
Understanding the Pathogenesis of Cardiac Complications in Patients with Propionic Acidemia and Exploring Therapeutic Alternatives for Those Who Are Not Eligible or Are Waiting for Liver Transplantation

 ,
,  ,
,  , ,
, ,  and
and 
Abstract
:1. Introduction
2. Aim of the Review
- −
- The current knowledge of the pathogenetic mechanisms responsible for cardiac complications in PA;
- −
- The therapeutic options for the prevention or treatment of cardiac complications in PA.
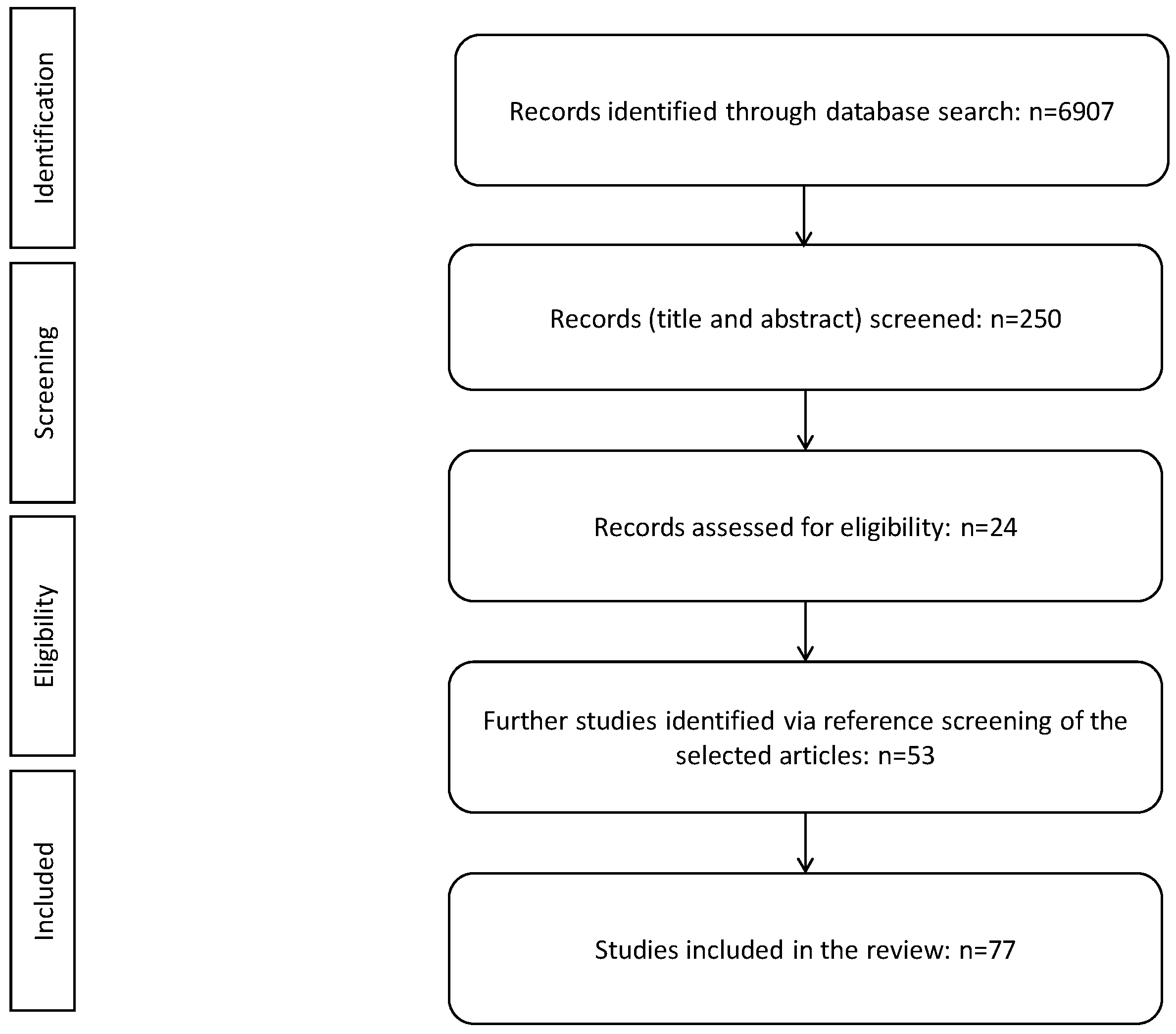
3. Material and Methods
4. Results
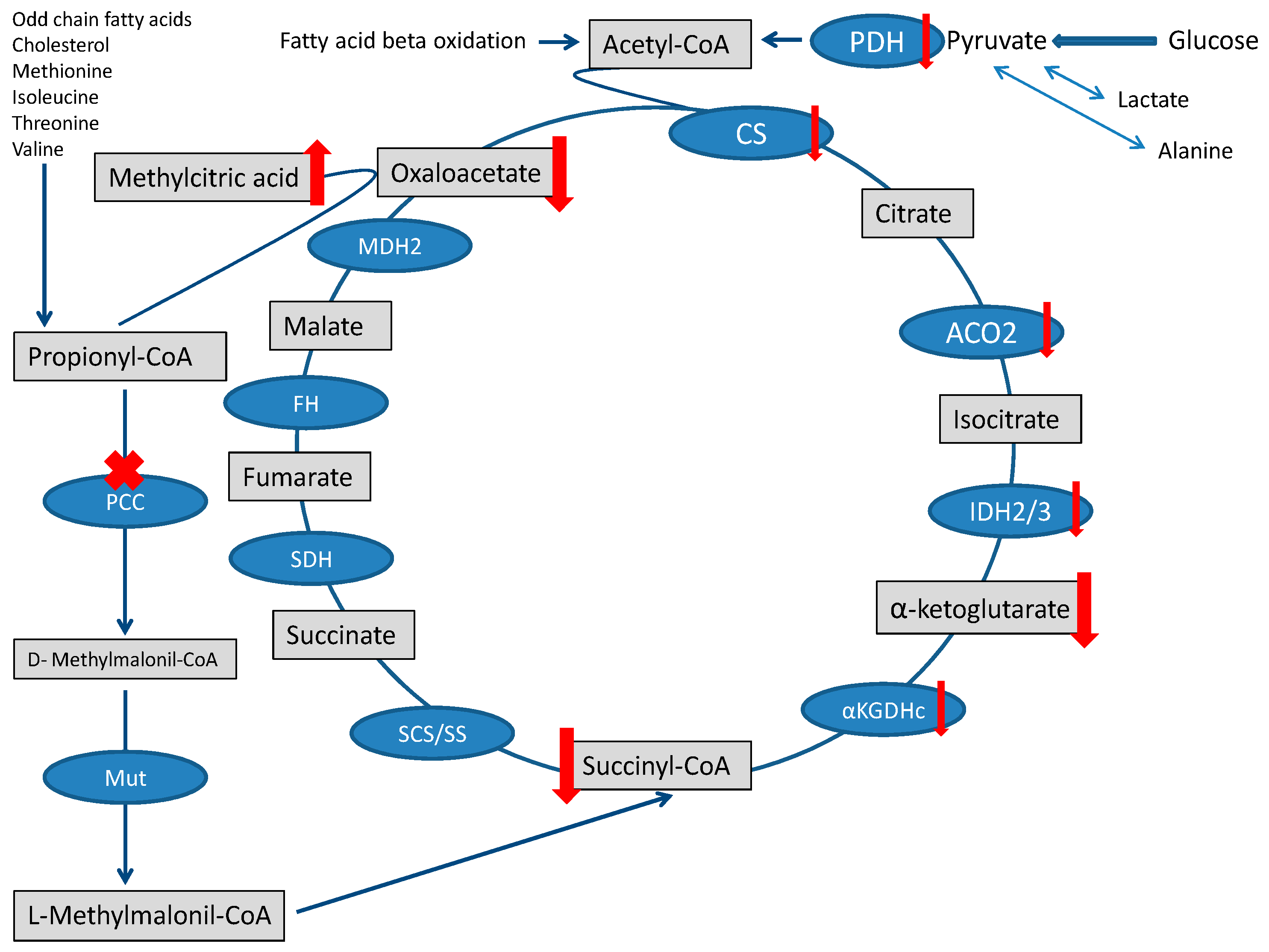
4.1. Impaired Substrate Delivery to TCA Cycle and TCA Dysfunction
4.2. Secondary Mitochondrial Electron Transport (mtETC) Chain Dysfunction and Oxidative Stress
4.3. Coenzyme Q10 Deficiency
4.4. Metabolic Reprogramming
4.5. Carnitine Deficiency
4.6. Cardiac Excitation-Contraction Coupling Alteration
4.7. Genetics
4.8. Epigenetics
4.9. MicroRNAs
4.10. Micronutrients Deficiencies
4.10.1. Selenium
4.10.2. Iron
4.10.3. Vitamin D
4.10.4. Zinc
4.10.5. Thiamine
4.10.6. Riboflavin
4.10.7. Biotin
4.11. The Renin–Angiotensin–Aldosterone System
4.12. Increased Sympathetic Activation
5. Discussion
6. Conclusions and Future Directions
Author Contributions
Funding
Conflicts of Interest
Abbreviations

{kind=link}
{kind=link}
| ACE | angiotensin-converting enzyme |
| ACEi | ACE inhibitors |
| aLQTS | acquired long QT syndrome |
| ARBs | angiotensin II receptor blockers |
| BB | beta-blockers |
| BNP | brain natriuretic peptide |
| CM | cardiomyopathy |
| CoQ10 | Coenzyme Q10 |
| CV | cardiovascular |
| D,L-BHB | D,L-beta-hydroxybutyrate |
| DCM | dilated cardiomyopathy |
| FS | fractional shortening |
| HAT | histone acetyltransferases |
| HDACs | histone deacetylase |
| HF | heart failure |
| HFrEF | HF with reduced LVEF |
| ID | iron deficiency |
| Iv | intravenous |
| iPSCs | induced pluripotent stem cells |
| LT | liver transplantation |
| LV | left ventricular |
| LVEF | left ventricular ejection fraction |
| miRNAs | microRNAs |
| MRA | mineralcorticoid receptor antagonists |
| mtETC | mitochondrial electron transport chain |
| NT-ProBNP | amino-terminal fragment of the BNP |
| OXPHOS | oxidative phosphorylation |
| PA | propionic acidemia |
| PCC | propionyl-CoA carboxylase |
| RAAS | renin–angiotensin–aldosterone system |
| ROS | reactive oxygen species |
| SCA | sudden cardiac arrest |
| SGLT2 | Na+-glucose cotransporter-2 |
| TCA | tricarboxylic acid |
References
- Wongkittichote, P.; Ah Mew, N.; Chapman, K.A. Propionyl-CoA carboxylase—A review. Mol. Genet. Metab. 2017, 122, 145–152. [Google Scholar] [CrossRef]
- Baumgartner, M.R.; Hörster, F.; Dionisi-Vici, C.; Haliloglu, G.; Karall, D.; Chapman, K.A.; Huemer, M.; Hochuli, M.; Assoun, M.; Ballhausen, D.; et al. Proposed guidelines for the diagnosis and management of methylmalonic and propionic acidemia. Orphanet. J. Rare Dis. 2014, 9, 130. [Google Scholar] [CrossRef]
- Kölker, S.; Valayannopoulos, V.; Burlina, A.B.; Sykut-Cegielska, J.; Wijburg, F.A.; Teles, E.L.; Zeman, J.; Dionisi-Vici, C.; Barić, I.; Karall, D.; et al. The phenotypic spectrum of organic acidurias and urea cycle disorders. Part 2: The evolving clinical phenotype. J. Inherit. Metab. Dis. 2015, 38, 1059–1074. [Google Scholar]
- Forny, P.; Hörster, F.; Ballhausen, D.; Chakrapani, A.; Chapman, K.A.; Dionisi-Vici, C.; Dixon, M.; Grünert, S.C.; Grunewald, S.; Haliloglu, G.; et al. Guidelines for the diagnosis and management of methylmalonic acidaemia and propionic acidaemia: First revision. J. Inherit. Metab. Dis. 2021, 44, 566–592. [Google Scholar] [CrossRef]
- Massoud, A.F.; Leonard, J.V. Cardiomyopathy in propionic acidaemia. Eur. J. Pediatr. 1993, 152, 441–445. [Google Scholar] [CrossRef]
- Romano, S.; Valayannopoulos, V.; Touati, G.; Jais, J.P.; Rabier, D.; de Keyzer, Y.; Bonnet, D.; de Lonlay, P. Cardiomyopathies in propionic aciduria are reversible after liver transplantation. J. Pediatr. 2010, 156, 128–134. [Google Scholar] [CrossRef] [PubMed]
- Kovacevic, A.; Garbade, S.F.; Hoffmann, G.F.; Gorenflo, M.; Kölker, S.; Staufner, C. Cardiac phenotype in propionic acidemia—Results of an observational monocentric study. Mol. Genet. Metab. 2020, 130, 41–48. [Google Scholar] [CrossRef] [PubMed]
- Baumgartner, D.; Scholl-Bürgi, S.; Sass, J.O.; Sperl, W.; Schweigmann, U.; Stein, J.I.; Karall, D. Prolonged QTc intervals and decreased left ventricular contractility in patients with propionic acidemia. J. Pediatr. 2007, 150, 192–197.e1. [Google Scholar] [CrossRef]
- Mardach, R.; Verity, M.A.; Cederbaum, S.D. Clinical, pathological, and biochemical studies in a patient with propionic acidemia and fatal cardiomyopathy. Mol. Genet. Metab. 2005, 85, 286–290. [Google Scholar] [CrossRef] [PubMed]
- Lee, T.M.; Addonizio, L.J.; Barshop, B.A.; Chung, W.K. Unusual presentation of propionic acidaemia as isolated cardiomyopathy. J. Inherit. Metab. Dis. 2009, 32 (Suppl. S1), S97–S101. [Google Scholar] [CrossRef]
- Grünert, S.C.; Müllerleile, S.; De Silva, L.; Barth, M.; Walter, M.; Walter, K.; Meissner, T.; Lindner, M.; Ensenauer, R.; Santer, R.; et al. Propionic acidemia: Clinical course and outcome in 55 pediatric and adolescent patients. Orphanet J. Rare Dis. 2013, 8, 6. [Google Scholar] [CrossRef] [PubMed]
- Jameson, E.; Walter, J. Cardiac arrest secondary to long QTc in a child with propionic acidemia. Pediatr. Cardiol. 2008, 29, 969–970. [Google Scholar] [CrossRef]
- Tan, N.S.; Bajaj, R.R.; Morel, C.; Singh, S.M. Metabolic cardiomyopathy from propionic acidemia precipitating cardiac arrest in a 25-year-old man. CMAJ 2018, 190, E883–E887. [Google Scholar] [CrossRef]
- McDonagh, T.A.; Metra, M.; Adamo, M.; Gardner, R.S.; Baumbach, A.; Böhm, M.; Burri, H.; Butler, J.; Čelutkienė, J.; Chioncel, O.; et al. 2021 ESC Guidelines for the diagnosis and treatment of acute and chronic heart failure. Eur. Heart J. 2021, 42, 3599–3726. [Google Scholar] [CrossRef] [PubMed]
- Heidenreich, P.A.; Bozkurt, B.; Aguilar, D.; Allen, L.A.; Byun, J.J.; Colvin, M.M.; Deswal, A.; Drazner, M.H.; Dunlay, S.M.; Evers, L.R.; et al. 2022 AHA/ACC/HFSA Guideline for the Management of Heart Failure: A Report of the American College of Cardiology/American Heart Association Joint Committee on Clinical Practice Guidelines. Circulation 2022, 145, e895–e1032. [Google Scholar] [CrossRef]
- Kirk, R.; Dipchand, A.I.; Rosenthal, D.N.; Addonizio, L.; Burch, M.; Chrisant, M.; Dubin, A.; Everitt, M.; Gajarski, R.; Mertens, L.; et al. The International Society for Heart and Lung Transplantation Guidelines for the management of pediatric heart failure: Executive summary. J. Heart Lung Transplant. 2014, 33, 888–909. [Google Scholar] [CrossRef]
- Zhou, G.-P.; Jiang, Y.-Z.; Wu, S.-S.; Kong, Y.-Y.; Sun, L.-Y.; Zhu, Z.-J. Liver Transplantation for Propionic Acidemia: Evidence From a Systematic Review and Meta-analysis. Transplantation 2021, 105, 2272–2282. [Google Scholar] [CrossRef] [PubMed]
- Berry, G.T.; Blume, E.D.; Wessel, A.; Singh, T.; Hecht, L.; Marsden, D.; Sahai, I.; Elisofon, S.; Ferguson, M.; Kim, H.B.; et al. The re-occurrence of cardiomyopathy in propionic acidemia after liver transplantation. JIMD Rep. 2020, 54, 3–8. [Google Scholar] [CrossRef]
- Hejazi, Y.; Hijazi, Z.M.; Al-Saloos, H.; Omran, T.B. The re-occurrence of dilated cardiomyopathy in propionic acidemia after liver transplantation requiring heart transplant, first case from Middle East. Cardiol. Young 2023, 33, 86–89. [Google Scholar] [CrossRef]
- Jafri, M.S.; Dudycha, S.J.; O’Rourke, B. Cardiac energy metabolism: Models of cellular respiration. Annu. Rev. Biomed. Eng. 2001, 3, 57–81. [Google Scholar] [CrossRef]
- Maines, E.; Catesini, G.; Boenzi, S.; Mosca, A.; Candusso, M.; Dello Strologo, L.; Martinelli, D.; Maiorana, A.; Liguori, A.; Olivieri, G.; et al. Plasma methylcitric acid and its correlations with other disease biomarkers: The impact in the follow up of patients with propionic and methylmalonic acidemia. J. Inherit. Metab. Dis. 2020, 43, 1173–1185. [Google Scholar] [CrossRef]
- Filipowicz, H.R.; Ernst, S.L.; Ashurst, C.L.; Pasquali, M.; Longo, N. Metabolic changes associated with hyperammonemia in patients with propionic acidemia. Mol. Genet. Metab. 2006, 88, 123–130. [Google Scholar] [CrossRef]
- Roginski, A.C.; Wajner, A.; Cecatto, C.; Wajner, S.M.; Castilho, R.F.; Wajner, M.; Amaral, A.U. Disturbance of bioenergetics and calcium homeostasis provoked by metabolites accumulating in propionic acidemia in heart mitochondria of developing rats. Biochim. Biophys. Acta Mol. Basis Dis. 2020, 1866, 165682. [Google Scholar] [CrossRef] [PubMed]
- Wang, Y.; Christopher, B.A.; Wilson, K.A.; Muoio, D.; McGarrah, R.W.; Brunengraber, H.; Zhang, G.F. Propionate-induced changes in cardiac metabolism, notably CoA trapping, are not altered by l-carnitine. Am. J. Physiol. Endocrinol. Metab. 2018, 315, E622–E633. [Google Scholar] [CrossRef] [PubMed]
- Bordugo, A.; Salvetti, E.; Rodella, G.; Piazza, M.; Dianin, A.; Amoruso, A.; Piacentini, G.; Pane, M.; Torriani, S.; Vitulo, N.; et al. Assessing Gut Microbiota in an Infant with Congenital Propionic Acidemia before and after Probiotic Supplementation. Microorganisms 2021, 9, 2599. [Google Scholar] [CrossRef] [PubMed]
- Siekmeyer, M.; Petzold-Quinque, S.; Terpe, F.; Beblo, S.; Gebhardt, R.; Schlensog-Schuster, F.; Kiess, W.; Siekmeyer, W. Citric acid as the last therapeutic approach in an acute life-threatening metabolic decompensation of propionic acidaemia. J. Pediatr. Endocrinol. Metab. 2013, 26, 569–574. [Google Scholar] [CrossRef]
- Longo, N.; Price, L.B.; Gappmaier, E.; Cantor, N.L.; Ernst, S.L.; Bailey, C.; Pasquali, M. Anaplerotic therapy in propionic acidemia. Mol. Genet. Metab. 2017, 122, 51–59. [Google Scholar] [CrossRef]
- De Keyzer, Y.; Valayannopoulos, V.; Benoist, J.F.; Batteux, F.; Lacaille, F.; Hubert, L.; Chrétien, D.; Chadefeaux-Vekemans, B.; Niaudet, P.; Touati, G.; et al. Multiple OXPHOS deficiency in the liver, kidney, heart, and skeletal muscle of patients with methylmalonic aciduria and propionic aciduria. Pediatr. Res. 2009, 66, 91–95. [Google Scholar] [CrossRef]
- Baruteau, J.; Hargreaves, I.; Krywawych, S.; Chalasani, A.; Land, J.M.; Davison, J.E.; Kwok, M.K.; Christov, G.; Karimova, A.; Ashworth, M.; et al. Successful reversal of propionic acidaemia associated cardiomyopathy: Evidence for low myocardial coenzyme Q10 status and secondary mitochondrial dysfunction as an underlying pathophysiological mechanism. Mitochondrion 2014, 17, 150–156. [Google Scholar] [CrossRef]
- Gallego-Villar, L.; Rivera-Barahona, A.; Cuevas-Martín, C.; Guenzel, A.; Pérez, B.; Barry, M.A.; Murphy, M.P.; Logan, A.; Gonzalez-Quintana, A.; Martín, M.A.; et al. In vivo evidence of mitochondrial dysfunction and altered redox homeostasis in a genetic mouse model of propionic acidemia: Implications for the pathophysiology of this disorder. Free Radic. Biol. Med. 2016, 96, 1–12. [Google Scholar] [CrossRef] [PubMed]
- Alonso-Barroso, E.; Pérez, B.; Desviat, L.R.; Richard, E. Cardiomyocytes Derived from Induced Pluripotent Stem Cells as a Disease Model for Propionic Acidemia. Int. J. Mol. Sci. 2021, 22, 1161. [Google Scholar] [CrossRef]
- Fragaki, K.; Cano, A.; Benoist, J.F.; Rigal, O.; Chaussenot, A.; Rouzier, C.; Bannwarth, S.; Caruba, C.; Chabrol, B.; Paquis-Flucklinger, V. Fatal heart failure associated with CoQ10 and multiple OXPHOS deficiency in a child with propionic acidemia. Mitochondrion 2011, 11, 533–536. [Google Scholar] [CrossRef] [PubMed]
- Gallego-Villar, L.; Pérez-Cerdá, C.; Pérez, B.; Abia, D.; Ugarte, M.; Richard, E.; Desviat, L.R. Functional characterization of novel genotypes and cellular oxidative stress studies in propionic acidemia. J. Inherit. Metab. Dis. 2013, 36, 731–740. [Google Scholar] [CrossRef] [PubMed]
- Mc Guire, P.J.; Parikh, A.; Diaz, G.A. Profiling of oxidative stress in patients with inborn errors of metabolism. Mol. Genet. Metab. 2009, 98, 173–180. [Google Scholar] [CrossRef]
- Schwab, M.A.; Sauer, S.W.; Okun, J.G.; Nijtmans, L.G.; Rodenburg, R.J.; van den Heuvel, L.P.; Dröse, S.; Brandt, U.; Hoffmann, G.F.; Ter Laak, H.; et al. Secondary mitochondrial dysfunction in propionic aciduria: A pathogenic role for endogenous mitochondrial toxins. Biochem. J. 2006, 398, 107–112. [Google Scholar] [CrossRef] [PubMed]
- Takimoto, E.; Kass, D.A. Role of oxidative stress in cardiac hypertrophy and remodeling. Hypertension 2007, 49, 241–248. [Google Scholar] [CrossRef]
- Rivera-Barahona, A.; Alonso-Barroso, E.; Pérez, B.; Murphy, M.P.; Richard, E.; Desviat, L.R. Treatment with antioxidants ameliorates oxidative damage in a mouse model of propionic acidemia. Mol. Genet. Metab. 2017, 122, 43–50. [Google Scholar] [CrossRef]
- Gallego-Villar, L.; Pérez, B.; Ugarte, M.; Desviat, L.R.; Richard, E. Antioxidants successfully reduce ROS production in propionic acidemia fibroblasts. Biochem. Biophys. Res. Commun. 2014, 452, 457–461. [Google Scholar] [CrossRef]
- Gottwald, E.M.; Duss, M.; Bugarski, M.; Haenni, D.; Schuh, C.D.; Landau, E.M.; Hall, A.M. The targeted anti-oxidant MitoQ causes mitochondrial swelling and depolarization in kidney tissue. Physiol. Rep. 2018, 6, e13667. [Google Scholar] [CrossRef]
- Pokrzywinski, K.L.; Biel, T.G.; Kryndushkin, D.; Rao, V.A. Therapeutic Targeting of the Mitochondria Initiates Excessive Superoxide Production and Mitochondrial Depolarization Causing Decreased mtDNA Integrity. PLoS ONE. 2016, 11, e0168283. [Google Scholar] [CrossRef]
- Pala, R.; Beyaz, F.; Tuzcu, M.; Er, B.; Sahin, N.; Cinar, V.; Sahin, K. The effects of coenzyme Q10 on oxidative stress and heat shock proteins in rats subjected to acute and chronic exercise. J. Exerc. Nutr. Biochem. 2018, 22, 14–20. [Google Scholar] [CrossRef] [PubMed]
- McMurray, J.J.; Dunselman, P.; Wedel, H.; Cleland, J.G.; Lindberg, M.; Hjalmarson, A.; Kjekshus, J.; Waagstein, F.; Apetrei, E.; Barrios, V.; et al. Coenzyme Q10, rosuvastatin, and clinical outcomes in heart failure: A pre-specified substudy of CORONA (controlled rosuvastatin multinational study in heart failure). J. Am. Coll. Cardiol. 2010, 56, 1196–1204. [Google Scholar] [CrossRef] [PubMed]
- Bomer, N.; Pavez-Giani, M.G.; Grote Beverborg, N.; Cleland, J.G.F.; van Veldhuisen, D.J.; van der Meer, P. Micronutrient deficiencies in heart failure: Mitochondrial dysfunction as a common pathophysiological mechanism? J. Intern. Med. 2022, 291, 713–731. [Google Scholar] [CrossRef]
- Morisco, C.; Trimarco, B.; Condorelli, M. Effect of coenzyme Q10 therapy in patients with congestive heart failure: A long-term multicenter randomized study. Clin. Investig. 1993, 71 (Suppl. S8), S134–S136. [Google Scholar] [CrossRef] [PubMed]
- Alehagen, U.; Aaseth, J.; Johansson, P. Reduced Cardiovascular Mortality 10 Years after Supplementation with Selenium and Coenzyme Q10 for Four Years: Follow-Up Results of a Prospective Randomized Double-Blind Placebo-Controlled Trial in Elderly Citizens. PLoS ONE. 2015, 10, e0141641. [Google Scholar] [CrossRef]
- Neubauer, S. The failing heart--an engine out of fuel. N. Engl. J. Med. 2007, 356, 1140–1151. [Google Scholar] [CrossRef]
- Van Rijt, W.J.; Van Hove, J.L.K.; Vaz, F.M.; Havinga, R.; Allersma, D.P.; Zijp, T.R.; Bedoyan, J.K.; Heiner-Fokkema, M.R.; Reijngoud, D.J.; Geraghty, M.T.; et al. Enantiomer-specific pharmacokinetics of, D,L-3-hydroxybutyrate: Implications for the treatment of multiple acyl-CoA dehydrogenase deficiency. J. Inherit. Metab. Dis. 2021, 44, 926–938. [Google Scholar] [CrossRef]
- Valayannopoulos, V.; Bajolle, F.; Arnoux, J.B.; Dubois, S.; Sannier, N.; Baussan, C.; Petit, F.; Labrune, P.; Rabier, D.; Ottolenghi, C.; et al. Successful treatment of severe cardiomyopathy in glycogen storage disease type III With, D,L-3-hydroxybutyrate, ketogenic and high-protein diet. Pediatr. Res. 2011, 70, 638–641. [Google Scholar] [CrossRef]
- Kölker, S.; Burgard, P.; Sauer, S.W.; Okun, J.G. Current concepts in organic acidurias: Understanding intra- and extracerebral disease manifestation. J. Inherit. Metab. Dis. 2013, 36, 635–644. [Google Scholar] [CrossRef]
- He, W.; Wang, Y.; Xie, E.J.; Barry, M.A.; Zhang, G.F. Metabolic perturbations mediated by propionyl-CoA accumulation in organs of mouse model of propionic acidemia. Mol. Genet. Metab. 2021, 134, 257–266. [Google Scholar] [CrossRef]
- Rijlaarsdam, R.S.; van Spronsen, F.J.; Bink-Boelkens, M.T.; Reijngoud, D.J.; Wanders, R.J.; Niezen-Koning, K.E.; Van der Sluijs, F.H.; Dorland, B.; Beaufort-Krol, G.C. Ventricular fibrillation without overt cardiomyopathy as first presentation of organic cation transporter 2-deficiency in adolescence. Pacing Clin. Electrophysiol. 2004, 27, 675–676. [Google Scholar] [CrossRef]
- Sutton, V.R.; Chapman, K.A.; Gropman, A.L.; MacLeod, E.; Stagni, K.; Summar, M.L.; Ueda, K.; Ah Mew, N.; Franks, J.; Island, E.; et al. Chronic management and health supervision of individuals with propionic acidemia. Mol. Genet. Metab. 2012, 105, 26–33. [Google Scholar] [CrossRef]
- Eisner, D.A.; Caldwell, J.L.; Kistamás, K.; Trafford, A.W. Calcium and Excitation-Contraction Coupling in the Heart. Circ. Res. 2017, 121, 181–195. [Google Scholar] [CrossRef] [PubMed]
- Tamayo, M.; Fulgencio-Covián, A.; Navarro-García, J.A.; Val-Blasco, A.; Ruiz-Hurtado, G.; Gil-Fernández, M.; Martín-Nunes, L.; Lopez, J.A.; Desviat, L.R.; Delgado, C.; et al. Intracellular calcium mishandling leads to cardiac dysfunction and ventricular arrhythmias in a mouse model of propionic acidemia. Biochim. Biophys. Acta Mol. Basis Dis. 2020, 1866, 165586. [Google Scholar] [CrossRef]
- Landstrom, A.P.; Dobrev, D.; Wehrens, X.H.T. Calcium Signaling and Cardiac Arrhythmias. Circ. Res. 2017, 120, 1969–1993. [Google Scholar] [CrossRef]
- Bodi, I.; Grünert, S.C.; Becker, N.; Stoelzle-Feix, S.; Spiekerkoetter, U.; Zehender, M.; Bugger, H.; Bode, C.; Odening, K.E. Mechanisms of acquired long QT syndrome in patients with propionic academia. Heart Rhythm. 2016, 13, 1335–1345. [Google Scholar] [CrossRef] [PubMed]
- Liu, C.F.; Tang, W.H.W. Epigenetics in Cardiac Hypertrophy and Heart Failure. JACC Basic Transl. Sci. 2019, 4, 976–993. [Google Scholar] [CrossRef] [PubMed]
- Park, K.C.; Krywawych, S.; Richard, E.; Desviat, L.R.; Swietach, P. Cardiac Complications of Propionic and Other Inherited Organic Acidemias. Front. Cardiovasc. Med. 2020, 7, 617451. [Google Scholar] [CrossRef]
- Sabari, B.R.; Zhang, D.; Allis, C.D.; Zhao, Y. Metabolic regulation of gene expression through histone acylations. Nat. Rev. Mol. Cell Biol. 2017, 18, 90–101. [Google Scholar] [CrossRef]
- Waldecker, M.; Kautenburger, T.; Daumann, H.; Busch, C.; Schrenk, D. Inhibition of histone-deacetylase activity by short-chain fatty acids and some polyphenol metabolites formed in the colon. J. Nutr. Biochem. 2008, 19, 587–593. [Google Scholar] [CrossRef]
- Bagchi, R.A.; Weeks, K.L. Histone deacetylases in cardiovascular and metabolic diseases. J. Mol. Cell. Cardiol. 2019, 130, 151–159. [Google Scholar] [CrossRef]
- Kebede, A.F.; Nieborak, A.; Shahidian, L.Z.; Le Gras, S.; Richter, F.; Gómez, D.A.; Baltissen, M.P.; Meszaros, G.; Magliarelli, H.F.; Taudt, A.; et al. Histone propionylation is a mark of active chromatin. Nat. Struct. Mol. Biol. 2017, 24, 1048–1056. [Google Scholar] [CrossRef]
- Jonas, S.; Izaurralde, E. Towards a molecular understanding of microRNA-mediated gene silencing. Nat. Rev. Genet. 2015, 16, 421–433. [Google Scholar] [CrossRef] [PubMed]
- Christian, P.; Su, Q. MicroRNA regulation of mitochondrial and ER stress signaling pathways: Implications for lipoprotein metabolism in metabolic syndrome. Am. J. Physiol. Endocrinol. Metab. 2014, 307, E729–E737. [Google Scholar] [CrossRef]
- Yang, B.F.; Lu, Y.J.; Wang, Z.G. MicroRNAs and apoptosis: Implications in the molecular therapy of human disease. Clin. Exp. Pharmacol. Physiol. 2009, 36, 951–960. [Google Scholar] [CrossRef]
- Van Rooij, E.; Olson, E.N. MicroRNA therapeutics for cardiovascular disease: Opportunities and obstacles. Nat. Rev. Drug Discov. 2012, 11, 860–872. [Google Scholar] [CrossRef]
- Rivera-Barahona, A.; Fulgencio-Covián, A.; Pérez-Cerdá, C.; Ramos, R.; Barry, M.A.; Ugarte, M.; Pérez, B.; Richard, E.; Desviat, L.R. Dysregulated miRNAs and their pathogenic implications for the neurometabolic disease propionic acidemia. Sci. Rep. 2017, 7, 5727. [Google Scholar] [CrossRef] [PubMed]
- Fulgencio-Covián, A.; Alonso-Barroso, E.; Guenzel, A.J.; Rivera-Barahona, A.; Ugarte, M.; Pérez, B.; Barry, M.A.; Pérez-Cerdá, C.; Richard, E.; Desviat, L.R. Pathogenic implications of dysregulated miRNAs in propionic acidemia related cardiomyopathy. Transl. Res. 2020, 218, 43–56. [Google Scholar] [CrossRef] [PubMed]
- Cicero, A.F.G.; Colletti, A.; von Haehling, S.; Vinereanu, D.; Bielecka-Dabrowa, A.; Sahebkar, A.; Toth, P.P.; Reiner, Ž.; Wong, N.D.; Mikhailidis, D.P.; et al. Nutraceutical support in heart failure: A position paper of the International Lipid Expert Panel (ILEP). Nutr. Res. Rev. 2020, 33, 155–179. [Google Scholar] [CrossRef]
- Yang, G.Q.; Ge, K.Y.; Chen, J.S.; Chen, X.S. Selenium-related endemic diseases and the daily selenium requirement of humans. World Rev. Nutr. Diet. 1988, 55, 98–152. [Google Scholar]
- Loscalzo, J. Keshan disease, selenium deficiency, and the selenoproteome. N. Engl. J. Med. 2014, 370, 1756–1760. [Google Scholar] [CrossRef]
- Bomer, N.; Grote Beverborg, N.; Hoes, M.F.; Streng, K.W.; Vermeer, M.; Dokter, M.M.; IJmker, J.; Anker, S.D.; Cleland, J.G.F.; Hillege, H.L.; et al. Selenium and outcome in heart failure. Eur. J. Heart Fail. 2020, 22, 1415–1423. [Google Scholar] [CrossRef]
- Garakyaraghi, M.; Bahrami, P.; Sadeghi, M.; Rabiei, K. Combination effects of selenium and coenzyme Q10 on left ventricular systolic function in patients with heart failure. Iran. Heart J. 2015, 15, 6–12. [Google Scholar]
- Jenkins, D.J.A.; Kitts, D.; Giovannucci, E.L.; Sahye-Pudaruth, S.; Paquette, M.; Blanco Mejia, S.; Patel, D.; Kavanagh, M.; Tsirakis, T.; Kendall, C.W.C.; et al. Selenium, antioxidants, cardiovascular disease, and all-cause mortality: A systematic review and meta-analysis of randomized controlled trials. Am. J. Clin. Nutr. 2020, 112, 1642–1652. [Google Scholar] [CrossRef]
- Kuria, A.; Tian, H.; Li, M.; Wang, Y.; Aaseth, J.O.; Zang, J.; Cao, Y. Selenium status in the body and cardiovascular disease: A systematic review and meta-analysis. Crit. Rev. Food Sci. Nutr. 2021, 61, 3616–3625. [Google Scholar] [CrossRef] [PubMed]
- Hoes, M.F.; Grote Beverborg, N.; Kijlstra, J.D.; Kuipers, J.; Swinkels, D.W.; Giepmans, B.N.G.; Rodenburg, R.J.; van Veldhuisen, D.J.; de Boer, R.A.; van der Meer, P. Iron deficiency impairs contractility of human cardiomyocytes through decreased mitochondrial function. Eur. J. Heart Fail. 2018, 20, 910–919. [Google Scholar] [CrossRef]
- Van der Ent, M.; Jeneson, J.A.; Remme, W.J.; Berger, R.; Ciampricotti, R.; Visser, F. A non-invasive selective assessment of type I fibre mitochondrial function using 31P NMR spectroscopy. Evidence for impaired oxidative phosphorylation rate in skeletal muscle in patients with chronic heart failure. Eur. Heart J. 1998, 19, 124–131. [Google Scholar] [CrossRef]
- Weiss, K.; Schär, M.; Panjrath, G.S.; Zhang, Y.; Sharma, K.; Bottomley, P.A.; Golozar, A.; Steinberg, A.; Gerstenblith, G.; Russell, S.D.; et al. Fatigability, exercise intolerance, and abnormal skeletal muscle energetics in heart failure. Circ. Heart Fail. 2017, 10, e004129. [Google Scholar] [CrossRef] [PubMed]
- Melenovsky, V.; Hlavata, K.; Sedivy, P.; Dezortova, M.; Borlaug, B.A.; Petrak, J.; Kautzner, J.; Hajek, M. Skeletal muscle abnormalities and iron deficiency in chronic heart failure (an exercise (31)P magnetic resonance spectroscopy study of calf muscle). Circ. Heart Fail. 2018, 11, e004800. [Google Scholar] [CrossRef]
- Anker, S.D.; Comin Colet, J.; Filippatos, G.; Willenheimer, R.; Dickstein, K.; Drexler, H.; Lüscher, T.F.; Bart, B.; Banasiak, W.; Niegowska, J.; et al. Ferric carboxymaltose in patients with heart failure and iron deficiency. N. Engl. J. Med. 2009, 361, 2436–2448. [Google Scholar] [CrossRef]
- Van Veldhuisen, D.J.; Ponikowski, P.; van der Meer, P.; Metra, M.; Böhm, M.; Doletsky, A.; Voors, A.A.; Macdougall, I.C.; Anker, S.D.; Roubert, B.; et al. Effect of ferric carboxymaltose on exercise capacity in patients with chronic heart failure and iron deficiency. Circulation 2017, 136, 1374–1383. [Google Scholar] [CrossRef] [PubMed]
- Ponikowski, P.; Van Veldhuisen, D.J.; Comin-Colet, J.; Ertl, G.; Komajda, M.; Mareev, V.; McDonagh, T.; Parkhomenko, A.; Tavazzi, L.; Levesque, V.; et al. Beneficial effects of longterm intravenous iron therapy with ferric carboxymaltose in patients with symptomatic heart failure and iron deficiency. Eur. Heart J. 2014, 36, 657–668. [Google Scholar] [CrossRef]
- Maiya, S.; Sullivan, I.; Allgrove, J.; Yates, R.; Malone, M.; Brain, C.; Archer, N.; Mok, Q.; Daubeney, P.; Tulloh, R.; et al. Hypocalcaemia and vitamin D deficiency: An important, but preventable, cause of life-threatening infant heart failure. Heart 2008, 94, 581–584. [Google Scholar] [CrossRef] [PubMed]
- Busa, V.; Dardeir, A.; Marudhai, S.; Patel, M.; Valaiyaduppu Subas, S.; Ghani, M.R.; Cancarevic, I. Role of Vitamin D Supplementation in Heart Failure Patients With Vitamin D Deficiency and Its Effects on Clinical Outcomes: A Literature Review. Cureus 2020, 12, e10840. [Google Scholar] [CrossRef]
- Rosenblum, H.; Wessler, J.D.; Gupta, A.; Maurer, M.S.; Bikdeli, B. Zinc Deficiency and Heart Failure: A Systematic Review of the Current Literature. J. Card. Fail. 2020, 26, 180–189. [Google Scholar] [CrossRef]
- Frustaci, A.; Sabbioni, E.; Fortaner, S.; Farina, M.; del Torchio, R.; Tafani, M.; Morgante, E.; Ciriolo, M.R.; Russo, M.A.; Chimenti, C. Selenium- and zinc-deficient cardiomyopathy in human intestinal malabsorption: Preliminary results of selenium/zinc infusion. Eur. J. Heart Fail. 2012, 14, 202–210. [Google Scholar] [CrossRef] [PubMed]
- Yoshihisa, A.; Abe, S.; Kiko, T.; Kimishima, Y.; Sato, Y.; Watanabe, S.; Kanno, Y.; Miyata-Tatsumi, M.; Misaka, T.; Sato, T.; et al. Association of Serum Zinc Level with Prognosis in Patients with Heart Failure. J. Card. Fail. 2018, 24, 375–383. [Google Scholar] [CrossRef]
- Rosenblum, H.; Bikdeli, B.; Wessler, J.; Gupta, A.; Jacoby, D.L. Zinc Deficiency as a Reversible Cause of Heart Failure. Tex. Heart Inst. J. 2020, 47, 152–154. [Google Scholar] [CrossRef]
- Kattoor, A.J.; Goel, A.; Mehta, J.L. Thiamine Therapy for Heart Failure: A Promise or Fiction? Cardiovasc. Drugs Ther. 2018, 32, 313–317. [Google Scholar] [CrossRef]
- Jain, A.; Mehta, R.; Al-Ani, M.; Hill, J.A.; Winchester, D.E. Determining the role of thiamine deficiency in systolic heart failure: A meta-analysis and systematic review. J. Card. Fail. 2015, 21, 1000–1007. [Google Scholar] [CrossRef]
- Piquereau, J.; Boitard, S.E.; Ventura-Clapier, R.; Mericskay, M. Metabolic Therapy of Heart Failure: Is There a Future for B Vitamins? Int. J. Mol. Sci. 2021, 23, 30. [Google Scholar] [CrossRef]
- Wang, G.; Li, W.; Lu, X.; Zhao, X. Riboflavin alleviates cardiac failure in Type I diabetic cardiomyopathy. Heart Int. 2011, 6, e21. [Google Scholar] [CrossRef]
- Keith, M.E.; Walsh, N.A.; Darling, P.B.; Hanninen, S.A.; Thirugnanam, S.; Leong-Poi, H.; Barr, A.; Sole, M.J. B-vitamin deficiency in hospitalized patients with heart failure. J. Am. Diet. Assoc. 2009, 109, 1406–1410. [Google Scholar] [CrossRef]
- Jurecki, E.; Ueda, K.; Frazier, D.; Rohr, F.; Thompson, A.; Hussa, C.; Obernolte, L.; Reineking, B.; Roberts, A.M.; Yannicelli, S.; et al. Nutrition management guideline for propionic acidemia: An evidence- and consensus-based approach. Mol. Genet. Metab. 2019, 126, 341–354. [Google Scholar] [CrossRef]
- Yannicelli, S. Nutrition therapy of organic acidaemias with amino acid-based formulas: Emphasis on methylmalonic and propionic acidaemia. J. Inherit. Metab. Dis. 2006, 29, 281–287. [Google Scholar] [CrossRef]
- Jia, G.; Aroor, A.R.; Hill, M.A.; Sowers, J.R. Role of Renin-Angiotensin-Aldosterone System Activation in Promoting Cardiovascular Fibrosis and Stiffness. Hypertension 2018, 72, 537–548. [Google Scholar] [CrossRef]
- Borghi, C.; Boschi, S.; Ambrosioni, E.; Melandri, G.; Branzi, A.; Magnani, B. Evidence of a partial escape of renin-angiotensin-aldosterone blockade in patients with acute myocardial infarction treated with ACE inhibitors. J. Clin. Pharmacol. 1993, 33, 40–45. [Google Scholar] [CrossRef]
- Dyck, J.R.B.; Sossalla, S.; Hamdani, N.; Coronel, R.; Weber, N.C.; Light, P.E.; Zuurbier, C.J. Cardiac mechanisms of the beneficial effects of SGLT2 inhibitors in heart failure: Evidence for potential off-target effects. J. Mol. Cell. Cardiol. 2022, 167, 17–31. [Google Scholar] [CrossRef]
- FDA Drug Safety Communication: FDA Warns That SGLT2 Inhibitors for Diabetes May Result in a Serious Condition of Too Much Acid in the Blood. Available online: https://www.fda.gov/drugs/drug-safety-and-availability/fda-revises-labels-sglt2-inhibitors-diabetes-include-warnings-about-too-much-acid-blood-and-serious (accessed on 9 February 2022).
- European Medicines Agency. Review of Diabetes Medicines Called SGLT2 Inhibitors Started. Risk of Diabetic Ketoacidosis to Be Examined. Available online: http://www.ema.europa.eu/ema/index.jsp?curl=pages/medicines/human/referrals/SGLT2_inhibitors/human_referral_prac_000052.jsp&mid=WC0b01ac05805c516f (accessed on 9 February 2022).
- Zeppenfeld, K.; Tfelt-Hansen, J.; de Riva, M.; Winkel, B.G.; Behr, E.R.; Blom, N.A.; Charron, P.; Corrado, D.; Dagres, N.; de Chillou, C.; et al. 2022 ESC Guidelines for the management of patients with ventricular arrhythmias and the prevention of sudden cardiac death. Eur. Heart J. 2022. Epub ahead of print. [Google Scholar] [CrossRef]
- Duras, E.; İrdem, A.; Özkaya, O. Long QT syndrome diagnosed in two sisters with propionic acidemia: A case report. J. Pediatr. Endocrinol. Metab. 2017, 30, 1133–1136. [Google Scholar] [CrossRef]
- Fu, L.; Huang, M.; Chen, S. Primary carnitine deficiency and cardiomyopathy. Korean Circ. J. 2013, 43, 785–792. [Google Scholar] [CrossRef] [PubMed]
- Antozzi, C.; Zeviani, M. Cardiomyopathies in disorders of oxidative metabolism. Cardiovasc. Res. 1997, 35, 184–199. [Google Scholar] [CrossRef] [PubMed]
- Guenzel, A.J.; Hofherr, S.E.; Hillestad, M.; Barry, M.; Weaver, E.; Venezia, S.; Kraus, J.P.; Matern, D.; Barry, M.A. Generation of a hypomorphic model of propionic acidemia amenable to gene therapy testing. Mol Ther. 2013, 21, 1316–1323. [Google Scholar] [CrossRef] [PubMed]
- Wessels, A.; Sedmera, D. Developmental anatomy of the heart: A tale of mice and man. Physiol. Genom. 2003, 15, 165–176. [Google Scholar] [CrossRef]
- Breckenridge, R. Heart failure and mouse models. Dis. Model Mech. 2010, 3, 138–143. [Google Scholar] [CrossRef]


| Accepted | Potential Harmful | References | |
|---|---|---|---|
| ACE-inhibitors (ACE-i) | X | [7] | |
| Mineralcorticoid receptor antagonists (MRA) | X | - | |
| Angiotensin receptor-neprilysin inhibitor sacubitril/valsartan | X | - | |
| Na+-glucose cotransporter-2 (SGLT2) inhibitors | X | [99,100] | |
| Beta-blockers (BBs) | X | [7,102] |
| Heart Failure in General | References | Propionic Acidemia | References | |
|---|---|---|---|---|
| Coenzyme Q10 | Not conclusive results. | [15,43] | Weak evidence | [25,29] |
| Vitamin D | Lack of evidence. Vitamin D deficiency has been described in some cases of reversible HF associated with severe hypocalcemia, but routine supplementation has not proven beneficial. | [15,83] | No data | - |
| Thiamine | Lack of evidence. Thiamine deficiency is recognized as a cause of HF, but routine supplementation has not proven beneficial. | [14,15] | No data | - |
| Carnitine | Lack of evidence | [15] | Carnitine supplementation is recommended to maintain its plasmatic level within the normal range, thus improving the metabolic stability of patients | [4] |
| Vitamin E | Potentially harmful. | [15] | No data | - |
| Selenium | Selenium deficiency is recognized as a cause of HF. A common consensus is building on the idea that only patients with selenium deficiency could benefit from selenium supplements. | [14,43,75] | No data | - |
| Iron | Iron deficiency has been associated with clinical instability. Treatment is recommended in the case of iron depletion. Routine oral supplementation has not proven beneficial. | [14,15] | No data | - |
| Zinc | Routine supplementation has not proven beneficial. Little evidence supports zinc supplementation in the case of zinc deficiency. | [15,86,88] | No data | - |
| Riboflavin | Multivitamins have not proven beneficial. | [15] | No data | - |
| Biotin | Multivitamins have not proven beneficial. | [15] | Its use in the chronic management of PA patients lacks evidence of efficacy | [57] |
Disclaimer/Publisher’s Note: The statements, opinions and data contained in all publications are solely those of the individual author(s) and contributor(s) and not of MDPI and/or the editor(s). MDPI and/or the editor(s) disclaim responsibility for any injury to people or property resulting from any ideas, methods, instructions or products referred to in the content. |
© 2023 by the authors. Licensee MDPI, Basel, Switzerland. This article is an open access article distributed under the terms and conditions of the Creative Commons Attribution (CC BY) license (https://creativecommons.org/licenses/by/4.0/).
Share and Cite
Maines, E.; Moretti, M.; Vitturi, N.; Gugelmo, G.; Fasan, I.; Lenzini, L.; Piccoli, G.; Gragnaniello, V.; Maiorana, A.; Soffiati, M.; et al. Understanding the Pathogenesis of Cardiac Complications in Patients with Propionic Acidemia and Exploring Therapeutic Alternatives for Those Who Are Not Eligible or Are Waiting for Liver Transplantation. Metabolites 2023, 13, 563. https://doi.org/10.3390/metabo13040563
Maines E, Moretti M, Vitturi N, Gugelmo G, Fasan I, Lenzini L, Piccoli G, Gragnaniello V, Maiorana A, Soffiati M, et al. Understanding the Pathogenesis of Cardiac Complications in Patients with Propionic Acidemia and Exploring Therapeutic Alternatives for Those Who Are Not Eligible or Are Waiting for Liver Transplantation. Metabolites. 2023; 13(4):563. https://doi.org/10.3390/metabo13040563
Chicago/Turabian StyleMaines, Evelina, Michele Moretti, Nicola Vitturi, Giorgia Gugelmo, Ilaria Fasan, Livia Lenzini, Giovanni Piccoli, Vincenza Gragnaniello, Arianna Maiorana, Massimo Soffiati, and et al. 2023. "Understanding the Pathogenesis of Cardiac Complications in Patients with Propionic Acidemia and Exploring Therapeutic Alternatives for Those Who Are Not Eligible or Are Waiting for Liver Transplantation" Metabolites 13, no. 4: 563. https://doi.org/10.3390/metabo13040563
APA StyleMaines, E., Moretti, M., Vitturi, N., Gugelmo, G., Fasan, I., Lenzini, L., Piccoli, G., Gragnaniello, V., Maiorana, A., Soffiati, M., Burlina, A., & Franceschi, R. (2023). Understanding the Pathogenesis of Cardiac Complications in Patients with Propionic Acidemia and Exploring Therapeutic Alternatives for Those Who Are Not Eligible or Are Waiting for Liver Transplantation. Metabolites, 13(4), 563. https://doi.org/10.3390/metabo13040563