Neuroprotective Effects of Doxycycline in the R6/2 Mouse Model of Huntington’s Disease. Mol. Neurobiol. 2020, 57, 1889–1903. [Google Scholar] [CrossRef] [Green Version]
- Garrido-Mesa, N.; Zarzuelo, A.; Galvez, J. Minocycline: Far beyond an antibiotic. Br. J. Pharmacol. 2013, 169, 337–352. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Keller, A.F.; Gravel, M.; Kriz, J. Treatment with minocycline after disease onset alters astrocyte reactivity and increases mi-crogliosis in SOD1 mutant mice. Exp. Neurol. 2011, 228, 69–79. [Google Scholar] [CrossRef] [PubMed]
- Beers, D.R.; Henkel, J.S.; Zhao, W.; Wang, J.; Huang, A.; Wen, S.; Liao, B.; Appel, S.H. Endogenous regulatory T lymphocytes ameliorate amyotrophic lateral sclerosis in mice and correlate with disease progression in patients with amyotrophic lateral sclerosis. Brain 2011, 134, 1293–1314. [Google Scholar] [CrossRef] [PubMed]
- Kigerl, K.A.; Gensel, J.C.; Ankeny, D.P.; Alexander, J.K.; Donnelly, D.J.; Popovich, P.G. Identification of two distinct macrophage subsets with divergent effects causing either neurotoxicity or regen-eration in the injured mouse spinal cord. J. Neurosci. 2009, 29, 13435–13444. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Liang, Y.; Zhou, T.; Chen, Y.; Lin, D.; Jing, X.; Peng, S.; Zheng, D.; Zeng, Z.; Lei, M.; Wu, X.; et al. Rifampicin inhibits rotenone-induced microglial inflammation via enhancement of autophagy. Neurotoxicology 2017, 63, 137–145. [Google Scholar] [CrossRef]
- Nelson, P.T.; Braak, H.; Markesbery, W.R. Neuropathology and Cognitive Impairment in Alzheimer Disease: A Complex but Coherent Relationship. J. Neuropathol. Exp. Neurol. 2009, 68, 1–14. [Google Scholar] [CrossRef] [PubMed]
- Alexiou, A.; Kamal, M.A.; Ashraf, G.M. Editorial: The Alzheimer’s Disease Challenge. Front. Neurosci. 2019, 13, 768. [Google Scholar] [CrossRef] [Green Version]
- Martins, R.N.; Villemagne, V.; Sohrabi, H.R.; Chatterjee, P.; Shah, T.M.; Verdile, G.; Fraser, P.; Taddei, K.; Gupta, V.B.; Rainey-Smith, S.R.; et al. Alzheimer’s Disease: A Journey from Amyloid Peptides and Oxidative Stress, to Biomarker Technologies and Disease Prevention Strategies-Gains from AIBL and DIAN Cohort Studies. J. Alzheimers. Dis. 2018, 62, 965–992. [Google Scholar] [CrossRef] [Green Version]
- Morris, G.P.; Clark, I.A.; Vissel, B. Questions concerning the role of amyloid-beta in the definition, aetiology and diagnosis of Alzheimer’s disease. Acta Neuropathol. 2018, 136, 663–689. [Google Scholar] [CrossRef] [Green Version]
- Swerdlow, R.H. Mitochondria and Mitochondrial Cascades in Alzheimer’s Disease. J. Alzheimer’s Dis. 2018, 62, 1403–1416. [Google Scholar] [CrossRef] [Green Version]
- Anandatheerthavarada, H.K.; Biswas, G.; Robin, M.A.; Avadhani, N.G. Mitochondrial targeting and a novel transmembrane arrest of Alzheimer’s amyloid precursor protein impairs mitochondrial function in neuronal cells. J. Cell Biol. 2003, 161, 41–54. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Pagani, L.; Eckert, A. Amyloid-Beta Interaction with Mitochondria. Int. J. Alzheimer’s Dis. 2011, 2011, 925050. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Anandatheerthavarada, H.K.; Devi, L. Amyloid precursor protein and mitochondrial dysfunction in Alzheimer’s disease. Neuroscientist 2007, 13, 626–638. [Google Scholar] [CrossRef]
- Macdonald, R.; Barnes, K.; Hastings, C.; Mortiboys, H. Mitochondrial abnormalities in Parkinson’s disease and Alzheimer’s disease: Can mitochondria be targeted therapeutically? Biochem. Soc. Trans. 2018, 46, 891–909. [Google Scholar] [CrossRef]
- Mathew, A.; Lindsley, T.A.; Sheridan, A.; Bhoiwala, D.L.; Hushmendy, S.F.; Yager, E.J.; Ruggiero, E.A.; Crawford, D.R. Degraded Mitochondrial DNA is a Newly Identified Subtype of the Damage Associated Molecular Pattern (DAMP) Family and Possible Trigger of Neurodegeneration. J. Alzheimer’s Dis. 2012, 30, 617–627. [Google Scholar] [CrossRef]
- Abramova, N.E.; Davies, K.J.; Crawford, D.R. Polynucleotide degradation during early stage response to oxidative stress is specific to mitochondria. Free. Radic. Biol. Med. 2000, 28, 281–288. [Google Scholar] [CrossRef]
- Fauconnier, J. Mitochondria and proteostasis: It’s a kind of MAGIC. Cardiovasc. Res. 2018, 114, e68–e69. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Cummings, J.L.; Tong, G.; Ballard, C. Treatment Combinations for Alzheimer’s Disease: Current and Future Pharmacotherapy Options. J. Alzheimer’s Dis. 2019, 67, 779–794. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Airoldi, C.; Colombo, L.; Manzoni, C.; Sironi, E.; Natalello, A.; Doglia, S.M.; Forloni, G.; Tagliavini, F.; Del Favero, E.; Cantu, L.; et al. Tetracycline prevents Abeta oligomer toxicity through an atypical supramolecular interaction. Org. Biomol. Chem. 2011, 9, 463–472. [Google Scholar] [CrossRef] [PubMed]
- De Luigi, A.; Colombo, L.; Diomede, L.; Capobianco, R.; Mangieri, M.; Miccolo, C.; Limido, L.; Forloni, G.; Tagliavini, F.; Salmona, M. The Efficacy of Tetracyclines in Peripheral and Intracerebral Prion Infection. PLoS ONE 2008, 3, e1888. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Diomede, L.; Cassata, G.; Fiordaliso, F.; Salio, M.; Ami, D.; Natalello, A.; Doglia, S.M.; De Luigi, A.; Salmona, M. Tetracycline and its analogues protect Caenorhabditis elegans from beta amyloid-induced toxicity by targeting oligomers. Neurobiol. Dis. 2010, 40, 424–431. [Google Scholar] [CrossRef] [PubMed]
- Feng, B.Y.; Toyama, B.H.; Wille, H.; Colby, D.W.; Collins, S.R.; May, B.C.H.; Prusiner, S.B.; Weissman, J.; Shoichet, B.K. Small-molecule aggregates inhibit amyloid polymerization. Nat. Chem. Biol. 2008, 4, 197–199. [Google Scholar] [CrossRef] [Green Version]
- Loeb, M.B.; Molloy, D.W.; Smieja, M.; Standish, T.; Goldsmith, C.H.; Mahony, J.; Smith, S.; Borrie, M.; Decoteau, E.; Davidson, W.; et al. A randomized, controlled trial of doxycycline and rifampin for patients with Alzheimer’s disease. J. Am. Geriatr. Soc. 2004, 52, 381–387. [Google Scholar] [CrossRef]
- Molloy, D.W.; Standish, T.I.; Zhou, Q.; Guyatt, G.; DARAD Study Group. A multicenter, blinded, randomized, factorial controlled trial of doxycycline and rifampin for treatment of Alzheimer’s disease: The DARAD trial. Int. J. Geriatr. Psychiatry 2013, 28, 463–470. [Google Scholar] [CrossRef]
- Hunter, C.L.; Bachman, D.; Granholm, A.C. Minocycline prevents cholinergic loss in a mouse model of Down’s syndrome. Ann. Neurol. 2004, 56, 675–688. [Google Scholar] [CrossRef] [PubMed]
- Seabrook, T.J.; Jiang, L.; Maier, M.; Lemere, C.A. Minocycline affects microglia activation, Aβ deposition, and behavior in APP-tg mice. Glia 2006, 53, 776–782. [Google Scholar] [CrossRef]
- Cuello, A.C.; Ferretti, M.T.; Leon, W.C.; Iulita, M.F.; Melis, T.; Ducatenzeiler, A.; Bruno, M.A.; Canneva, F. Early-stage inflammation and experimental therapy in transgenic models of the Alzheimer-like amyloid pathology. Neurodegener. Dis. 2010, 7, 96–98. [Google Scholar] [CrossRef] [PubMed]
- Howard, R.; Zubko, O.; Gray, R.; Bradley, R.; Harper, E.; Kelly, L.; Pank, L.; O’Brien, J.; Fox, C.; Tabet, N.; et al. Minocycline 200 mg or 400 mg versus placebo for mild Alzheimer’s disease: The MADE Phase II, three-arm RCT. Effic. Mech. Eval. 2020, 7, 1–45. [Google Scholar] [CrossRef] [Green Version]
- Raza, C.; Anjum, R.; Shakeel, N.U.A. Parkinson’s disease: Mechanisms, translational models and management strategies. Life Sci. 2019, 226, 77–90. [Google Scholar] [CrossRef]
- Grünewald, A.; Kumar, K.R.; Sue, C.M. New insights into the complex role of mitochondria in Parkinson’s disease. Prog. Neurobiol. 2018, 177, 73–93. [Google Scholar] [CrossRef] [PubMed]
- Dominguez-Meijide, A.; Parrales, V.; Vasili, E.; González-Lizárraga, F.; König, A.; Lázaro, D.F.; Lannuzel, A.; Haik, S.; Del Bel, E.; Chehín, R.; et al. Doxycycline inhibits α-synuclein-associated pathologies in vitro and in vivo. Neurobiol. Dis. 2021, 151, 105256. [Google Scholar] [CrossRef] [PubMed]
- Volpicelli-Daley, L.A.; Luk, K.C.; Patel, T.P.; Tanik, S.A.; Riddle, D.M.; Stieber, A.; Meaney, D.F.; Trojanowski, J.Q.; Lee, V.M.-Y. Exogenous α-Synuclein Fibrils Induce Lewy Body Pathology Leading to Synaptic Dysfunction and Neuron Death. Neuron 2011, 72, 57–71. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Bortolanza, M.; Nascimento, G.C.; Socias, S.B.; Ploper, D.; Chehín, R.N.; Raisman-Vozari, R.; Del-Bel, E. Tetracycline repurposing in neurodegeneration: Focus on Parkinson’s disease. J. Neural Transm. 2018, 125, 1403–1415. [Google Scholar] [CrossRef] [PubMed]
- Codolo, G.; Plotegher, N.; Pozzobon, T.; Brucale, M.; Tessari, I.; Bubacco, L.; de Bernard, M. Triggering of inflammasome by aggregated alpha-synuclein, an inflammatory response in synucleinopathies. PLoS ONE 2013, 8, e55375. [Google Scholar] [CrossRef] [Green Version]
- Ludtmann, M.H.; Angelova, P.R.; Ninkina, N.N.; Gandhi, S.; Buchman, V.L.; Abramov, A.Y. Monomeric Alpha-Synuclein Exerts a Physiological Role on Brain ATP Synthase. J. Neurosci. 2016, 36, 10510–10521. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Lindström, V.; Gustafsson, G.; Sanders, L.H.; Howlett, E.H.; Sigvardson, J.; Kasrayan, A.; Ingelsson, M.; Bergström, J.; Erlandsson, A. Extensive uptake of α-synuclein oligomers in astrocytes results in sustained intracellular deposits and mitochondrial damage. Mol. Cell. Neurosci. 2017, 82, 143–156. [Google Scholar] [CrossRef]
- Liang, B.; Tamm, L.K. Solution NMR of SNAREs, complexin and alpha-synuclein in association with membrane-mimetics. Prog. Nucl. Magn. Reason. Spectrosc. 2018, 105, 41–53. [Google Scholar] [CrossRef]
- Yurtsever, İ.; Üstündağ, Ü.V.; Ünal, İ.; Ateş, P.S.; Emekli-Alturfan, E. Rifampicin decreases neuroinflammation to maintain mitochondrial function and calcium homeostasis in rote-none-treated zebrafish. Drug Chem. Toxicol. 2022, 45, 1544–1551. [Google Scholar] [CrossRef]
- Kaur, B.; Prakash, A. Ceftriaxone attenuates glutamate-mediated neuro-inflammation and restores BDNF in MPTP model of Parkinson’s disease in rats. Pathophysiology 2017, 24, 71–79. [Google Scholar] [CrossRef]
- Han, J.; Kim, S.J.; Ryu, M.J.; Jang, Y.; Lee, M.J.; Ju, X.; Lee, Y.L.; Cui, J.; Shong, M.; Heo, J.Y.; et al. Chloramphenicol Mitigates Oxidative Stress by Inhibiting Translation of Mitochondrial Complex I in Dopaminergic Neurons of Toxin-Induced Parkinson’s Disease Model. Oxidative Med. Cell. Longev. 2019, 2019, 4174803. [Google Scholar] [CrossRef] [PubMed]
- Egeberg, A.; Hansen, P.R.; Gislason, G.H.; Thyssen, J.P. Exploring the Association between Rosacea and Parkinson Disease: A Danish Nationwide Cohort Study. JAMA Neurol. 2016, 73, 529–534. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Thomas, J.; Metheny, R.; Karakiozis, J.; Wetzel, J.; Crout, R. Long-Term Sub-Antimicrobial Doxycycline (Periostat®) as Adjunctive Management in Adult Periodontitis: Effects on Subgingival Bacterial Population Dynamics. Adv. Dent. Res. 1998, 12, 32–39. [Google Scholar] [CrossRef]
- Walker, C.; Preshaw, P.M.; Novak, J.; Hefti, A.F.; Bradshaw, M.; Powala, C. Long-term treatment with sub-antimicrobial dose doxycycline has no antibacterial effect on intestinal flora. J. Clin. Periodontol. 2005, 32, 1163–1169. [Google Scholar] [CrossRef]
- Gonzalez-Alcocer, A.; Gopar-Cuevas, Y.; Soto-Dominguez, A.; Arias, M.d.J.L.; Saucedo-Cardenas, O.; de Oca-Luna, R.M.; Rodriguez-Rocha, H.; Garcia-Garcia, A. Peripheral tissular analysis of rapamycin’s effect as a neuroprotective agent in vivo. Naunyn-Schmiedeberg’s Arch. Pharmacol. 2022, 395, 1239–1255. [Google Scholar] [CrossRef] [PubMed]
- Bi, W.; Zhu, L.; Jing, X.; Zeng, Z.; Liang, Y.; Xu, A.; Liu, J.; Xiao, S.; Yang, L.; Shi, Q.; et al. Rifampicin improves neuronal apoptosis in LPS-stimulated co-cultured BV2 cells through inhibition of the TLR-4 pathway. Mol. Med. Rep. 2014, 10, 1793–1799. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Weng, J.-C.; Tikhonova, M.A.; Chen, J.-H.; Shen, M.-S.; Meng, W.-Y.; Chang, Y.-T.; Chen, K.-H.; Liang, K.-C.; Hung, C.-S.; Amstislavskaya, T.G.; et al. Ceftriaxone prevents the neurodegeneration and decreased neurogenesis seen in a Parkinson’s disease rat model: An immunohistochemical and MRI study. Behav. Brain Res. 2016, 305, 126–139. [Google Scholar] [CrossRef]
- Huang, C.-K.; Chang, Y.-T.; Amstislavskaya, T.G.; Tikhonova, M.A.; Lin, C.-L.; Hung, C.-S.; Lai, T.-J.; Ho, Y.-J. Synergistic effects of ceftriaxone and erythropoietin on neuronal and behavioral deficits in an MPTP-induced animal model of Parkinson’s disease dementia. Behav. Brain Res. 2015, 294, 198–207. [Google Scholar] [CrossRef] [PubMed]
- Xiong, R.; Zhou, W.; Siegel, D.; Kitson, R.R.; Freed, C.R.; Moody, C.J.; Ross, D. A Novel Hsp90 Inhibitor Activates Compensatory Heat Shock Protein Responses and Autophagy and Alleviates Mutant A53T alpha-Synuclein Toxicity. Mol. Pharmacol. 2015, 88, 1045–1054. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Barini, E.; Miccoli, A.; Tinarelli, F.; Mulholland, K.; Kadri, H.; Khanim, F.; Stojanovski, L.; Read, K.D.; Burness, K.; Blow, J.J.; et al. The Anthelmintic Drug Niclosamide and Its Analogues Activate the Parkinson’s Disease Associated Protein Kinase PINK1. Chembiochem 2018, 19, 425–429. [Google Scholar] [CrossRef] [PubMed]
- Kadri, H.; Lambourne, O.A.; Mehellou, Y. Niclosamide, a Drug with Many (Re)purposes. Chemmedchem 2018, 13, 1088–1091. [Google Scholar] [CrossRef] [PubMed]
- Hurkacz, M.; Dobrek, L.; Wiela-Hojenska, A. Antibiotics and the Nervous System—Which Face of Antibiotic Therapy Is Real, Dr. Jekyll (Neurotoxicity) or Mr. Hyde (Neuroprotection)? Molecules 2021, 26, 7456. [Google Scholar] [CrossRef] [PubMed]
- Wyant, K.J.; Ridder, A.J.; Dayalu, P. Huntington’s Disease-Update on Treatments. Curr. Neurol. Neurosci. Rep. 2017, 17, 33. [Google Scholar] [CrossRef] [PubMed]
- A novel gene containing a trinucleotide repeat that is expanded and unstable on Huntington’s disease chromosomes. The Hunting-ton’s Disease Collaborative Research Group. Cell 1993, 72, 971–983. [CrossRef]
- Saudou, F.; Humbert, S. The Biology of Huntingtin. Neuron 2016, 89, 910–926. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Cornett, J.; Cao, F.; Wang, C.-E.; Ross, C.A.; Bates, G.P.; Li, S.-H.; Li, X.-J. Polyglutamine expansion of huntingtin impairs its nuclear export. Nat. Genet. 2005, 37, 198–204. [Google Scholar] [CrossRef]
- Godin, J.D.; Colombo, K.; Molina-Calavita, M.; Keryer, G.; Zala, D.; Charrin, B.C.; Dietrich, P.; Volvert, M.-L.; Guillemot, F.; Dragatsis, I.; et al. Huntingtin Is Required for Mitotic Spindle Orientation and Mammalian Neurogenesis. Neuron 2010, 67, 392–406. [Google Scholar] [CrossRef] [Green Version]
- McKinstry, S.U.; Karadeniz, Y.B.; Worthington, A.K.; Hayrapetyan, V.Y.; Ozlu, M.I.; Serafin-Molina, K.; Risher, W.C.; Ustunkaya, T.; Dragatsis, I.; Zeitlin, S.; et al. Huntingtin Is Required for Normal Excitatory Synapse Development in Cortical and Striatal Circuits. J. Neurosci. 2014, 34, 9455–9472. [Google Scholar] [CrossRef] [Green Version]
- Hickman, S.; Izzy, S.; Sen, P.; Morsett, L.; El Khoury, J. Microglia in neurodegeneration. Nat. Neurosci. 2018, 21, 1359–1369. [Google Scholar] [CrossRef]
- Block, M.; Hong, J.-S. Chronic microglial activation and progressive dopaminergic neurotoxicity. Biochem. Soc. Trans. 2007, 35, 1127–1132. [Google Scholar] [CrossRef] [Green Version]
- Dik, B.; Coskun, D.; Bahcivan, E.; Er, A. Doxycycline and meloxicam can treat neuroinflammation by increasing activity of antioxidant enzymes in rat brain. Pak. J. Pharm. Sci. 2019, 32, 391–396. [Google Scholar]
- Forloni, G.; Colombo, L.; Girola, L.; Tagliavini, F.; Salmona, M. Anti-amyloidogenic activity of tetracyclines: Studies in vitro. FEBS Lett. 2001, 487, 404–407. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Brundula, V.; Rewcastle, N.B.; Metz, L.M.; Bernard, C.C.; Yong, V.W. Targeting leukocyte MMPs and transmigration: Minocycline as a potential therapy for multiple sclerosis. Brain 2002, 125, 1297–1308. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Shirendeb, U.; Reddy, A.P.; Manczak, M.; Calkins, M.J.; Mao, P.; Tagle, D.A.; Hemachandra Reddy, P. Abnormal mitochondrial dynamics, mitochondrial loss and mutant huntingtin oligomers in Huntington’s disease: Implications for selective neuronal damage. Hum. Mol. Genet. 2011, 20, 1438–1455. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Song, W.; Chen, J.; Petrilli, A.; Liot, G.; Klinglmayr, E.; Zhou, Y.; Poquiz, P.; Tjong, J.; Pouladi, M.A.; Hayden, M.R.; et al. Mutant huntingtin binds the mitochondrial fission GTPase dynamin-related protein-1 and increases its enzymatic activity. Nat. Med. 2011, 17, 377–382. [Google Scholar] [CrossRef] [Green Version]
- Chang, C.-R.; Blackstone, C. Dynamic regulation of mitochondrial fission through modification of the dynamin-related protein Drp1. Ann. N. Y. Acad. Sci. 2010, 1201, 34–39. [Google Scholar] [CrossRef] [Green Version]
- Suárez-Rivero, J.M.; Villanueva-Paz, M.; de la Cruz-Ojeda, P.; de la Mata, M.; Cotán, D.; Oropesa-Ávila, M.; de Lavera, I.; Álvarez-Córdoba, M.; Luzón-Hidalgo, R.; Sánchez-Alcázar, J.A. Mitochondrial Dynamics in Mitochondrial Diseases. Diseases 2016, 5, 1. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Jahani-Asl, A.; Cheung, E.C.C.; Neuspiel, M.; MacLaurin, J.G.; Fortin, A.; Park, D.S.; McBride, H.M.; Slack, R.S. Mitofusin 2 Protects Cerebellar Granule Neurons against Injury-induced Cell Death. J. Biol. Chem. 2007, 282, 23788–23798. [Google Scholar] [CrossRef] [Green Version]
- Khalil, B.; El Fissi, N.; Aouane, A.; Cabirol-Pol, M.-J.; Rival, T.; Liévens, J.-C. PINK1-induced mitophagy promotes neuroprotection in Huntington’s disease. Cell Death Dis. 2015, 6, e1617. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Martin, L.J. Biology of Mitochondria in Neurodegenerative Diseases. Prog. Mol. Biol. Transl. Sci. 2012, 107, 355–415. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Cui, L.; Jeong, H.; Borovecki, F.; Parkhurst, C.N.; Tanese, N. Transcriptional repression of PGC-1alpha by mutant huntingtin leads to mitochondrial dysfunction and neuro-degeneration. Cell 2006, 127, 59–69. [Google Scholar] [CrossRef] [Green Version]
- Villena, J.A. New insights into PGC-1 coactivators: Redefining their role in the regulation of mitochondrial function and beyond. FEBS J. 2015, 282, 647–672. [Google Scholar] [CrossRef] [PubMed]
- Herben-Dekker, M.; Van Oostrom, J.C.H.; Roos, R.A.C.; Jurgens, C.K.; Witjes-Ané, M.-N.W.; Kremer, H.P.H.; Leenders, K.L.; Spikman, J.M. Striatal metabolism and psychomotor speed as predictors of motor onset in Huntington’s disease. J. Neurol. 2014, 261, 1387–1397. [Google Scholar] [CrossRef] [PubMed]
- Jiang, H.; Nucifora Jr, F.C.; Ross, C.A.; DeFranco, D.B. Cell death triggered by polyglutamine-expanded huntingtin in a neuronal cell line is associated with degradation of CREB-binding protein. Hum. Mol. Genet. 2003, 12, 1–12. [Google Scholar] [CrossRef]
- Luthi-Carter, R.; Strand, A.; Peters, N.L.; Solano, S.M.; Hollingsworth, Z.R.; Menon, A.S.; Frey, A.S.; Spektor, B.S.; Penney, E.B.; Schilling, G.; et al. Decreased expression of striatal signaling genes in a mouse model of Huntington’s disease. Hum. Mol. Genet. 2000, 9, 1259–1271. [Google Scholar] [CrossRef] [Green Version]
- Giampa, C.; DeMarch, Z.; D’Angelo, V.; Morello, M.; Martorana, A.; Sancesario, G.; Bernardi, G.; Fusco, F.R. Striatal modulation of cAMP-response-element-binding protein (CREB) after excitotoxic lesions: Implications with neuronal vulnerability in Huntington’s disease. Eur. J. Neurosci. 2006, 23, 11–20. [Google Scholar] [CrossRef]
- Suárez-Rivero, J.M.; Pastor-Maldonado, C.J.; Povea-Cabello, S.; Álvarez-Córdoba, M.; Villalón-García, I.; Talaverón-Rey, M.; Suárez-Carrillo, A.; Munuera-Cabeza, M.; Reche-López, D.; Cilleros-Holgado, P.; et al. Activation of the Mitochondrial Unfolded Protein Response: A New Therapeutic Target? Biomedicines 2022, 10, 1611. [Google Scholar] [CrossRef] [PubMed]
- Zhang, X.; Odom, D.T.; Koo, S.H.; Conkright, M.D.; Canettieri, G.; Best, J.; Chen, H.; Jenner, R.; Herbolsheimer, E.; Jacobsen, E.; et al. Genome-wide analysis of cAMP-response element binding protein occupancy, phosphorylation, and target gene activation in human tissues. Proc. Natl. Acad. Sci. USA 2005, 102, 4459–4464. [Google Scholar] [CrossRef] [Green Version]
- Mantamadiotis, T.; Lemberger, T.; Bleckmann, S.C.; Kern, H.; Kretz, O.; Martin-Villalba, A.; Tronche, F.; Kellendonk, C.; Gau, D.; Kapfhammer, J.P.; et al. Disruption of CREB function in brain leads to neurodegeneration. Nat. Genet. 2002, 31, 47–54. [Google Scholar] [CrossRef]
- Díaz-Ruiz, C.; Parlato, R.; Aguado, F.; Ureña, J.M.; Burgaya, F.; Martínez, A.; Carmona, M.A.; Kreiner, G.; Bleckmann, S.; del Río, J.A.; et al. Regulation of neural migration by the CREB/CREM transcription factors and altered Dab1 levels in CREB/CREM mutants. Mol. Cell Neurosci. 2008, 39, 519–528. [Google Scholar] [CrossRef]
- Aguado, F.; Díaz-Ruiz, C.; Parlato, R.; Martínez, A.; Carmona, M.A.; Bleckmann, S.; Ureña, J.M.; Burgaya, F.; Jose, A.; Schütz, G.; et al. The CREB/CREM transcription factors negatively regulate early synaptogenesis and spontaneous network activity. J. Neurosci. 2009, 29, 328–333. [Google Scholar] [CrossRef] [Green Version]
- Bourtchuladze, R.; Frenguelli, B.; Blendy, J.; Cioffi, D.; Schutz, G.; Silva, A.J. Deficient long-term memory in mice with a targeted mutation of the cAMP-responsive element-binding protein. Cell 1994, 79, 59–68. [Google Scholar] [CrossRef]
- Zuccato, C.; Ciammola, A.; Rigamonti, D.; Leavitt, B.R.; Goffredo, D.; Conti, L.; MacDonald, M.E.; Friedlander, R.M.; Silani, V.; Hayden, M.R.; et al. Loss of huntingtin-mediated BDNF gene transcription in Huntington’s disease. Science 2001, 293, 493–498. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Dan, H.C.; Cooper, M.J.; Cogswell, P.C.; Duncan, J.A.; Ting JP, Y.; Baldwin, A.S. Akt-dependent regulation of NF-κB is controlled by mTOR and Raptor in association with IKK. Genes Dev. 2008, 22, 1490–1500. [Google Scholar] [CrossRef] [Green Version]
- Zeng, X.; Mao, X.; Huang, Z.; Wang, F.; Wu, G.; Qiao, S. Arginine enhances embryo implantation in rats through PI3K/PKB/mTOR/NO signaling pathway during early pregnancy. Reproduction 2013, 145, 1–7. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Thomas, M.; Ashizawa, T.; Jankovic, J. Minocycline in Huntington’s disease: A pilot study. Mov. Disord. 2004, 19, 692–695. [Google Scholar] [CrossRef]
- Cudkowicz, M.; Huntington Study Group DOMINO Investigators. A futility study of minocycline in Huntington’s disease. Mov. Disord. 2010, 25, 2219–2224. [Google Scholar] [CrossRef] [PubMed]
- Zhang, W.; Narayanan, M.; Friedlander, R.M. Additive neuroprotective effects of minocycline with creatine in a mouse model of ALS. Ann. Neurol. 2003, 53, 267–270. [Google Scholar] [CrossRef] [PubMed]
- Hulisz, D. Amyotrophic lateral sclerosis: Disease state overview. Am. J. Manag. Care 2018, 24, S320–S326. [Google Scholar] [PubMed]
- Rumfeldt, J.A.; Lepock, J.R.; Meiering, E.M. Unfolding and Folding Kinetics of Amyotrophic Lateral Sclerosis-Associated Mutant Cu, Zn Superoxide Dismutases. J. Mol. Biol. 2009, 385, 278–298. [Google Scholar] [CrossRef] [PubMed]
- Nandi, D.; Tahiliani, P.; Kumar, A.; Chandu, D. The ubiquitin-proteasome system. J. Biosci. 2006, 31, 137–155. [Google Scholar] [CrossRef] [PubMed]
- Evans, C.S.; Holzbaur, E.L. Autophagy and mitophagy in ALS. Neurobiol. Dis. 2018, 122, 35–40. [Google Scholar] [CrossRef]
- Rubinsztein, D.C. The roles of intracellular protein-degradation pathways in neurodegeneration. Nature 2006, 443, 780–786. [Google Scholar] [CrossRef]
- Taylor, J.P.; Brown, R.H., Jr.; Cleveland, D.W. Decoding ALS: From genes to mechanism. Nature 2016, 539, 197–206. [Google Scholar] [CrossRef] [Green Version]
- Chua, J.P.; De Calbiac, H.; Kabashi, E.; Barmada, S.J. Autophagy and ALS: Mechanistic insights and therapeutic implications. Autophagy 2021, 18, 254–282. [Google Scholar] [CrossRef] [PubMed]
- Yang, C.; Zhang, X. Research progress on vesicular trafficking in amyotrophic lateral sclerosis. J. Zhejiang Univ. (Medical Sci.) 2022, 51, 380–387. [Google Scholar] [CrossRef] [PubMed]
- Moore, A.S.; Holzbaur, E.L. Dynamic recruitment and activation of ALS-associated TBK1 with its target optineurin are re-quired for efficient mitophagy. Proc. Natl. Acad. Sci. USA 2016, 113, E3349–E3358. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Hara, T.; Nakamura, K.; Matsui, M.; Yamamoto, A.; Nakahara, Y.; Suzuki-Migishima, R.; Yokoyama, M.; Mishima, K.; Saito, I.; Okano, H.; et al. Suppression of basal autophagy in neural cells causes neurodegenerative disease in mice. Nature 2006, 441, 885–889. [Google Scholar] [CrossRef] [PubMed]
- Zhang, X.; Li, L.; Chen, S.; Yang, D.; Wang, Y.; Zhang, X.; Wang, Z.; Le, W. Rapamycin treatment augments motor neuron degeneration in SOD1(G93A) mouse model of amyotrophic lateral sclerosis. Autophagy 2011, 7, 412–425. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Castillo, K.; Nassif, M.; Valenzuela, V.; Rojas, F.; Matus, S.; Mercado, G.; Court, F.A.; Van Zundert, B.; Hetz, C. Trehalose delays the progression of amyotrophic lateral sclerosis by enhancing autophagy in motoneurons. Autophagy 2013, 9, 1308–1320. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Sun, J.; Zhan, Y.; Mariosa, D.; Larsson, H.; Almqvist, C.; Ingre, C.; Zagai, U.; Pawitan, Y.; Fang, F. Antibiotics use and risk of amyotrophic lateral sclerosis in Sweden. Eur. J. Neurol. 2019, 26, 1355–1361. [Google Scholar] [CrossRef] [PubMed]
- Prusiner, S. Novel proteinaceous infectious particles cause scrapie. Science 1982, 216, 136–144. [Google Scholar] [CrossRef] [Green Version]
- Prusiner, S.B.; Scott, M.R.; DeArmond, S.J.; Cohen, F.E. Prion Protein Biology. Cell 1998, 93, 337–348. [Google Scholar] [CrossRef] [Green Version]
- DeArmond, S.J.; Prusiner, S.B. Etiology and pathogenesis of prion diseases. Am. J. Pathol. 1995, 146, 785–811. [Google Scholar] [PubMed]
- Takada, L.T.; Geschwind, M.D. Prion diseases. Semin. Neurol. 2013, 33, 348–356. [Google Scholar] [CrossRef] [Green Version]
- Park, J.H.; Kim, B.H.; Park, S.J.; Jin, J.K.; Jeon, Y.C.; Wen, G.Y.; Shin, H.Y.; Carp, R.I.; Kim, Y.S. Association of endothelial nitric oxide synthase and mitochondrial dysfunction in the hippocampus of scrapie-infected mice. Hippocampus 2011, 21, 319–333. [Google Scholar] [CrossRef] [PubMed]
- Moon, J.-H.; Park, S.-Y. Prion peptide-mediated calcium level alteration governs neuronal cell damage through AMPK-autophagy flux. Cell Commun. Signal. 2020, 18, 109. [Google Scholar] [CrossRef] [PubMed]
- Ferreiro, E.; Oliveira, C.R.; Pereira, C.M. The release of calcium from the endoplasmic reticulum induced by amyloid-beta and prion peptides activates the mitochondrial apoptotic pathway. Neurobiol. Dis. 2008, 30, 331–342. [Google Scholar] [CrossRef] [Green Version]
- Choi, H.-S.; Choi, Y.-G.; Shin, H.-Y.; Oh, J.-M.; Park, J.-H.; Kim, J.-I.; Carp, R.I.; Choi, E.-K.; Kim, Y.-S. Dysfunction of mitochondrial dynamics in the brains of scrapie-infected mice. Biochem. Biophys. Res. Commun. 2014, 448, 157–162. [Google Scholar] [CrossRef] [PubMed]
- Yang, X.-D.; Shi, Q.; Sun, J.; Lv, Y.; Ma, Y.; Chen, C.; Xiao, K.; Zhou, W.; Dong, X.-P. Aberrant Alterations of Mitochondrial Factors Drp1 and Opa1 in the Brains of Scrapie Experiment Rodents. J. Mol. Neurosci. 2016, 61, 368–378. [Google Scholar] [CrossRef]
- Li, C.; Wang, D.; Wu, W.; Yang, W.; Shah, S.Z.A.; Zhao, Y.; Duan, Y.; Wang, L.; Zhou, X.; Zhao, D.; et al. DLP1-dependent mitochondrial fragmentation and redistribution mediate prion-associated mitochondrial dysfunction and neuronal death. Aging Cell 2017, 17, e12693. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Forloni, G.; Iussich, S.; Awan, T.; Colombo, L.; Angeretti, N.; Girola, L.; Bertani, I.; Poli, G.; Caramelli, M.; Bruzzone, M.G.; et al. Tetracyclines affect prion infectivity. Proc. Natl. Acad. Sci. USA 2002, 99, 10849–10854. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Hannaoui, S.; Gougerot, A.; Privat, N.; Levavasseur, E.; Bizat, N.; Hauw, J.-J.; Brandel, J.-P.; Haïk, S. Cycline Efficacy on the Propagation of Human Prions in Primary Cultured Neurons is Strain-Specific. J. Infect. Dis. 2013, 209, 1144–1148. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Haïk, S.; Marcon, G.; Mallet, A.; Tettamanti, M.; Welaratne, A.; Giaccone, G.; Azimi, S.; Pietrini, V.; Fabreguettes, J.-R.; Imperiale, D.; et al. Doxycycline in Creutzfeldt-Jakob disease: A phase 2, randomised, double-blind, placebo-controlled trial. Lancet Neurol. 2014, 13, 150–158. [Google Scholar] [CrossRef]
- Varges, D.; Manthey, H.; Heinemann, U.; Ponto, C.; Schmitz, M.; Schulz-Schaeffer, W.J.; Krasnianski, A.; Breithaupt, M.; Fincke, F.; Kramer, K.; et al. Doxycycline in early CJD: A double-blinded randomised phase II and observational study. J. Neurol. Neurosurg. Psychiatry 2016, 88, 119–125. [Google Scholar] [CrossRef] [Green Version]
- Beringue, V.; Adjou, K.T.; Lamoury, F.; Maignien, T.; Deslys, J.-P.; Race, R.; Dormont, D. Opposite Effects of Dextran Sulfate 500, the Polyene Antibiotic MS-8209, and Congo Red on Accumulation of the Protease-Resistant Isoform of PrP in the Spleens of Mice Inoculated Intraperitoneally with the Scrapie Agent. J. Virol. 2000, 74, 5432–5440. [Google Scholar] [CrossRef] [Green Version]
- Demaimay, R.; Race, R.; Chesebro, B. Effectiveness of polyene antibiotics in treatment of transmissible spongiform encepha-lopathy in transgenic mice expressing Syrian hamster PrP only in neurons. J. Virol. 1999, 73, 3511–3513. [Google Scholar] [CrossRef] [Green Version]
- Cortes, C.J.; Qin, K.; Cook, J.; Solanki, A.; Mastrianni, J.A. Rapamycin delays disease onset and prevents PrP plaque deposition in a mouse model of Gerstmann-Straussler-Scheinker disease. J. Neurosci. 2012, 32, 12396–12405. [Google Scholar] [CrossRef] [Green Version]
- Burtscher, J.; Romani, M.; Bernardo, G.; Popa, T.; Ziviani, E.; Hummel, F.C.; Sorrentino, V.; Millet, G.P. Boosting mitochondrial health to counteract neurodegeneration. Prog. Neurobiol. 2022, 215, 102289. [Google Scholar] [CrossRef]
- Gorman, G.S.; Chinnery, P.F.; DiMauro, S.; Hirano, M.; Koga, Y.; McFarland, R.; Suomalainen, A.; Thorburn, D.R.; Zeviani, M.; Turnbull, D.M. Mitochondrial diseases. Nat. Rev. Dis. Primers 2016, 2, 16080. [Google Scholar] [CrossRef]
- Zeviani, M.; Viscomi, C. Mitochondrial Neurodegeneration. Cells 2022, 11, 637. [Google Scholar] [CrossRef] [PubMed]
- Nelson, I.; Hanna, M.G.; Alsanjari, N.; Scaravilli, F.; Morgan-Hughes, J.A.; Harding, A.E. A new mitochondrial DNA mutation associated with progressive dementia and chorea: A clinical, pathological, and molecular genetic study. Ann. Neurol. 1995, 37, 400–403. [Google Scholar] [CrossRef] [PubMed]
- Chen, H.; Chan, D.C. Critical dependence of neurons on mitochondrial dynamics. Curr. Opin. Cell Biol. 2006, 18, 453–459. [Google Scholar] [CrossRef]
- Lake, N.J.; Bird, M.J.; Isohanni, P.; Paetau, A. Leigh syndrome: Neuropathology and pathogenesis. J. Neuropathol. Exp. Neurol. 2015, 74, 482–492. [Google Scholar] [CrossRef] [Green Version]
- Harding, B.N. Review Article: Progressive Neuronal Degeneration of Childhood with Liver Disease (Alpers-Huttenlocher Syndrome): A Personal Review. J. Child Neurol. 1990, 5, 273–287. [Google Scholar] [CrossRef]
- Gorman, G.S.; Schaefer, A.M.; Ng, Y.; Gomez, N.; Blakely, E.L.; Alston, C.L.; Feeney, C.; Horvath, R.; Yu-Wai-Man, P.; Chinnery, P.F.; et al. Prevalence of nuclear and mitochondrial DNA mutations related to adult mitochondrial disease. Ann. Neurol. 2015, 77, 753–759. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Russell, O.; Turnbull, D. Mitochondrial DNA disease—Molecular insights and potential routes to a cure. Exp. Cell Res. 2014, 325, 38–43. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Xiao, Y.; Xiong, T.; Meng, X.; Yu, D.; Xiao, Z.; Song, L. Different influences on mitochondrial function, oxidative stress and cytotoxicity of antibiotics on primary human neuron and cell lines. J. Biochem. Mol. Toxicol. 2018, 33, e22277. [Google Scholar] [CrossRef] [PubMed]
- Perry, E.A.; Bennett, C.F.; Luo, C.; Balsa, E.; Jedrychowski, M.; O’Malley, K.E.; Latorre-Muro, P.; Ladley, R.P.; Reda, K.; Wright, P.M.; et al. Tetracyclines promote survival and fitness in mitochondrial disease models. Nat. Metab. 2021, 3, 33–42. [Google Scholar] [CrossRef]
- Yun, J.; Finkel, T. Mitohormesis. Cell Metab. 2014, 19, 757–766. [Google Scholar] [CrossRef] [Green Version]
- Barcena, C.; Mayoral, P.; Quiros, P.M. Mitohormesis, an Antiaging Paradigm. Int. Rev. Cell Mol. Biol. 2018, 340, 35–77. [Google Scholar] [PubMed]
- Palmeira, C.M.; Teodoro, J.S.; Amorim, J.A.; Steegborn, C.; Sinclair, D.A.; Rolo, A.P. Mitohormesis and metabolic health: The interplay between ROS, cAMP and sirtuins. Free. Radic. Biol. Med. 2019, 141, 483–491. [Google Scholar] [CrossRef] [PubMed]
- Kenny, T.C.; Gomez, M.L.; Germain, D. Mitohormesis, UPRmt, and the Complexity of Mitochondrial DNA Landscapes in Cancer. Cancer Res. 2019, 79, 6057–6066. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Quirós, P.M.; Mottis, A.; Auwerx, J. Mitonuclear communication in homeostasis and stress. Nat. Rev. Mol. Cell Biol. 2016, 17, 213–226. [Google Scholar] [CrossRef] [PubMed]
- Ristow, M. Unraveling the Truth About Antioxidants: Mitohormesis explains ROS-induced health benefits. Nat. Med. 2014, 20, 709–711. [Google Scholar] [CrossRef] [PubMed]
- Suárez-Rivero, J.M.; Pastor-Maldonado, C.J.; Povea-Cabello, S.; Álvarez-Córdoba, M.; Villalón-García, I.; Talaverón-Rey, M.; Suárez-Carrillo, A.; Munuera-Cabeza, M.; Reche-López, D.; Cilleros-Holgado, P.; et al. UPRmt activation improves pathological alterations in cellular models of mitochondrial diseases. Orphanet J. Rare Dis. 2022, 17, 204. [Google Scholar] [CrossRef]
- Zhang, F.; Zhang, L.; Qi, Y.; Xu, H. Mitochondrial cAMP signaling. Cell. Mol. Life Sci. 2016, 73, 4577–4590. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Scarpulla, R.C.; Vega, R.B.; Kelly, D.P. Transcriptional integration of mitochondrial biogenesis. Trends Endocrinol. Metab. 2012, 23, 459–466. [Google Scholar] [CrossRef] [Green Version]
- Ventura-Clapier, R.; Garnier, A.; Veksler, V. Transcriptional control of mitochondrial biogenesis: The central role of PGC-1 alpha. Cardiovasc. Res. 2008, 79, 208–217. [Google Scholar] [CrossRef] [Green Version]
- Song, M.; Chen, Y.; Gong, G.; Murphy, E.; Rabinovitch, P.S.; Dorn, G.W. Super-Suppression of Mitochondrial Reactive Oxygen Species Signaling Impairs Compensatory Autophagy in Primary Mitophagic Cardiomyopathy. Circ. Res. 2014, 115, 348–353. [Google Scholar] [CrossRef] [Green Version]
- Dogan, S.A.; Cerutti, R.; Benincá, C.; Brea-Calvo, G.; Jacobs, H.T.; Zeviani, M.; Szibor, M.; Viscomi, C. Perturbed Redox Signaling Exacerbates a Mitochondrial Myopathy. Cell Metab. 2018, 28, 764–775.e5. [Google Scholar] [CrossRef] [Green Version]
- Cox, C.S.; McKay, S.E.; Holmbeck, M.A.; Christian, B.E.; Scortea, A.C.; Tsay, A.J.; Newman, L.E.; Shadel, G.S. Mitohormesis in Mice via Sustained Basal Activation of Mitochondrial and Antioxidant Signaling. Cell Metab. 2018, 28, 776–786.e5. [Google Scholar] [CrossRef] [Green Version]
- Lin, H.W.; Lee, R.H.C.; Lee, M.H.H.; Wu, C.Y.C.; Silva, A.C.; Possoit, H.E.; Hsieh, T.-H.; Minagar, A. Cerebral ischemia and neuroregeneration. Neural Regen. Res. 2018, 13, 373–385. [Google Scholar] [CrossRef]
- Hayashi, T.; Takada, K.; Matsuda, M. Post-transient ischemia increase in ubiquitin conjugates in the early reperfusion. Neuroreport 1992, 3, 519–520. [Google Scholar] [CrossRef] [PubMed]
- Kahl, A.; Blanco, I.; Jackman, K.; Baskar, J.; Milaganur Mohan, H.; Rodney-Sandy, R.; Zhang, S.; Iadecola, C.; Hochrainer, K. Cerebral ischemia induces the aggregation of proteins linked to neurodegenerative diseases. Sci. Rep. 2018, 8, 2701. [Google Scholar] [CrossRef] [Green Version]
- Yang, W.; Sheng, H.; Warner, D.S.; Paschen, W. Transient Focal Cerebral Ischemia Induces a Dramatic Activation of Small Ubiquitin-Like Modifier Conjugation. J. Cereb. Blood Flow Metab. 2008, 28, 892–896. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Pluta, R. Brain ischemia as a bridge to Alzheimer’s disease. Neural Regen. Res. 2022, 17, 791. [Google Scholar] [CrossRef]
- Pluta, R.; Januszewski, S.; Czuczwar, S.J. Brain Ischemia as a Prelude to Alzheimer’s Disease. Front. Aging Neurosci. 2021, 13, 636653. [Google Scholar] [CrossRef] [PubMed]
- Kaur, M.M.; Sharma, D.S. Mitochondrial repair as potential pharmacological target in cerebral ischemia. Mitochondrion 2022, 63, 23–31. [Google Scholar] [CrossRef]
- He, Z.; Ning, N.; Zhou, Q.; Khoshnam, S.E.; Farzaneh, M. Mitochondria as a therapeutic target for ischemic stroke. Free Radic. Biol. Med. 2019, 146, 45–58. [Google Scholar] [CrossRef]
- Yimer, E.M.; Hishe, H.Z.; Tuem, K.B. Repurposing of the β-Lactam Antibiotic, Ceftriaxone for Neurological Disorders: A Review. Front. Neurosci. 2019, 13, 236. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Lu, Y.; Zhou, M.; Li, Y.; Li, Y.; Hua, Y.; Fan, Y. Minocycline promotes functional recovery in ischemic stroke by modulating microglia polarization through STAT1/STAT6 pathways. Biochem. Pharmacol. 2021, 186, 114464. [Google Scholar] [CrossRef]
- Camargos, Q.M.; Silva, B.C.; Silva, D.G.; de Brito Toscano, E.C.; da Silva Oliveira, B.; Bellozi, P.M.Q.; de Oliveira Jardim, B.L.; Vieira, É.L.M.; De Oliveira, A.C.P.; Sousa, L.P.; et al. Minocycline treatment prevents depression and anxiety-like behaviors and promotes neuroprotection after experimental ischemic stroke. Brain Res. Bull. 2020, 155, 1–10. [Google Scholar] [CrossRef]
- Yamasaki, T.; Hatori, A.; Zhang, Y.; Mori, W.; Kurihara, Y.; Ogawa, M.; Wakizaka, H.; Rong, J.; Wang, L.; Liang, S.; et al. Neuroprotective effects of minocycline and KML29, a potent inhibitor of monoacylglycerol lipase, in an ex-perimental stroke model: A small-animal positron emission tomography study. Theranostics 2021, 11, 9492–9502. [Google Scholar] [CrossRef] [PubMed]
- Smaga, I.; Fierro, D.; Mesa, J.; Filip, M.; Knackstedt, L.A. Molecular changes evoked by the beta-lactam antibiotic ceftriaxone across rodent models of substance use disorder and neurological disease. Neurosci. Biobehav. Rev. 2020, 115, 116–130. [Google Scholar] [CrossRef] [PubMed]
- Krzyzanowska, W.; Pomierny, B.; Budziszewska, B.; Filip, M.; Pera, J. N-Acetylcysteine and Ceftriaxone as Preconditioning Strategies in Focal Brain Ischemia: Influence on Glutamate Transporters Expression. Neurotox. Res. 2016, 29, 539–550. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Wingo, T.S.; Liu, Y.; Gerasimov, E.S.; Vattathil, S.M.; Wynne, M.E.; Liu, J.; Lori, A.; Faundez, V.; Bennett, D.A.; Seyfried, N.T.; et al. Shared mechanisms across the major psychiatric and neurodegenerative diseases. Nat. Commun. 2022, 13, 4314. [Google Scholar] [CrossRef] [PubMed]
- Zhang, L.; Zheng, H.; Wu, R.; Zhu, F.; Kosten, T.R.; Zhang, X.-Y.; Zhao, J. Minocycline adjunctive treatment to risperidone for negative symptoms in schizophrenia: Association with pro-inflammatory cytokine levels. Prog. Neuro-Psychopharmacol. Biol. Psychiatry 2018, 85, 69–76. [Google Scholar] [CrossRef]
- Ben-Shachar, D. The bimodal mechanism of interaction between dopamine and mitochondria as reflected in Parkinson’s disease and in schizophrenia. J. Neural Transm. 2019, 127, 159–168. [Google Scholar] [CrossRef]
- Inta, D.; Lang, U.E.; Borgwardt, S.; Meyer-Lindenberg, A.; Gass, P. Microglia Activation and Schizophrenia: Lessons from the Effects of Minocycline on Postnatal Neurogenesis, Neuronal Survival and Synaptic Pruning. Schizophr. Bull. 2016, 43, 493–496. [Google Scholar] [CrossRef] [Green Version]
- Krynicki, C.R.; Dazzan, P.; Pariante, C.M.; Barnes, N.M.; Vincent, R.C.; Roberts, A.; Giordano, A.; Watson, A.; Suckling, J.; Barnes, T.R.; et al. Deconstructing depression and negative symptoms of schizophrenia; differential and longitudinal immune correlates, and response to minocycline treatment. Brain Behav. Immun. 2021, 91, 498–504. [Google Scholar] [CrossRef] [PubMed]
- Çakici, N.; Van Beveren, N.J.M.; Judge-Hundal, G.; Koola, M.M.; Sommer, I.E.C. An update on the efficacy of anti-inflammatory agents for patients with schizophrenia: A meta-analysis. Psychol. Med. 2019, 49, 2307–2319. [Google Scholar] [CrossRef] [Green Version]
- Park, H.-J.; Choi, I.; Leem, K.-H. Decreased Brain pH and Pathophysiology in Schizophrenia. Int. J. Mol. Sci. 2021, 22, 8358. [Google Scholar] [CrossRef] [PubMed]
- Roberts, R.C. Mitochondrial dysfunction in schizophrenia: With a focus on postmortem studies. Mitochondrion 2020, 56, 91–101. [Google Scholar] [CrossRef]
- Kelly, D.L.; Sullivan, K.M.; McEvoy, J.P.; McMahon, R.P.; Wehring, H.J.; Gold, J.M.; Liu, F.; Warfel, D.; Vyas, G.; Richardson, C.M.; et al. Adjunctive Minocycline in Clozapine-Treated Schizophrenia Patients with Persistent Symptoms. J. Clin. Psychopharmacol. 2015, 35, 374–381. [Google Scholar] [CrossRef]
- Goldsmith, D.R.; Haroon, E.; Miller, A.H.; Addington, J.; Bearden, C.; Cadenhead, K.; Cannon, T.; Cornblatt, B.; Mathalon, D.; McGlashan, T.; et al. Association of baseline inflammatory markers and the development of negative symptoms in individuals at clinical high risk for psychosis. Brain, Behav. Immun. 2019, 76, 268–274. [Google Scholar] [CrossRef]
- Khandaker, G.M.; Pearson, R.M.; Zammit, S.; Lewis, G.; Jones, P.B. Association of serum interleukin 6 and C-reactive protein in childhood with depression and psychosis in young adult life: A population-based longitudinal study. JAMA Psychiatry 2014, 71, 1121–1128. [Google Scholar] [CrossRef] [Green Version]
- Sellgren, C.M.; Gracias, J.; Watmuff, B.; Biag, J.D.; Thanos, J.M.; Whittredge, P.B.; Fu, T.; Worringer, K.; Brown, H.E.; Wang, J.; et al. Increased synapse elimination by microglia in schizophrenia patient-derived models of synaptic pruning. Nat. Neurosci. 2019, 22, 374–385. [Google Scholar] [CrossRef]
- Xiang, Y.Q.; Zheng, W.; Wang, S.B.; Yang, X.H.; Cai, D.B.; Ng, C.H.; Ungvari, G.S.; Kelly, D.L.; Xu, W.Y.; Xiang, Y.T. Adjunctive minocycline for schizophrenia: A meta-analysis of randomized controlled trials. Eur. Neuropsy-chopharmacol. 2017, 27, 8–18. [Google Scholar] [CrossRef] [PubMed]
- Zhang, L.; Zheng, H.; Wu, R.; Kosten, T.R.; Zhang, X.-Y.; Zhao, J. The effect of minocycline on amelioration of cognitive deficits and pro-inflammatory cytokines levels in patients with schizophrenia. Schizophr. Res. 2019, 212, 92–98. [Google Scholar] [CrossRef] [PubMed]
- Solmi, M.; Veronese, N.; Thapa, N.; Facchini, S.; Stubbs, B.; Fornaro, M.; Carvalho, A.F.; Correll, C.U. Systematic review and meta-analysis of the efficacy and safety of minocycline in schizophrenia. CNS Spectrums 2017, 22, 415–426. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Wehring, H.J.; Elsobky, T.; McEvoy, J.P.; Vyas, G.; Richardson, C.M.; McMahon, R.P.; DiPaula, B.A.; Liu, F.; Sullivan, K.; Buchanan, R.W.; et al. Adjunctive Minocycline in Clozapine-Treated Patients with Schizophrenia: Analyzing the Effects of Minocycline on Clozapine Plasma Levels. Psychiatr. Q. 2018, 89, 73–80. [Google Scholar] [CrossRef]
- Weiser, M.; Levi, L.; Burshtein, S.; Chiriță, R.; Cirjaliu, D.; Gonen, I.; Yolken, R.; Davidson, M.; Zamora, D.; Davis, J.M. The effect of minocycline on symptoms in schizophrenia: Results from a randomized controlled trial. Schizophr. Res. 2019, 206, 325–332. [Google Scholar] [CrossRef]
- Deakin, B.; Suckling, J.; Barnes, T.R.; Byrne, K.; Chaudhry, I.B.; Dazzan, P.; Drake, R.J.; Giordano, A.; Husain, N.; Jones, P.B.; et al. The benefit of minocycline on negative symptoms of schizophrenia in patients with recent-onset psychosis (BeneMin): A randomised, double-blind, placebo-controlled trial. Lancet Psychiatry 2018, 5, 885–894. [Google Scholar] [CrossRef] [Green Version]
- Ben-Azu, B.; Omogbiya, I.A.; Aderibigbe, A.O.; Umukoro, S.; Ajayi, A.M.; Iwalewa, E.O. Doxycycline prevents and reverses schizophrenic-like behaviors induced by ketamine in mice via modulation of oxidative, nitrergic and cholinergic pathways. Brain Res. Bull. 2018, 139, 114–124. [Google Scholar] [CrossRef] [PubMed]
- Hellmann-Regen, J.; Clemens, V.; Grözinger, M.; Kornhuber, J.; Reif, A.; Prvulovic, D.; Goya-Maldonado, R.; Wiltfang, J.; Gruber, O.; Schüle, C.; et al. Effect of Minocycline on Depressive Symptoms in Patients with Treatment-Resistant Depression: A Randomized Clinical Trial. JAMA Netw. Open 2022, 5, e2230367. [Google Scholar] [CrossRef] [PubMed]
- Mello, B.S.; Chaves Filho, A.J.; Custódio, C.S.; de Araújo Rodrigues, P.; Carletti, J.V.; Vasconcelos, S.M.; de Sousa, F.C.; Sanders, L.L.; Macedo, D.S. Doxycycline at subantimicrobial dose combined with escitalopram reverses depressive-like behavior and neuroinflammatory hippocampal alterations in the lipopolysaccharide model of depression. J. Affect. Disord. 2021, 292, 733–745. [Google Scholar] [CrossRef]
- Dean, O.M.; Kanchanatawan, B.; Ashton, M.; Mohebbi, M.; Ng, C.H.; Maes, M.; Berk, L.; Sughondhabirom, A.; Tangwongchai, S.; Singh, A.B.; et al. Adjunctive minocycline treatment for major depressive disorder: A proof of concept trial. Aust. N. Z. J. Psychiatry 2017, 51, 829–840. [Google Scholar] [CrossRef] [PubMed]
- Zhang, C.; Zhang, Y.-P.; Li, Y.-Y.; Liu, B.-P.; Wang, H.-Y.; Li, K.-W.; Zhao, S.; Song, C. Minocycline ameliorates depressive behaviors and neuro-immune dysfunction induced by chronic unpredictable mild stress in the rat. Behav. Brain Res. 2018, 356, 348–357. [Google Scholar] [CrossRef]
- Soczynska, J.K.; Kennedy, S.H.; Alsuwaidan, M.; Mansur, R.B.; Li, M.; McAndrews, M.P.; Brietzke, E.; Woldeyohannes, H.O.; Taylor, V.H.; McIntyre, R.S. A pilot, open-label, 8-week study evaluating the efficacy, safety and tolerability of adjunctive minocycline for the treatment of bipolar I/II depression. Bipolar Disord. 2017, 19, 198–213. [Google Scholar] [CrossRef]



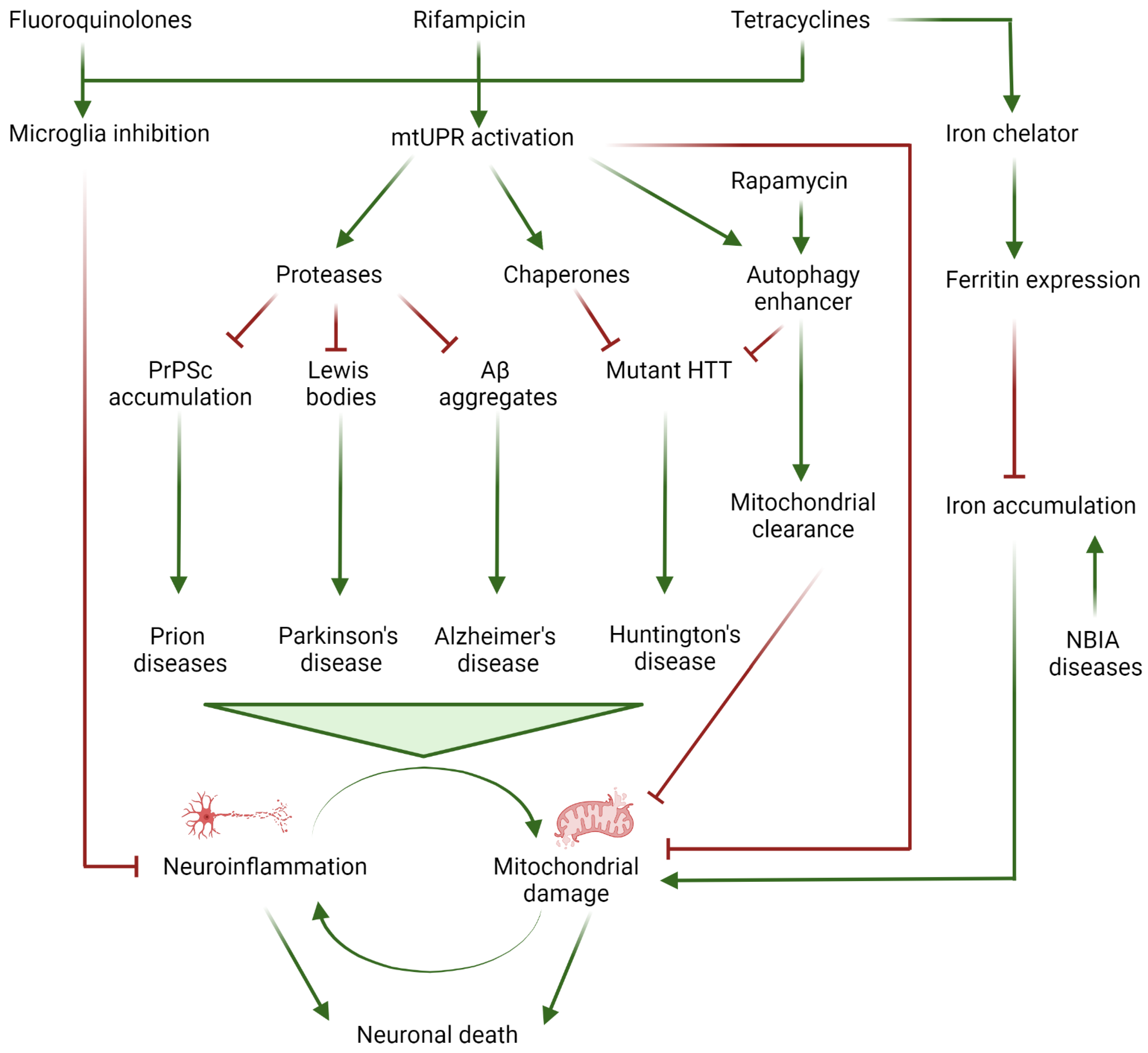{kind=link}
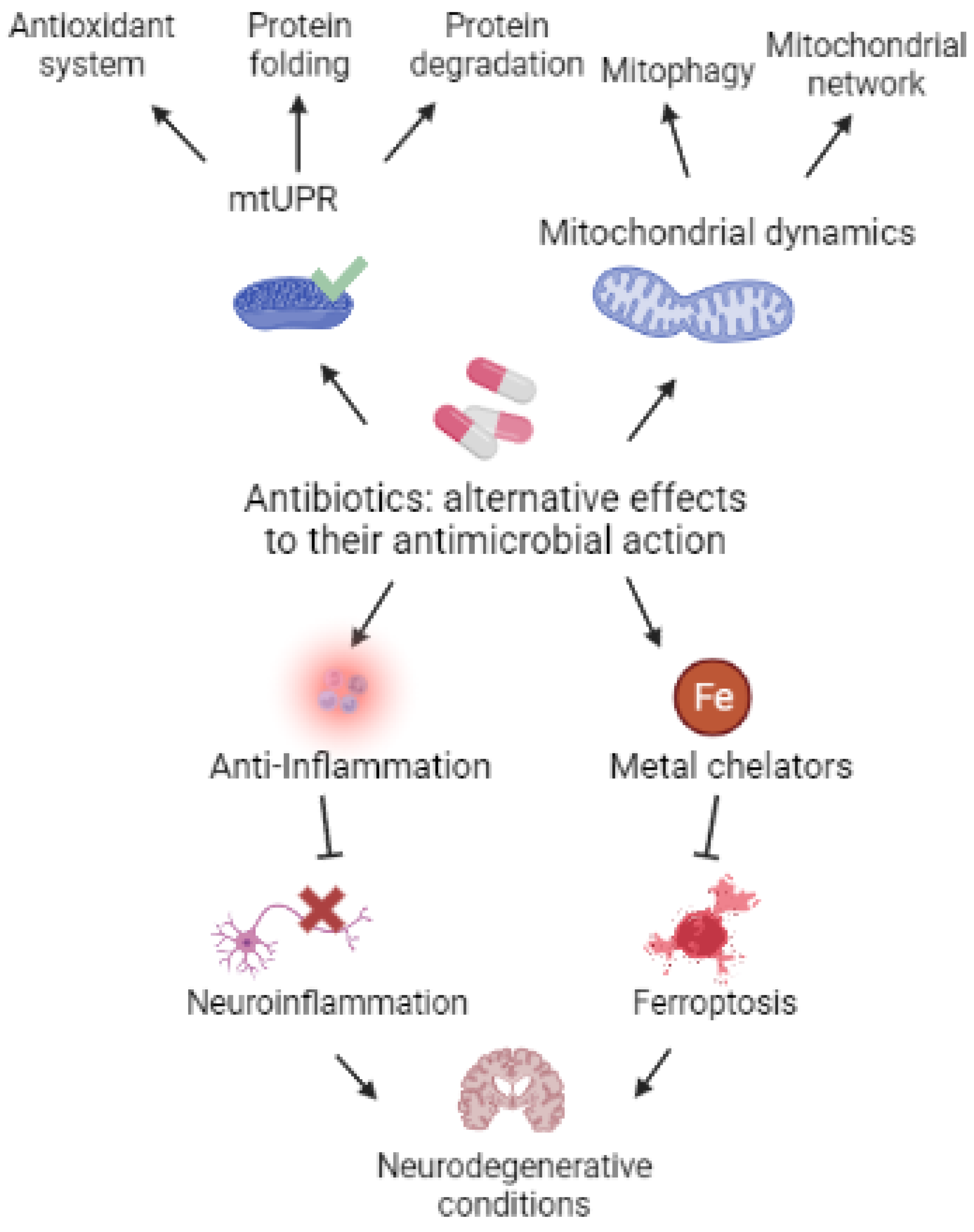{kind=link}
| Antibiotic | Standard Application | Potential Effect | Clinical Trials | Side Effects |
|---|---|---|---|---|
| Tetracyclines family [47] | Broad spectrum bacteriostatic Bacterial ribosome inhibitor | Anti-inflammatory (Microglial M1 inhibition) [48] α-synuclein complex [49,50], prion [51,52], and β-amyloid peptide [53] aggregation inhibition Neuritogenesis promoter [50] UPRmt activator [54] Increases mitochondrial protease activity [55] Iron chelator [56] | Huntington’s disease [57] Aneurysms and cerebral arteriovenous malformations [58] Pancreatic cancer [59] Degradation and permeability of collagen membrane [60] Alzheimer’s disease [61,62] | Gastrointestinal and skin adverse effects, drug-induced lupus, hypersensitive syndrome reaction [63] |
| Fluoroquinolones family [64] | Broad spectrum bactericidal Multidrug resistant bacteria | Anti-inflammatory (TLR4/NF-κB pathway) [65,66,67] | Bladder cancer [68] Crohn’s disease [69] Chronic obstructive pulmonary disease [70] | Tendinopathy [71], aortic diseases [72], gastrointestinal effects [73], psychiatric adverse reactions [74], seizures, confusion/encephalopathy [75,76], dysglycemia [77] |
| Rifampicin [78] | Gram positive bactericidal Tuberculosis, leprosy, and legionnaire’s disease treatment | Anti-inflammatory (TLR4 and NLRP3 pathway) [79,80] UPRmt activator [80] Chaperone enhancer [80] α-synuclein sumoylation [80] Improves autophagy flux [81] | Alzheimer’s disease [61] Diabetes [82] Metabolism homeostasis [83] | Cutaneous reactions, gastrointestinal effects, hepatitis, and thrombocytopenia [84] |
| Rapamycin [85] | Natural anti-fungal antibiotic used as an immunosuppressor | mTOR inhibitor (Autophagy enhancer) [86,87,88,89] Reduce α-synuclein aggregation [90,91] Mitochondrial clearance [92] | Alzheimer’s disease [93] ALS [94] Aging [95] Myelodysplastic syndrome [96] Metabolism homeostasis [97] | Anemia, hyperglycemia, dyslipidemia, renal, pulmonary, and dermatologic adverse effects, angioedema, osteonecrosis, and lymphedema [98] |
| Ceftriaxone [99] | Post-surgery infections Multidrug resistant bacteria | α-synuclein aggregation inhibitor [90,100,101] Improves glutamate homeostasis [102,103,104,105] Anti-inflammatory (TLR4/NF-κB pathway) [104] Reduces levodopa side effects [102] Promotes neurogenesis [106,107] | ALS [108,109] Bipolar disorder [110] Refractory psychosis [111] | Gastrointestinal, skin and vascular disorders [112] |
| Geldanamycin [113] | Antibiotic and a potent antitumor compound | Increases chaperone activity [90,114] | N.A. | Gastrointestinal, hepatic, and eye disorders [115] |
| Amphotericin B [116] | Fungicide | Reduces prion aggregation [117] | N.A. | Nephrotoxicity, anemia, and cardiomyopathy [118] |
Disclaimer/Publisher’s Note: The statements, opinions and data contained in all publications are solely those of the individual author(s) and contributor(s) and not of MDPI and/or the editor(s). MDPI and/or the editor(s) disclaim responsibility for any injury to people or property resulting from any ideas, methods, instructions or products referred to in the content. |
© 2023 by the authors. Licensee MDPI, Basel, Switzerland. This article is an open access article distributed under the terms and conditions of the Creative Commons Attribution (CC BY) license (https://creativecommons.org/licenses/by/4.0/).
Share and Cite
Suárez-Rivero, J.M.; López-Pérez, J.; Muela-Zarzuela, I.; Pastor-Maldonado, C.; Cilleros-Holgado, P.; Gómez-Fernández, D.; Álvarez-Córdoba, M.; Munuera-Cabeza, M.; Talaverón-Rey, M.; Povea-Cabello, S.; et al. Neurodegeneration, Mitochondria, and Antibiotics. Metabolites 2023, 13, 416. https://doi.org/10.3390/metabo13030416
Suárez-Rivero JM, López-Pérez J, Muela-Zarzuela I, Pastor-Maldonado C, Cilleros-Holgado P, Gómez-Fernández D, Álvarez-Córdoba M, Munuera-Cabeza M, Talaverón-Rey M, Povea-Cabello S, et al. Neurodegeneration, Mitochondria, and Antibiotics. Metabolites. 2023; 13(3):416. https://doi.org/10.3390/metabo13030416
Chicago/Turabian StyleSuárez-Rivero, Juan M., Juan López-Pérez, Inés Muela-Zarzuela, Carmen Pastor-Maldonado, Paula Cilleros-Holgado, David Gómez-Fernández, Mónica Álvarez-Córdoba, Manuel Munuera-Cabeza, Marta Talaverón-Rey, Suleva Povea-Cabello, and et al. 2023. "Neurodegeneration, Mitochondria, and Antibiotics" Metabolites 13, no. 3: 416. https://doi.org/10.3390/metabo13030416
APA StyleSuárez-Rivero, J. M., López-Pérez, J., Muela-Zarzuela, I., Pastor-Maldonado, C., Cilleros-Holgado, P., Gómez-Fernández, D., Álvarez-Córdoba, M., Munuera-Cabeza, M., Talaverón-Rey, M., Povea-Cabello, S., Suárez-Carrillo, A., Piñero-Pérez, R., Reche-López, D., Romero-Domínguez, J. M., & Sánchez-Alcázar, J. A. (2023). Neurodegeneration, Mitochondria, and Antibiotics. Metabolites, 13(3), 416. https://doi.org/10.3390/metabo13030416