


Computational Insights into Natural Antischistosomal Metabolites as SmHDAC8 Inhibitors: Molecular Docking, ADMET Profiling, and Molecular Dynamics Simulation

 , , ,
, , , 


Abstract
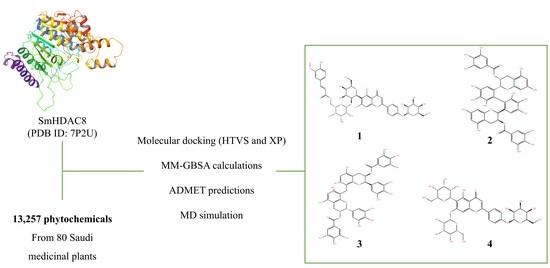
:
1. Introduction
2. Methods
2.1. Protein Retrieval and Preparation
2.2. Grid Generation of Protein Receptor
2.3. Ligands Preparation
2.4. Molecular Docking and MM-GBSA Binding Free Energy Calculation
2.5. In Silico ADME Prediction
2.6. Molecular Dynamics (MD) Simulations
3. Results
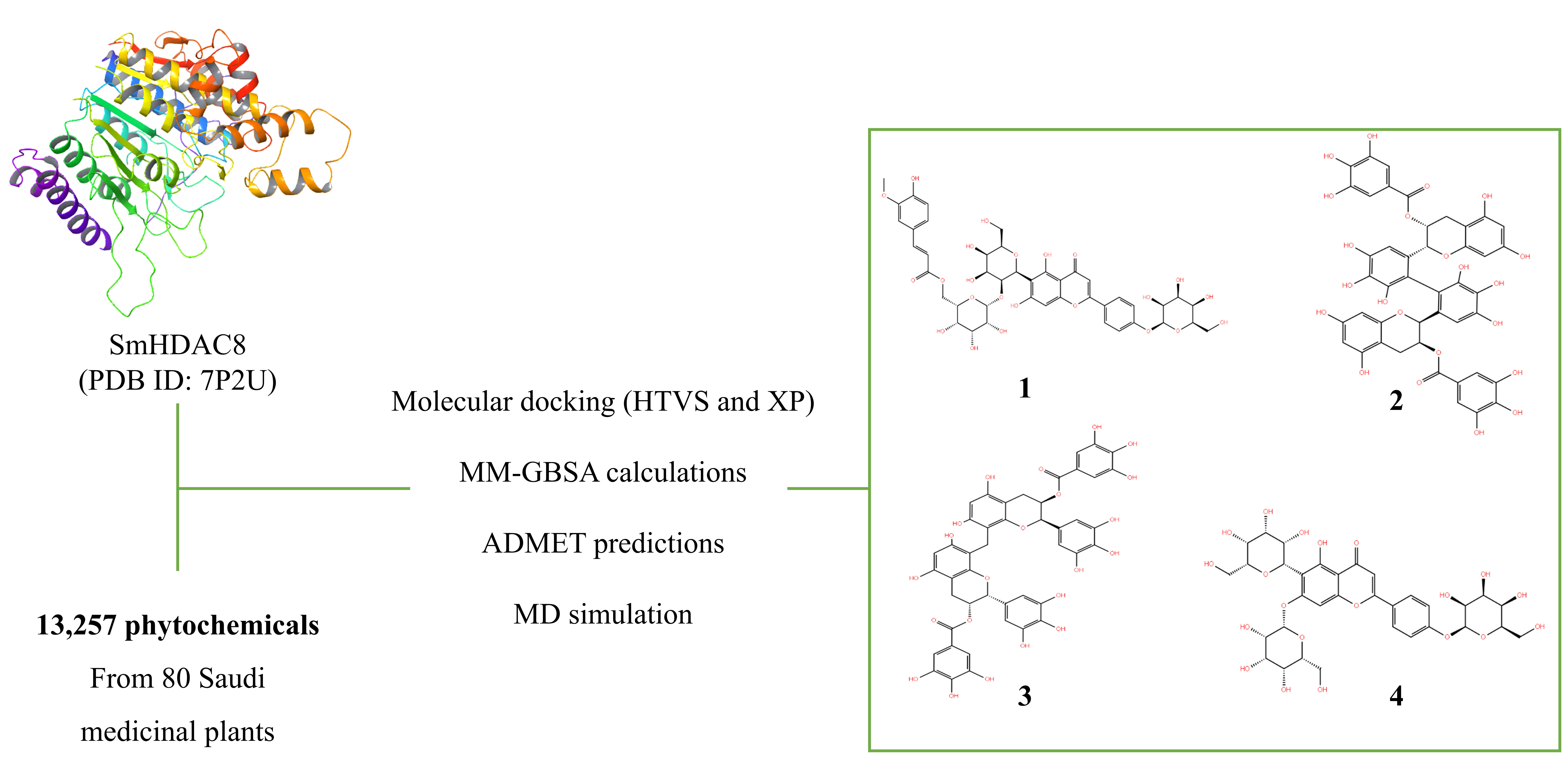
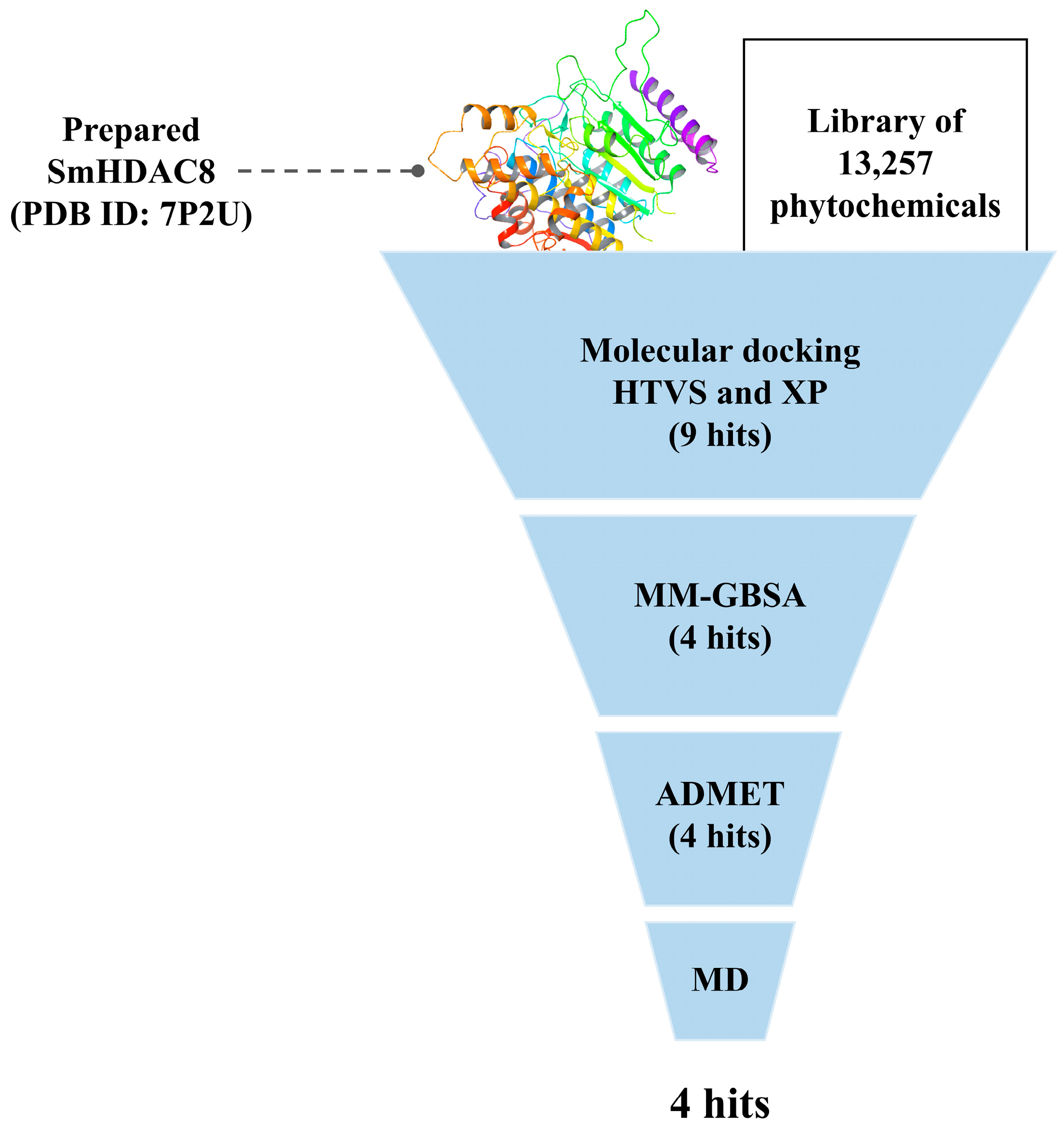
3.1. Molecular Docking and Free-Binding Energy Prediction
3.2. ADMET Prediction
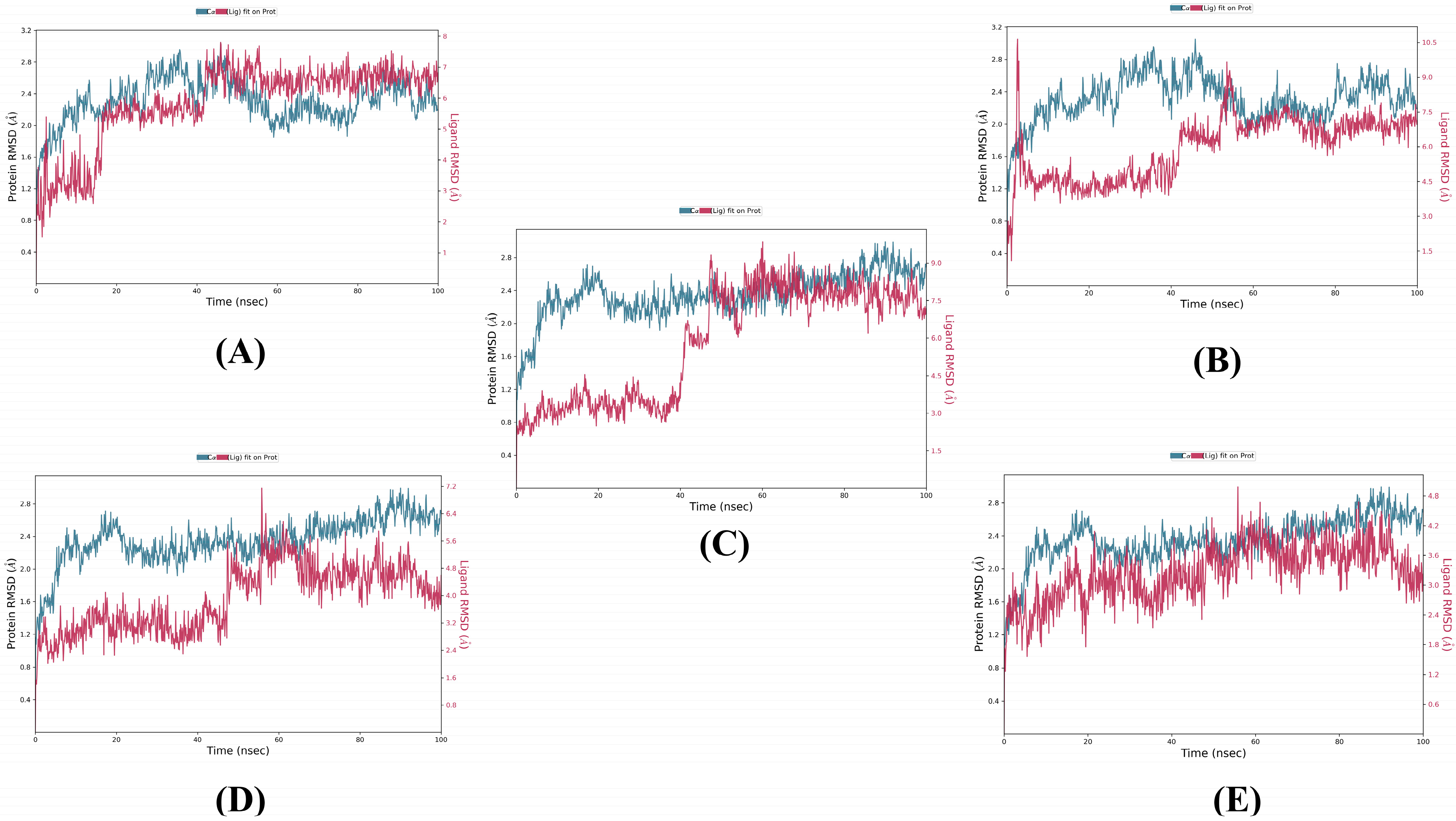
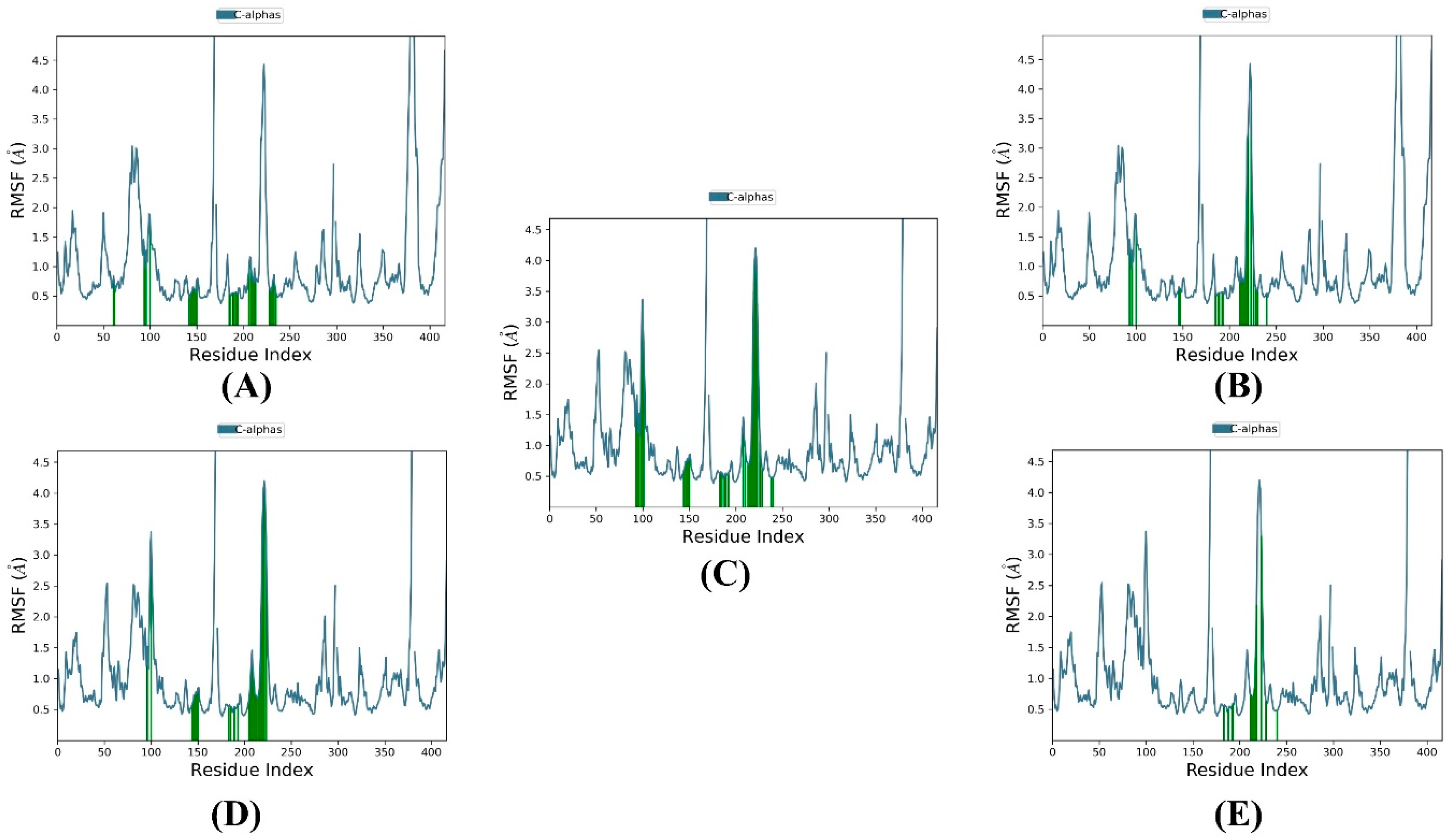
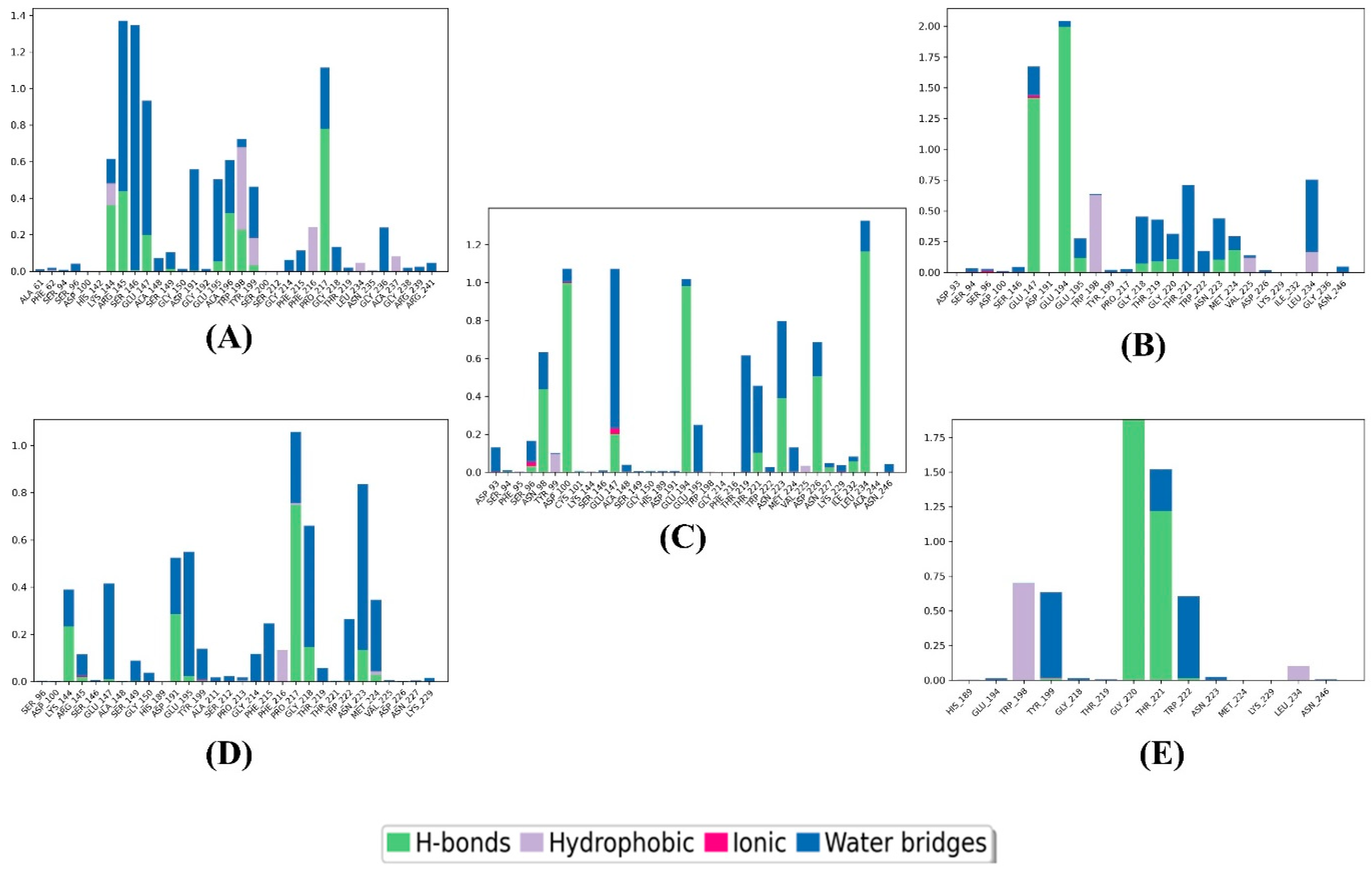
3.3. MD Simulation
4. Discussion
5. Conclusions
Author Contributions
Funding
Institutional Review Board Statement
Informed Consent Statement
Data Availability Statement
Conflicts of Interest
References
- Gundamaraju, R. Novel antipathy for schistosomiasis-the most lethal ailment of the tropical region. Asian Pac. J. Trop. Biomed. 2014, 4 (Suppl. 1), S43–S45. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Mathavan, I.; Liu, L.J.; Robinson, S.W.; El-Sakkary, N.; Elatico, A.J.J.; Gomez, D.; Nellas, R.; Owens, R.J.; Zuercher, W.; Navratilova, I.; et al. Identification of Inhibitors of the Schistosoma mansoni VKR2 Kinase Domain. ACS Med. Chem. Lett. 2022, 13, 1715–1722. [Google Scholar] [CrossRef] [PubMed]
- Verjee, M.A. Schistosomiasis: Still a Cause of Significant Morbidity and Mortality. Res. Rep. Trop. Med. 2019, 10, 153–163. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Santos, J.C.; Pereira, C.L.D.; Domingues, A.L.C.; Lopes, E.P. Noninvasive diagnosis of periportal fibrosis in schistosomiasis mansoni: A comprehensive review. World J. Hepatol. 2022, 14, 696–707. [Google Scholar] [CrossRef]
- Molehin, A.J. Schistosomiasis vaccine development: Update on human clinical trials. J. Biomed. Sci. 2020, 27, 28. [Google Scholar] [CrossRef]
- Eissa, M.M.; El-Azzouni, M.Z.; El-Khordagui, L.K.; Abdel Bary, A.; El-Moslemany, R.M.; Abdel Salam, S.A. Evaluation of prophylactic efficacy and safety of praziquantel-miltefosine nanocombination in experimental Schistosomiasis mansoni. Acta Trop. 2020, 212, 105714. [Google Scholar] [CrossRef]
- Saccoccia, F.; Pozzetti, L.; Gimmelli, R.; Butini, S.; Guidi, A.; Papoff, G.; Giannaccari, M.; Brogi, S.; Scognamiglio, V.; Gemma, S.; et al. Crystal structures of Schistosoma mansoni histone deacetylase 8 reveal a novel binding site for allosteric inhibitors. J. Biol. Chem. 2022, 298, 102375. [Google Scholar] [CrossRef]
- Heimburg, T.; Chakrabarti, A.; Lancelot, J.; Marek, M.; Melesina, J.; Hauser, A.T.; Shaik, T.B.; Duclaud, S.; Robaa, D.; Erdmann, F.; et al. Structure-Based Design and Synthesis of Novel Inhibitors Targeting HDAC8 from Schistosoma mansoni for the Treatment of Schistosomiasis. J. Med. Chem. 2016, 59, 2423–2435. [Google Scholar] [CrossRef] [Green Version]
- Haberland, M.; Montgomery, R.L.; Olson, E.N. The many roles of histone deacetylases in development and physiology: Implications for disease and therapy. Nat. Rev. Genet. 2009, 10, 32–42. [Google Scholar] [CrossRef] [Green Version]
- Chakrabarti, A.; Oehme, I.; Witt, O.; Oliveira, G.; Sippl, W.; Romier, C.; Pierce, R.J.; Jung, M. HDAC8: A multifaceted target for therapeutic interventions. Trends Pharmacol. Sci. 2015, 36, 481–492. [Google Scholar] [CrossRef] [Green Version]
- Wolfson, N.A.; Ann Pitcairn, C.; Fierke, C.A. HDAC8 Substrates: Histones and Beyond. Biopolymers 2013, 99, 112. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Gregoretti, I.V.; Lee, Y.M.; Goodson, H.V. Molecular evolution of the histone deacetylase family: Functional implications of phylogenetic analysis. J. Mol. Biol. 2004, 338, 17–31. [Google Scholar] [CrossRef] [PubMed]
- Dubois, F.; Caby, S.; Oger, F.; Cosseau, C.; Capron, M.; Grunau, C.; Dissous, C.; Pierce, R.J. Histone deacetylase inhibitors induce apoptosis, histone hyperacetylation and up-regulation of gene transcription in Schistosoma mansoni. Mol. Biochem. Parasitol. 2009, 168, 7–15. [Google Scholar] [CrossRef] [PubMed]
- Lancelot, J.; Caby, S.; Dubois-Abdesselem, F.; Vanderstraete, M.; Trolet, J.; Oliveira, G.; Bracher, F.; Jung, M.; Pierce, R.J. Schistosoma mansoni Sirtuins: Characterization and Potential as Chemotherapeutic Targets. PLoS Negl. Trop. Dis. 2013, 7, e2428. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Saccoccia, F.; Brindisi, M.; Gimmelli, R.; Relitti, N.; Guidi, A.; Saraswati, A.P.; Cavella, C.; Brogi, S.; Chemi, G.; Butini, S.; et al. Screening and Phenotypical Characterization of Schistosoma mansoni Histone Deacetylase 8 (SmHDAC8) Inhibitors as Multistage Antischistosomal Agents. ACS Infect. Dis. 2020, 6, 100–113. [Google Scholar] [CrossRef]
- Oger, F.; Dubois, F.; Caby, S.; Noël, C.; Cornette, J.; Bertin, B.; Capron, M.; Pierce, R.J. The class I histone deacetylases of the platyhelminth parasite Schistosoma mansoni. Biochem. Biophys. Res. Commun. 2008, 377, 1079–1084. [Google Scholar] [CrossRef]
- Nakagawa, M.; Oda, Y.; Eguchi, T.; Aishima, S.I.; Yao, T.; Hosoi, F.; Basaki, Y.; Ono, M.; Kuwano, M.; Tanaka, M.; et al. Expression profile of class I histone deacetylases in human cancer tissues. Oncol. Rep. 2007, 18, 769–774. [Google Scholar] [CrossRef]
- Hu, E.; Chen, Z.; Fredrickson, T.; Zhu, Y.; Kirkpatrick, R.; Zhang, G.F.; Johanson, K.; Sung, C.M.; Liu, R.; Winkler, J. Cloning and characterization of a novel human class I histone deacetylase that functions as a transcription repressor. J. Biol. Chem. 2000, 275, 15254–15264. [Google Scholar] [CrossRef] [Green Version]
- Mosquera-yuqui, F.; Lopez-guerra, N.; Moncayo-palacio, E.A. Targeting the 3CLpro and RdRp of SARS-CoV-2 with phytochemicals from medicinal plants of the Andean Region: Molecular docking and molecular dynamics simulations. J. Biomol. Struct. Dyn. 2020, 40, 2010–2023. [Google Scholar] [CrossRef]
- Ndegwa, F.K.; Kondam, C.; Aboagye, S.Y.; Margaret, E.; Mbugua, P.K.; Okemo, P.O.; Williams, D.L.; Hagen, T.J. Traditional Kenyan herbal medicine: Exploring natural products’ therapeutics against schistosomiasis. J. Helminthol. 2022, 96, e16. [Google Scholar] [CrossRef]
- Eltaib, L.; Alzain, A.A. Targeting the omicron variant of SARS-CoV-2 with phytochemicals from Saudi medicinal plants: Molecular docking combined with molecular dynamics investigations. J. Biomol. Struct. Dyn. 2022, 1–13. [Google Scholar] [CrossRef] [PubMed]
- Mtemeli, F.L.; Ndlovu, J.; Mugumbate, G.; Makwikwi, T.; Shoko, R. Advances in schistosomiasis drug discovery based on natural products. All Life 2022, 15, 608–622. [Google Scholar] [CrossRef]
- Moreira-Filho, J.T.; Silva, A.C.; Dantas, R.F.; Gomes, B.F.; Souza Neto, L.R.; Brandao-Neto, J.; Owens, R.J.; Furnham, N.; Neves, B.J.; Silva-Junior, F.P.; et al. Schistosomiasis Drug Discovery in the Era of Automation and Artificial Intelligence. Front. Immunol. 2021, 12, 642383. [Google Scholar] [CrossRef] [PubMed]
- Fu, T.T.; Tu, G.; Ping, M.; Zheng, G.X.; Yang, F.Y.; Yang, J.Y.; Zhang, Y.; Yao, X.J.; Xue, W.W.; Zhu, F. Subtype-selective mechanisms of negative allosteric modulators binding to group I metabotropic glutamate receptors. Acta Pharmacol. Sin. 2021, 42, 1354–1367. [Google Scholar] [CrossRef]
- Alzain, A.A.; Elbadwi, F.A. Identification of novel TMPRSS2 inhibitors for COVID-19 using e-pharmacophore modelling, molecular docking, molecular dynamics and quantum mechanics studies. Inform. Med. Unlocked 2021, 26, 100758. [Google Scholar] [CrossRef]
- Friesner, R.A.; Murphy, R.B.; Repasky, M.P.; Frye, L.L.; Greenwood, J.R.; Halgren, T.A.; Sanschagrin, P.C.; Mainz, D.T. Extra precision glide: Docking and scoring incorporating a model of hydrophobic enclosure for protein-ligand complexes. J. Med. Chem. 2006, 49, 6177–6196. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Alzain, A.A.; Ismail, A.; Fadlelmola, M.; Mohamed, M.A.; Mahjoub, M.; Makki, A.A.; Elsaman, T. De novo design of novel spike glycoprotein inhibitors using e-pharmacophore modeling, molecular hybridization, ADMET, quantum mechanics and molecular dynamics studies for COVID-19. Pak. J. Pharm. Sci. 2022, 35, 313–321. [Google Scholar]
- Elbadwi, F.A.; Khairy, E.A.; Alsamani, F.O.; Mahadi, M.A.; Abdalrahman, S.E.; Alsharf, Z.; Ahmed, M.; Elsayed, I.; Ibraheem, W.; Alzain, A.A. Informatics in Medicine Unlocked Identification of novel transmembrane Protease Serine Type 2 drug candidates for COVID-19 using computational studies. Inform. Med. Unlocked 2021, 26, 100725. [Google Scholar] [CrossRef]
- Bowers, K.; Chow, E.; Huageng, X.; Dror, R.; Eastwood, M.; Gregersen, B.; Klepeis, J.; Kolossváry, I.; Moraes, M.; Sacerdoti, F.; et al. Scalable Algorithms for Molecular Dynamics Simulations on Commodity Clusters; D. E. Shaw Research: New York, NY, USA, 2006; ISBN 0-7695-2700-0. [Google Scholar]
- Marek, M.; Kannan, S.; Hauser, A.T.; Moraes Mourão, M.; Caby, S.; Cura, V.; Stolfa, D.A.; Schmidtkunz, K.; Lancelot, J.; Andrade, L.; et al. Structural Basis for the Inhibition of Histone Deacetylase 8 (HDAC8), a Key Epigenetic Player in the Blood Fluke Schistosoma mansoni. PLoS Pathog. 2013, 9, e1003645. [Google Scholar] [CrossRef]
- Abela-Ridder, B.; Biswas, G.; Mbabazi, P.S.; Craven, M.; Gerber, A.; Hartenstein, L.; Vlcek, J.; Malecela, M.N.; Polo, M.R.; Tiendrebeogo, A.; et al. Ending the Neglect to Attain the Sustainable Development Goals: A Road Map for Neglected Tropical Diseases 2021–2030; WHO: Geneva, Switzerland, 2020; ISBN 9789240010352. [Google Scholar]
- Simoben, C.V.; Robaa, D.; Chakrabarti, A.; Schmidtkunz, K.; Marek, M.; Lancelot, J.; Kannan, S.; Melesina, J.; Shaik, T.B.; Pierce, R.J.; et al. A novel class of Schistosoma mansoni histone deacetylase 8 (HDAC8) inhibitors identified by structure-based virtual screening and in vitro testing. Molecules 2018, 23, 566. [Google Scholar] [CrossRef] [Green Version]
- Ghazy, E.; Abdelsalam, M.; Robaa, D.; Pierce, R.J.; Sippl, W. Histone Deacetylase (HDAC) Inhibitors for the Treatment of Schistosomiasis. Pharmaceuticals 2022, 15, 80. [Google Scholar] [CrossRef] [PubMed]
- Alzain, A.A. Insights from computational studies on the potential of natural compounds as inhibitors against SARS-CoV-2 spike omicron variant. SAR QSAR Environ. Res. 2022, 33, 953–968. [Google Scholar] [CrossRef] [PubMed]
- Maguire, J.H. Trematodes (Schistosomes and Liver, Intestinal, and Lung Flukes). In Mandell, Douglas, and Bennett’s Principles and Practice of Infectious Diseases; Bennett, J.E., Dolin, R., Blaser, M.J., Eds.; Elsevier: Amsterdam, The Netherlands, 2020; pp. 3451–3462. ISBN 9780323550277. [Google Scholar]
- Zeiger, E. The test that changed the world: The Ames test and the regulation of chemicals. Mutat. Res. Toxicol. Environ. Mutagen. 2019, 841, 43–48. [Google Scholar] [CrossRef] [PubMed]
- Aghajani, J.; Farnia, P.; Farnia, P.; Ghanavi, J.; Velayati, A.A. Molecular Dynamic Simulations and Molecular Docking as a Potential Way for Designed New Inhibitor Drug without Resistance. Tanaffos 2022, 21, 1–14. [Google Scholar] [PubMed]
- Jordaan, M.A.; Ebenezer, O.; Damoyi, N.; Shapi, M. Virtual screening, molecular docking studies and DFT calculations of FDA approved compounds similar to the non-nucleoside reverse transcriptase inhibitor (NNRTI) efavirenz. Heliyon 2020, 6, e04642. [Google Scholar] [CrossRef] [PubMed]
- Kumar, A.; Rathi, E.; Kini, S.G. E-pharmacophore modelling, virtual screening, molecular dynamics simulations and in-silico ADME analysis for identification of potential E6 inhibitors against cervical cancer. J. Mol. Struct. 2019, 1189, 299–306. [Google Scholar] [CrossRef]
- Falcone Ferreyra, M.L.; Rius, S.P.; Casati, P. Flavonoids: Biosynthesis, biological functions, and biotechnological applications. Front. Plant Sci. 2012, 3, 222. [Google Scholar] [CrossRef] [Green Version]
- Panche, A.N.; Diwan, A.D.; Chandra, S.R. Flavonoids: An overview. J. Nutr. Sci. 2016, 5, e47. [Google Scholar] [CrossRef] [Green Version]
- Dias, M.C.; Pinto, D.C.G.A.; Silva, A.M.S. Plant flavonoids: Chemical characteristics and biological activity. Molecules 2021, 26, 5377. [Google Scholar] [CrossRef]
- Abou-Zaid, M.M.; Lombardo, D.A.; Kite, G.C.; Grayer, R.J.; Veitch, N.C. Acylated flavone C-glycosides from Cucumis sativus. Phytochemistry 2001, 58, 167–172. [Google Scholar] [CrossRef]
- Krauze-Baranowska, M.; Cisowski, W. High-performance liquid chromatographic determination of flavone C-glycosides in some species of the Cucurbitaceae family. J. Chromatogr. A 1994, 675, 240–243. [Google Scholar] [CrossRef]
- Jo, H.E.; Son, S.Y.; Lee, C.H. Comparison of Metabolome and Functional Properties of Three Korean Cucumber Cultivars. Front. Plant Sci. 2022, 13, 1174. [Google Scholar] [CrossRef] [PubMed]
- Marinova, K.; Kleinschmidt, K.; Weissenböck, G.; Klein, M. Flavonoid biosynthesis in barley primary leaves requires the presence of the vacuole and controls the activity of vacuolar flavonoid transport. Plant Physiol. 2007, 144, 432–444. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Shankar, E.; Goel, A.; Gupta, K.; Gupta, S. Plant flavone apigenin: An emerging anticancer agent. Curr. Pharmacol. Rep. 2017, 3, 423–446. [Google Scholar] [CrossRef] [PubMed]
- NONAKA, G.; KAWAHARA, O.; NISHIOKA, I. Tannins and Related Compounds. XV. A New Class of Dimeric Flavan-3-ol Gallates, Theasinensins A and B, and Proanthocyanidin Gallates from Green Tea Leaf. (1). Chem. Pharm. Bull. 1983, 31, 3906–3914. [Google Scholar] [CrossRef] [Green Version]
- HASHIMOTO, F.; NONAKA, G.; NISHIOKA, I. Tannins and Related Compounds. XC.: 8-C-Ascorbyl (-)-Epigallocatechin 3-O-Gallate and Novel Dimeric Flavan -3-ols, Oolonghomobisflavans A and B, from Oolong Tea. (3). Chem. Pharm. Bull. 1989, 37, 3255–3263. [Google Scholar] [CrossRef]
- Shaker, E.M.; Al-Shaibani, K.T.M.; Al-Abodi, H.R. jameel Effect of alcohol extract of green tea plant camellia sinensis as a therapeutic treatment of parasite entamoeba histolytica. Plant Arch. 2018, 18, 953–959. [Google Scholar]
- Hajihossein, R.; Eslamirad, Z.; Rafiei, F.; Naderi, G.; Assadi, M. Anti-acanthamoeba effect of Camellia sinensis extract (black and green tea) in vitro. J. Med. Plants 2020, 19, 163–169. [Google Scholar] [CrossRef]
- Fakae, L.B.; Stevenson, C.W.; Zhu, X.-Q.; Elsheikha, H.M. In vitro activity of Camellia sinensis (green tea) against trophozoites and cysts of Acanthamoeba castellanii. Int. J. Parasitol. Drugs Drug Resist. 2020, 13, 59–72. [Google Scholar] [CrossRef]
- Garcia, R.A. Catechins as Emerging and Promising Antiparasitic Agents. Biomed. J. Sci. Tech. Res. 2020, 30, 23065–23071. [Google Scholar] [CrossRef]
- Paveto, C.; Güida, M.C.; Esteva, M.I.; Martino, V.; Coussio, J.; Flawiá, M.M.; Torres, H.N. Anti-Trypanosoma cruzi activity of green tea (Camellia sinensis) catechins. Antimicrob. Agents Chemother. 2004, 48, 69–74. [Google Scholar] [CrossRef] [PubMed] [Green Version]







{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
| Lotus ID | Docking Score | MM-GBSA dG Binding Energy |
|---|---|---|
| LTS0233470 (compound 1) | −10.782 | −55.14 |
| LTS0020703 (compound 2) | −10.375 | −55.69 |
| LTS0033093 (compound 3) | −10.114 | −56.02 |
| LTS0028823 (compound 4) | −8.046 | −54.66 |
| LTS0172554 | −8.021 | −42.4 |
| LTS0270922 | −7.627 | −28.38 |
| LTS0108335 | −7.292 | −30.11 |
| LTS0146028 | −7.271 | −33.35 |
| LTS0031359 | −7.054 | −31.26 |
| Co-crystalized ligand (PDB ID 7P2U) | −5.441 | −53.16 |
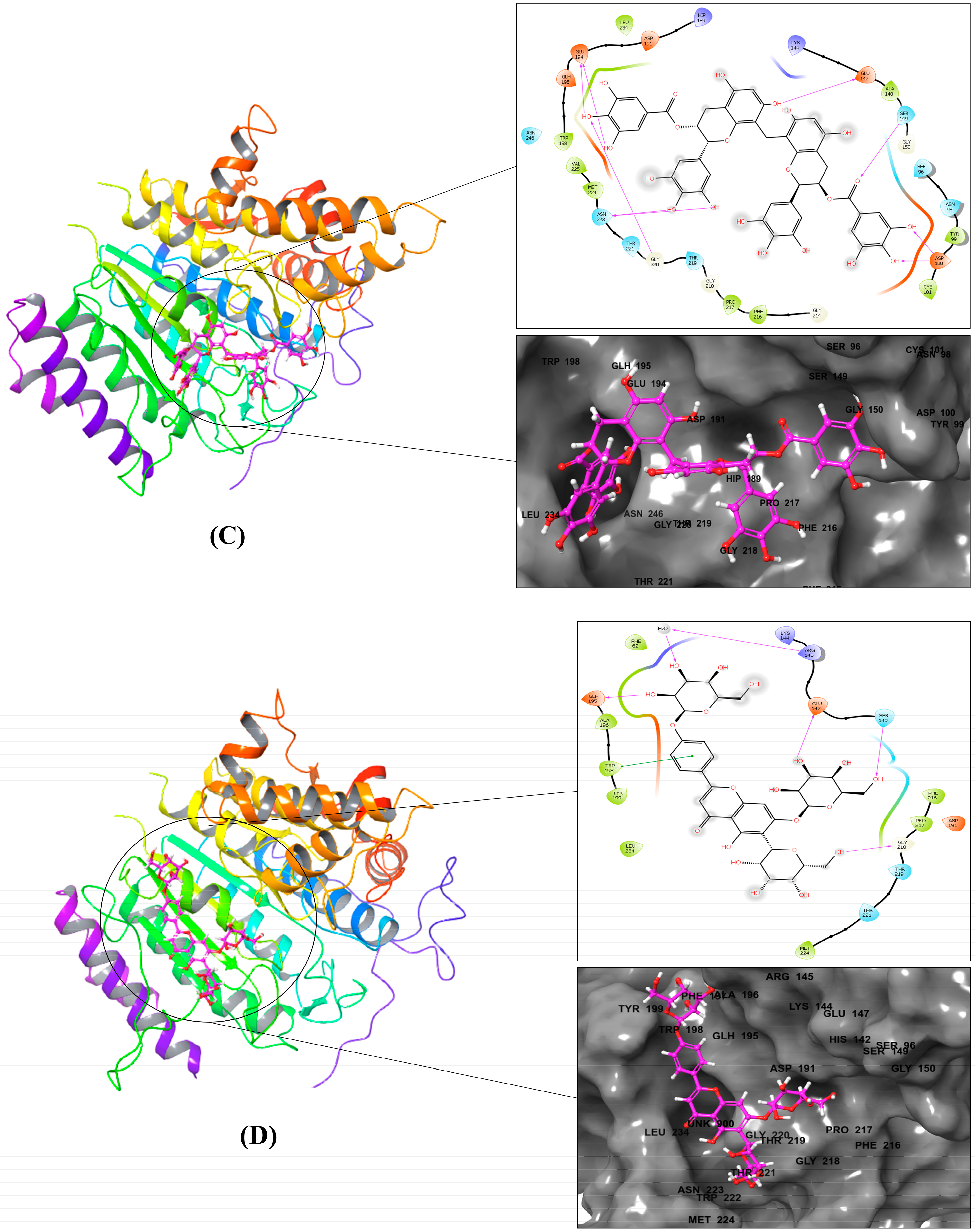
| Compound | Pi–pi Stacking Interaction | H-Bond Interaction | Hydrophobic Interaction | Other Interactions |
|---|---|---|---|---|
| 1 | - | LYS144, GLU147, ASP191, GLH195, ALA196, PRO217, THR219, ARG239 | PHE62, ALA196, PHE197, TRP198, TYR199, PHE216, PRO217 | Charged (positive): ARG139, LYS144, ARG145 Charged (negative): GLU147, ASP191, GLH195 Polar: SER149, THR219 |
| 2 | TRP198 | SER94, SER146, GLU194, GLH195, GLY220 | PHE95, ALA196, TRP198, TYR199, LEU234 | Charged (positive): LYS144, ARG145, HIP189 Charged (negative): GLU66, GLU147, ASP191, GLU194, GLH195 Polar: SER94, SER146, THR219, THR221, ASN223, ASN246 |
| 3 | - | ASP100, GLU147, SER149, GLU194, GLY220, ASN223 | TYR99, CYS101, ALA148, TRP198, PHE216, PRO217, MET224, VAL225, LEU234 | Charged (positive): LYS144, HIP189 Charged (negative): ASP100, GLU147, ASP191, GLU194, GLH195 Polar: SER96, ASN98, SER149, THR219, THR221, ASN223, ASN246 |
| 4 | TRP198 | ARG145, GLU147, SER149, GLH195, GLY218 | PHE62, ALA196, TRP198, TYR199, PHE216, PRO217, MET224, LEU234 | Charged (positive): LYS144, ARG145 Charged (negative): GLU147, ASP191, GLH195 Polar: SER149, THR219, THR221 |
| Compound | Absorption | Distribution | Metabolism | Excretion | Toxicity | ||||||
|---|---|---|---|---|---|---|---|---|---|---|---|
| CYP | |||||||||||
| 2D6 | 3A4 | 2D6 | 3A4 | 1A2 | 2C9 | ||||||
| Water Solubility | Blood-Brain Barrier Permeability | Substrate | Inhibitor | Renal OCT2 Substrate | AMES Toxicity | Hepatotoxicity | |||||
| Numeric (log mol/L) | Numeric (log BB) | Categorical (Yes/No) | Categorical (Yes/No) | Categorical (Yes/No) | |||||||
| 1 | −2.892 | −3.828 | No | No | No | No | No | No | No | No | No |
| 2 | −2.892 | −4.312 | No | No | No | No | No | No | Yes | No | No |
| 3 | −2.892 | −3.417 | No | No | No | No | No | No | No | No | No |
| 4 | −2.892 | 0.869 | No | No | No | No | No | No | No | No | No |
| Reference | −2.892 | −0.273 | No | No | No | No | Yes | No | No | Yes | No |
Disclaimer/Publisher’s Note: The statements, opinions and data contained in all publications are solely those of the individual author(s) and contributor(s) and not of MDPI and/or the editor(s). MDPI and/or the editor(s) disclaim responsibility for any injury to people or property resulting from any ideas, methods, instructions or products referred to in the content. |
© 2023 by the authors. Licensee MDPI, Basel, Switzerland. This article is an open access article distributed under the terms and conditions of the Creative Commons Attribution (CC BY) license (https://creativecommons.org/licenses/by/4.0/).
Share and Cite
Alzain, A.A.; Mukhtar, R.M.; Abdelmoniem, N.; Elbadwi, F.A.; Hussien, A.; Garelnabi, E.A.E.; Osman, W.; Sherif, A.E.; Khedr, A.I.M.; Ghazawi, K.F.; et al. Computational Insights into Natural Antischistosomal Metabolites as SmHDAC8 Inhibitors: Molecular Docking, ADMET Profiling, and Molecular Dynamics Simulation. Metabolites 2023, 13, 658. https://doi.org/10.3390/metabo13050658
Alzain AA, Mukhtar RM, Abdelmoniem N, Elbadwi FA, Hussien A, Garelnabi EAE, Osman W, Sherif AE, Khedr AIM, Ghazawi KF, et al. Computational Insights into Natural Antischistosomal Metabolites as SmHDAC8 Inhibitors: Molecular Docking, ADMET Profiling, and Molecular Dynamics Simulation. Metabolites. 2023; 13(5):658. https://doi.org/10.3390/metabo13050658
Chicago/Turabian StyleAlzain, Abdulrahim A., Rua M. Mukhtar, Nihal Abdelmoniem, Fatima A. Elbadwi, Amira Hussien, Elrashied A. E. Garelnabi, Wadah Osman, Asmaa E. Sherif, Amgad I. M. Khedr, Kholoud F. Ghazawi, and et al. 2023. "Computational Insights into Natural Antischistosomal Metabolites as SmHDAC8 Inhibitors: Molecular Docking, ADMET Profiling, and Molecular Dynamics Simulation" Metabolites 13, no. 5: 658. https://doi.org/10.3390/metabo13050658
APA StyleAlzain, A. A., Mukhtar, R. M., Abdelmoniem, N., Elbadwi, F. A., Hussien, A., Garelnabi, E. A. E., Osman, W., Sherif, A. E., Khedr, A. I. M., Ghazawi, K. F., Samman, W. A., Ibrahim, S. R. M., Mohamed, G. A., & Ashour, A. (2023). Computational Insights into Natural Antischistosomal Metabolites as SmHDAC8 Inhibitors: Molecular Docking, ADMET Profiling, and Molecular Dynamics Simulation. Metabolites, 13(5), 658. https://doi.org/10.3390/metabo13050658