
A Holistic Approach from Systems Biology Reveals the Direct Influence of the Quorum-Sensing Phenomenon on Pseudomonas aeruginosa Metabolism to Pyoverdine Biosynthesis

 ,
, 
Abstract
:1. Introduction
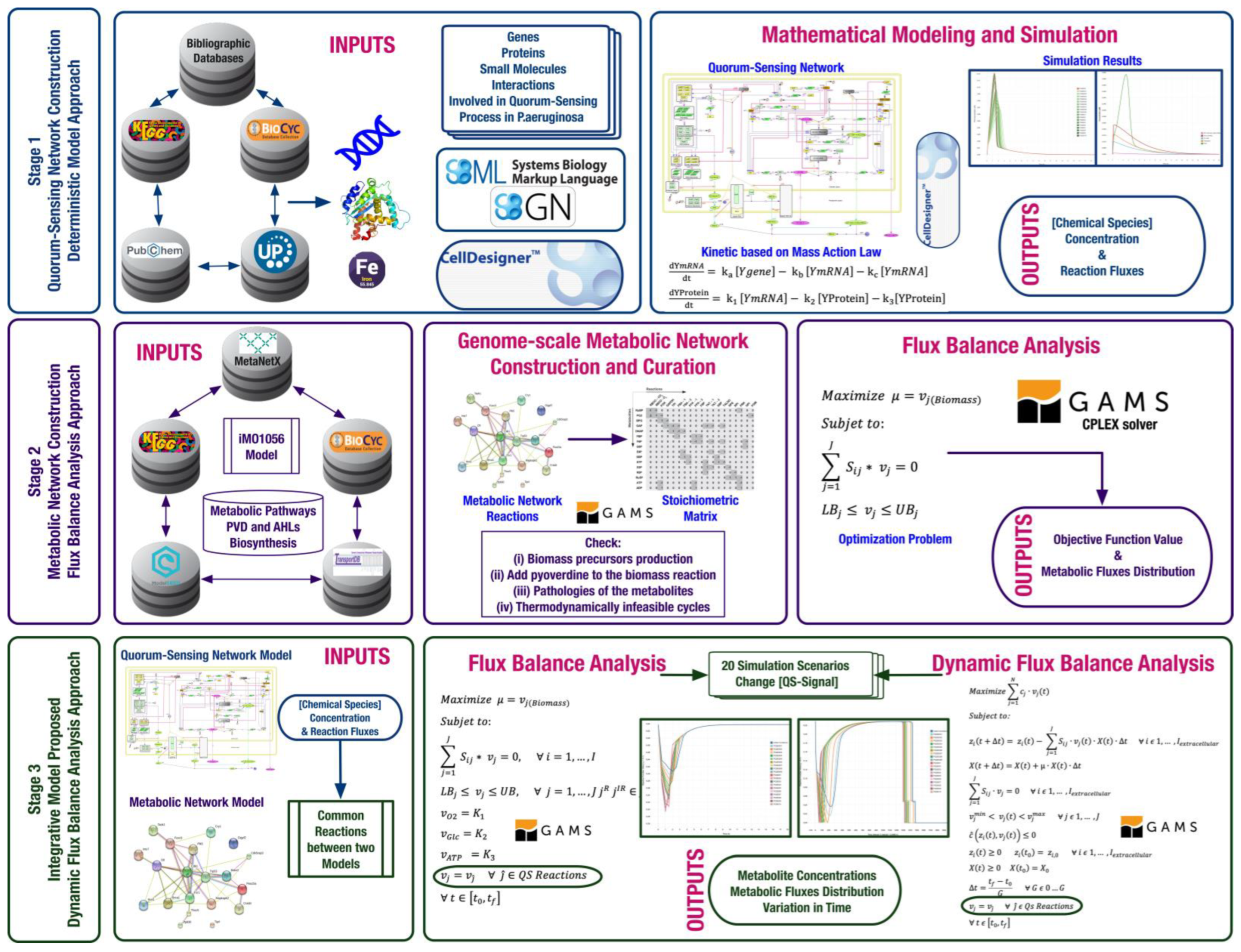
2. Materials and Methods
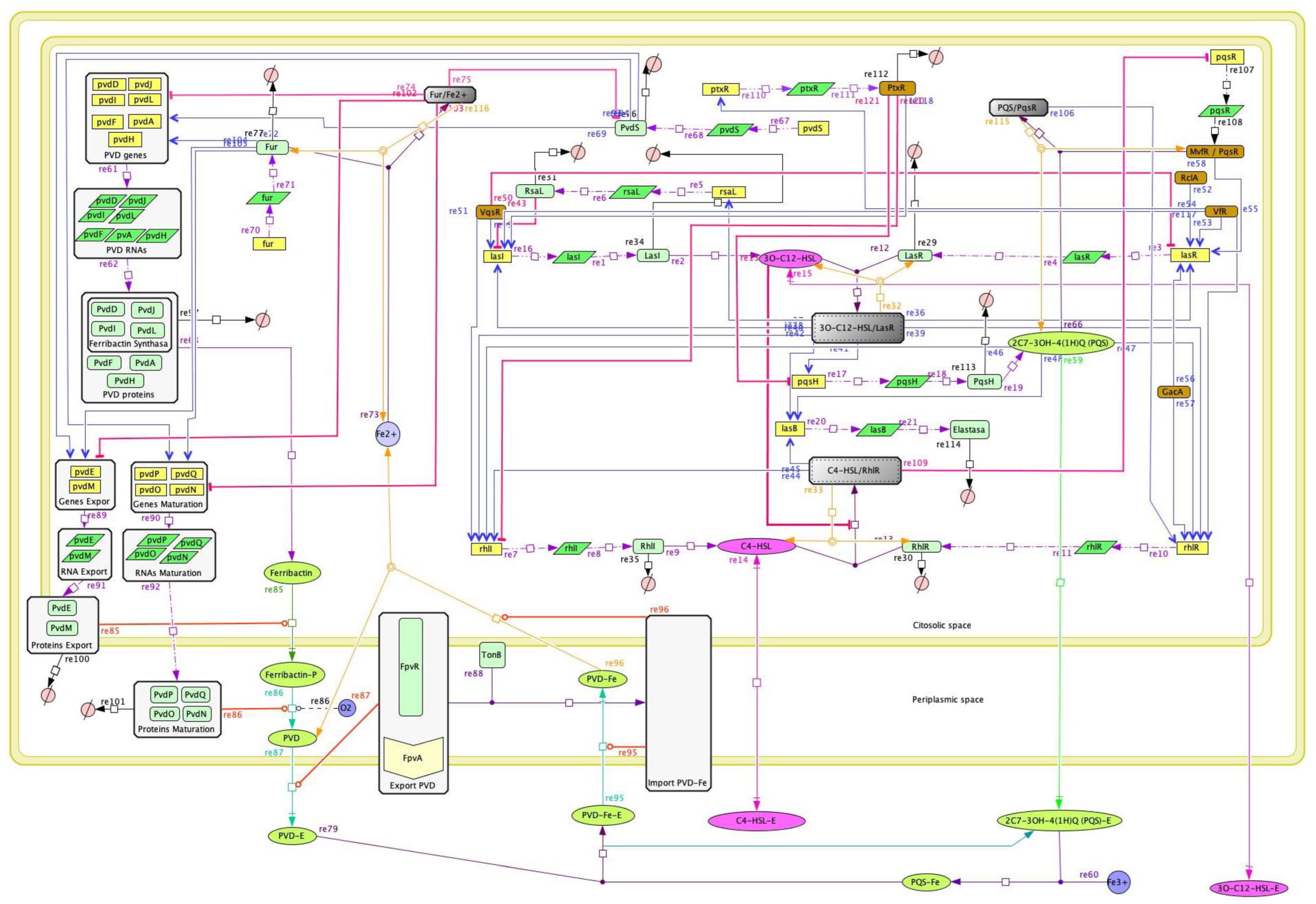
2.1. Construction and Modeling of the Quorum-Sensing Gene Regulatory Network
Simulation Scenario Conditions for the Quorum-Sensing Network
2.2. Construction of the Pseudomonas aeruginosa Metabolic Network
2.2.1. Curation of the Pseudomonas aeruginosa Metabolic Network
2.2.2. Modeling of the Pseudomonas aeruginosa Metabolic Network Using a Steady-State FBA Approximation
2.3. Combining the QS Gene Regulatory Network and the Pseudomonas aeruginosa Metabolic Network Models into an Integrative Model
2.3.1. Design of Simulation Scenarios
2.3.2. Simulation Using the Multi-Stage FBA Approximation
2.3.3. Simulation Using the DFBA Approximation
2.4. Cultures of Pseudomonas aeruginosa Strain PAO1
2.4.1. Evaluation of Bacterial Growth
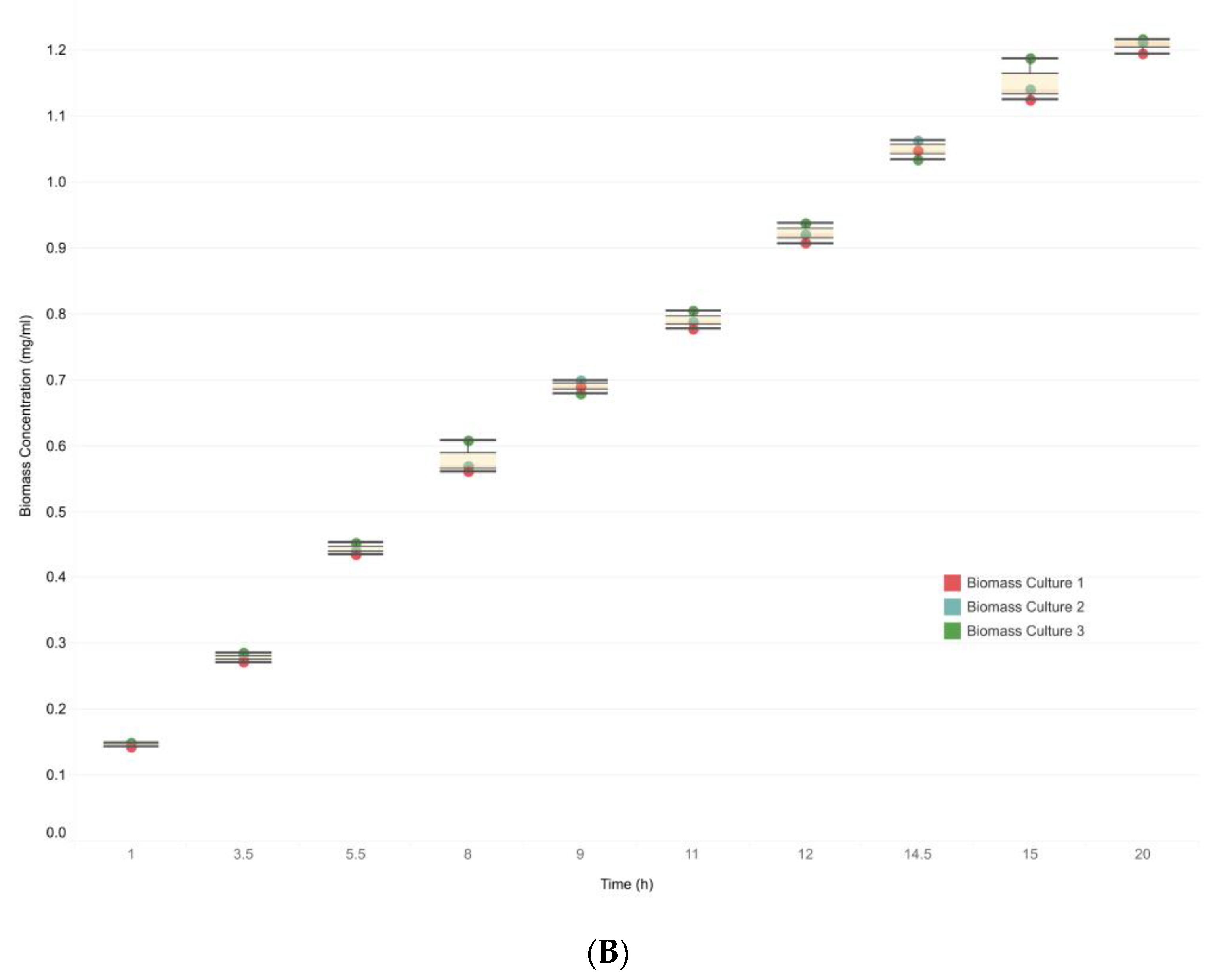
2.4.2. Evaluation of Biomass Production
3. Results
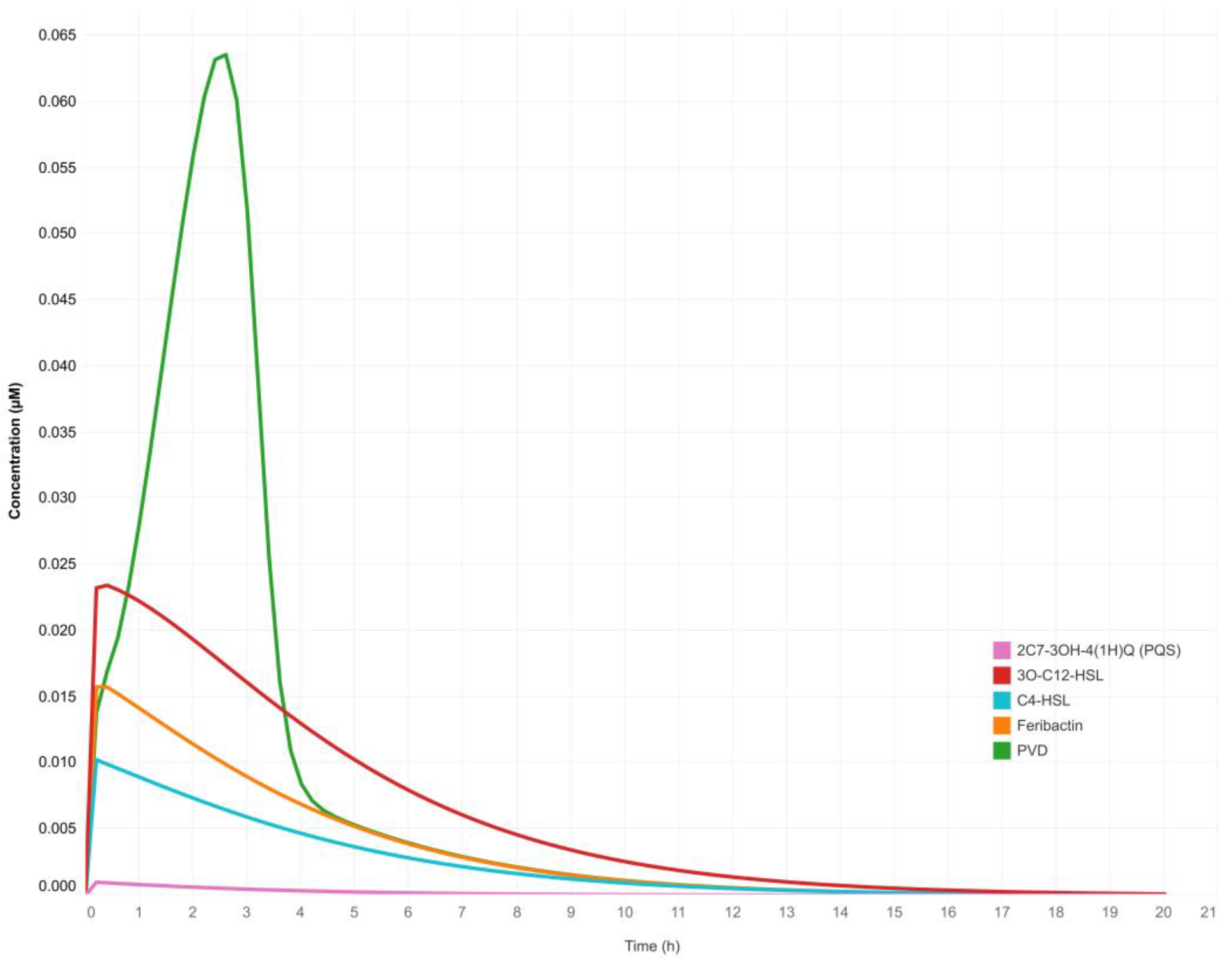
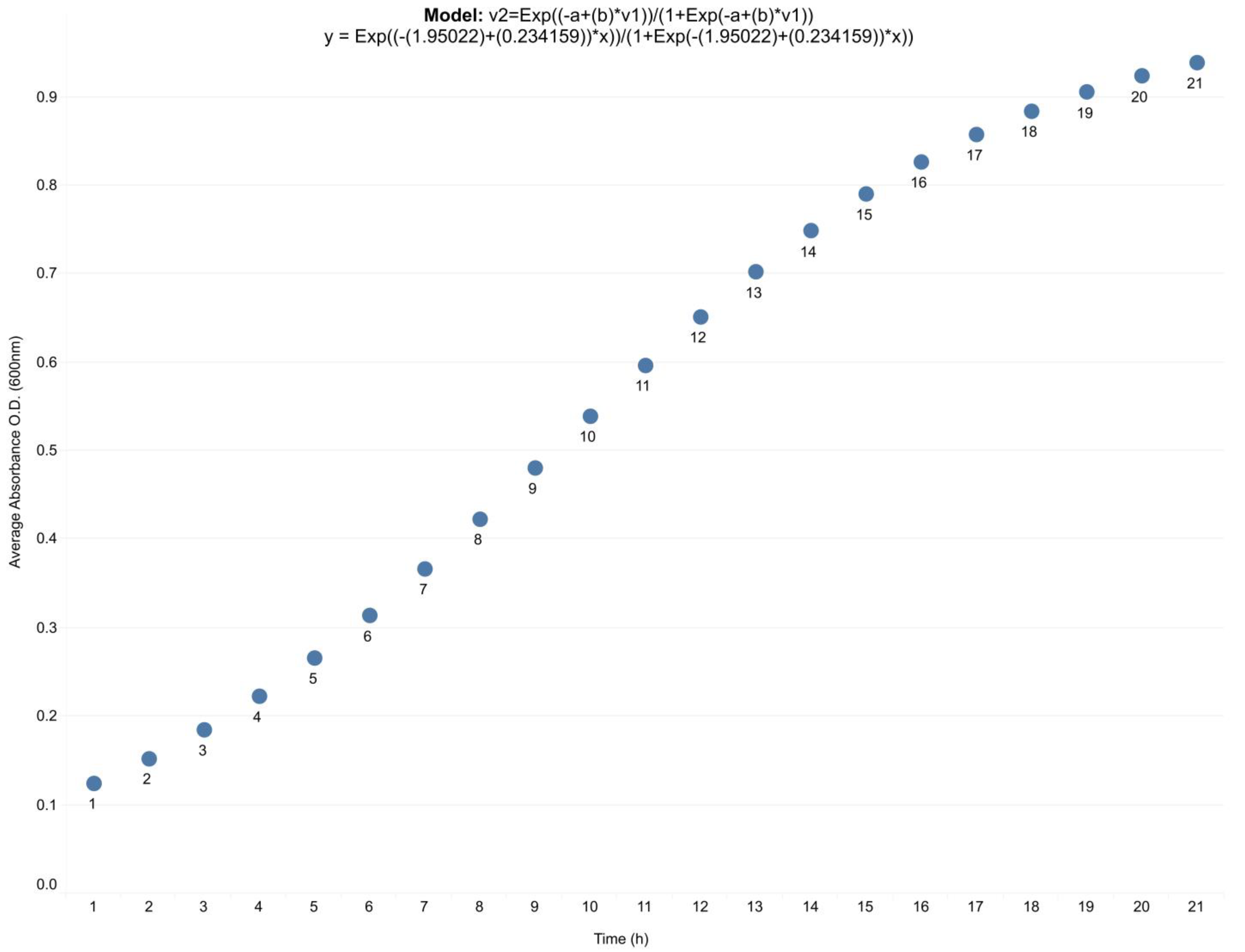
3.1. The Quorum-Sensing Gene Regulatory Network for Pyoverdine Expression in Pseudomonas aeruginosa: A Deterministic Model

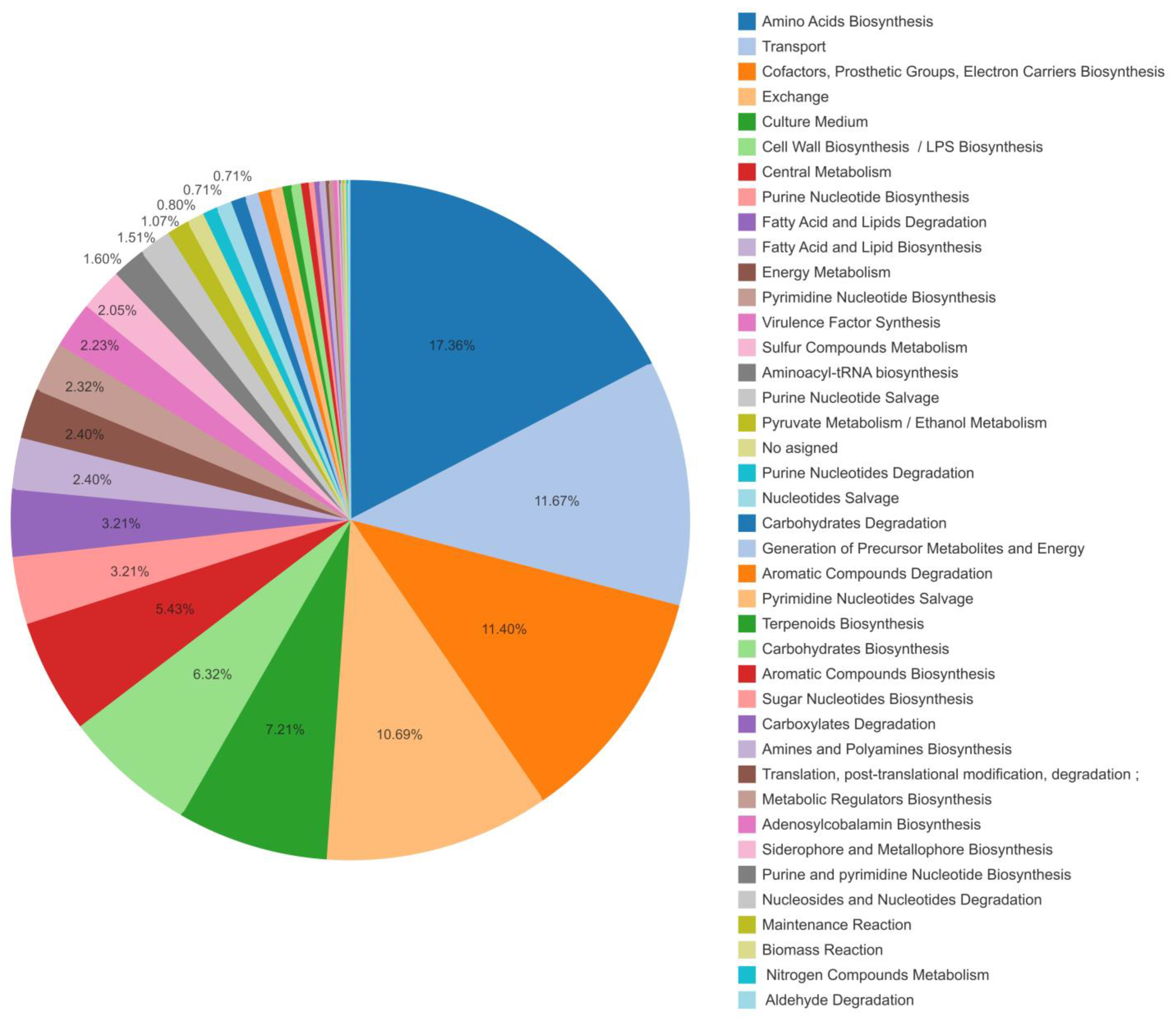
3.2. Pseudomonas aeruginosa Metabolic Network Model CCBM1146: An Improved Version of the Genome-Scale Metabolic Model iMO1056
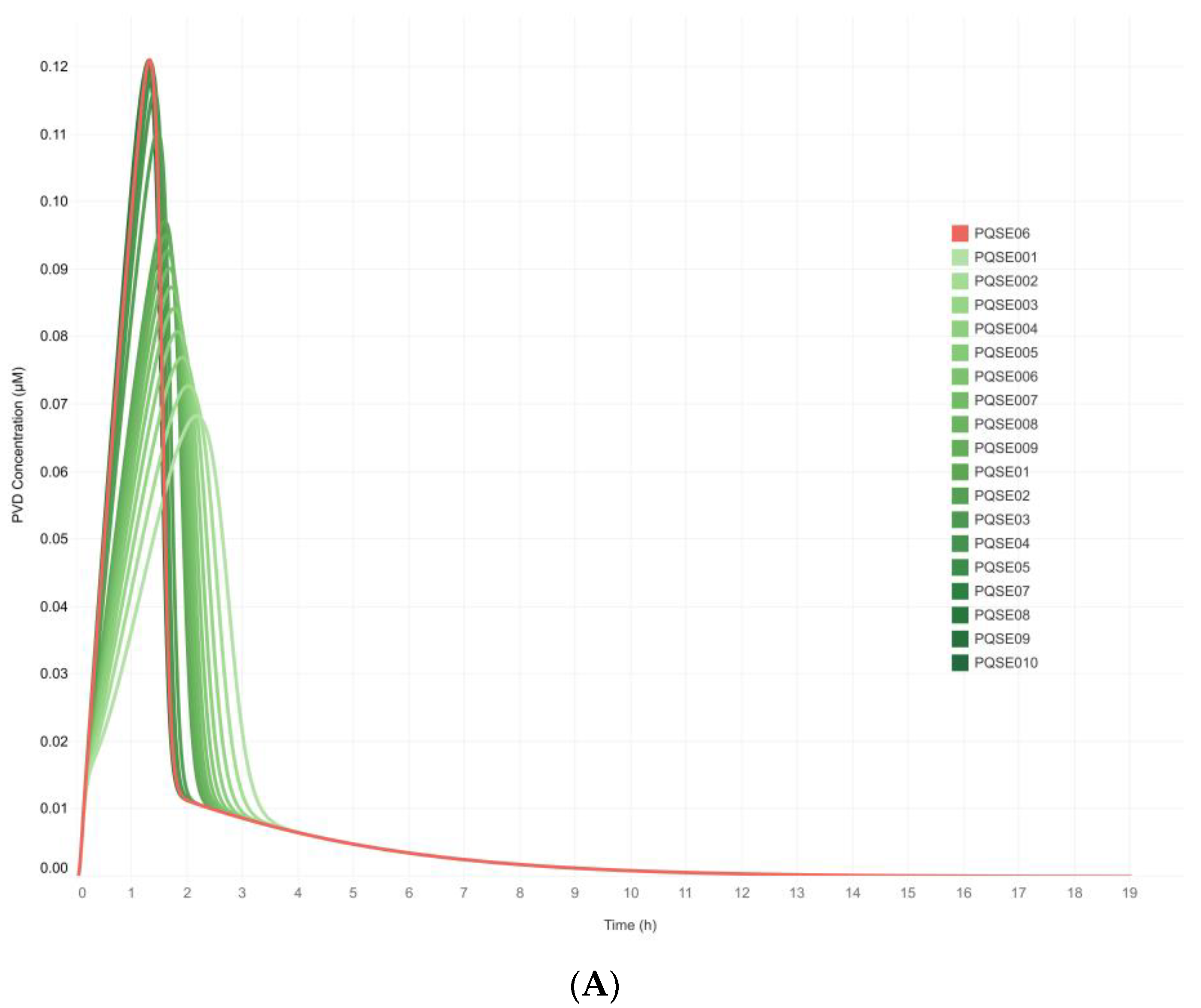
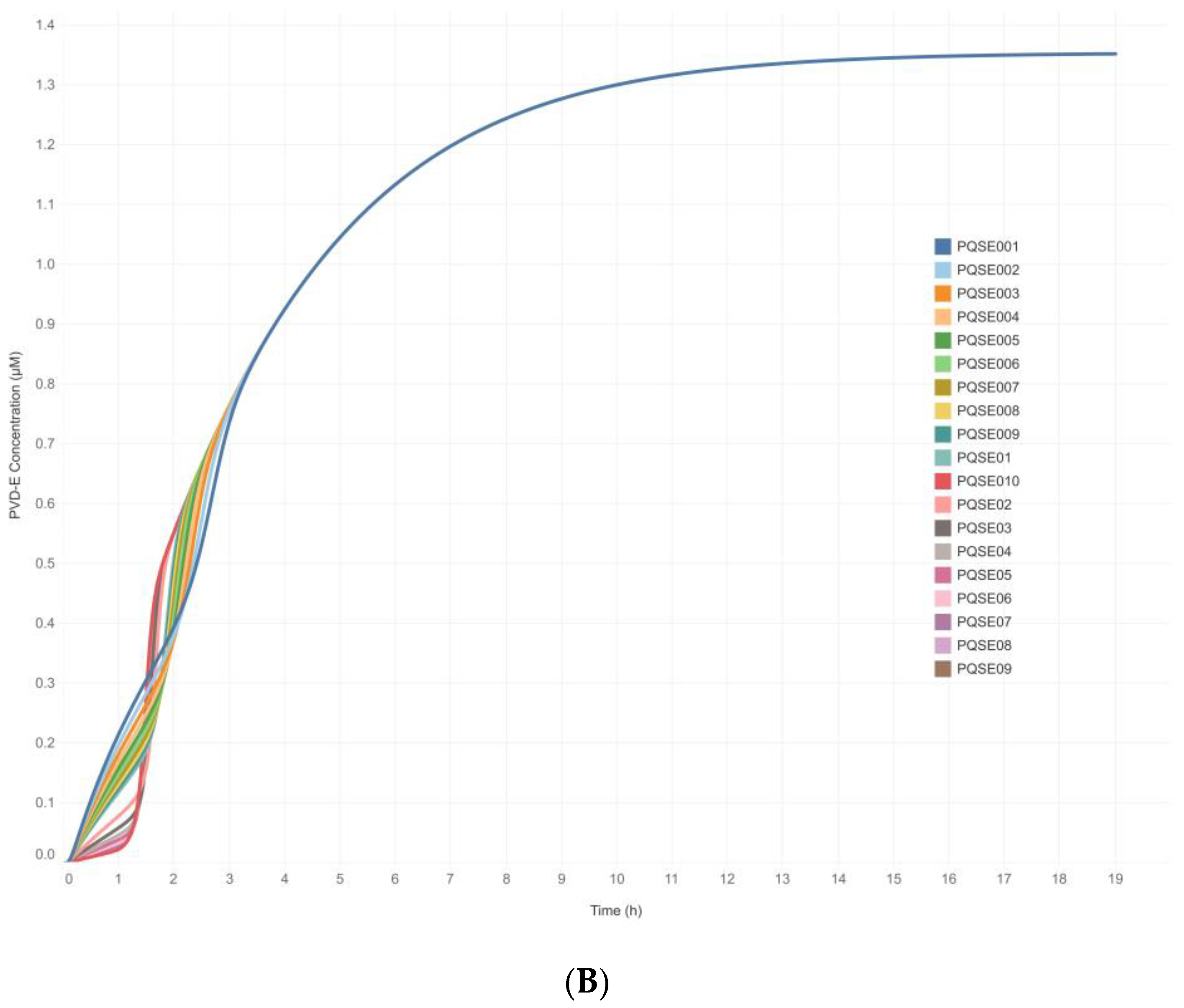
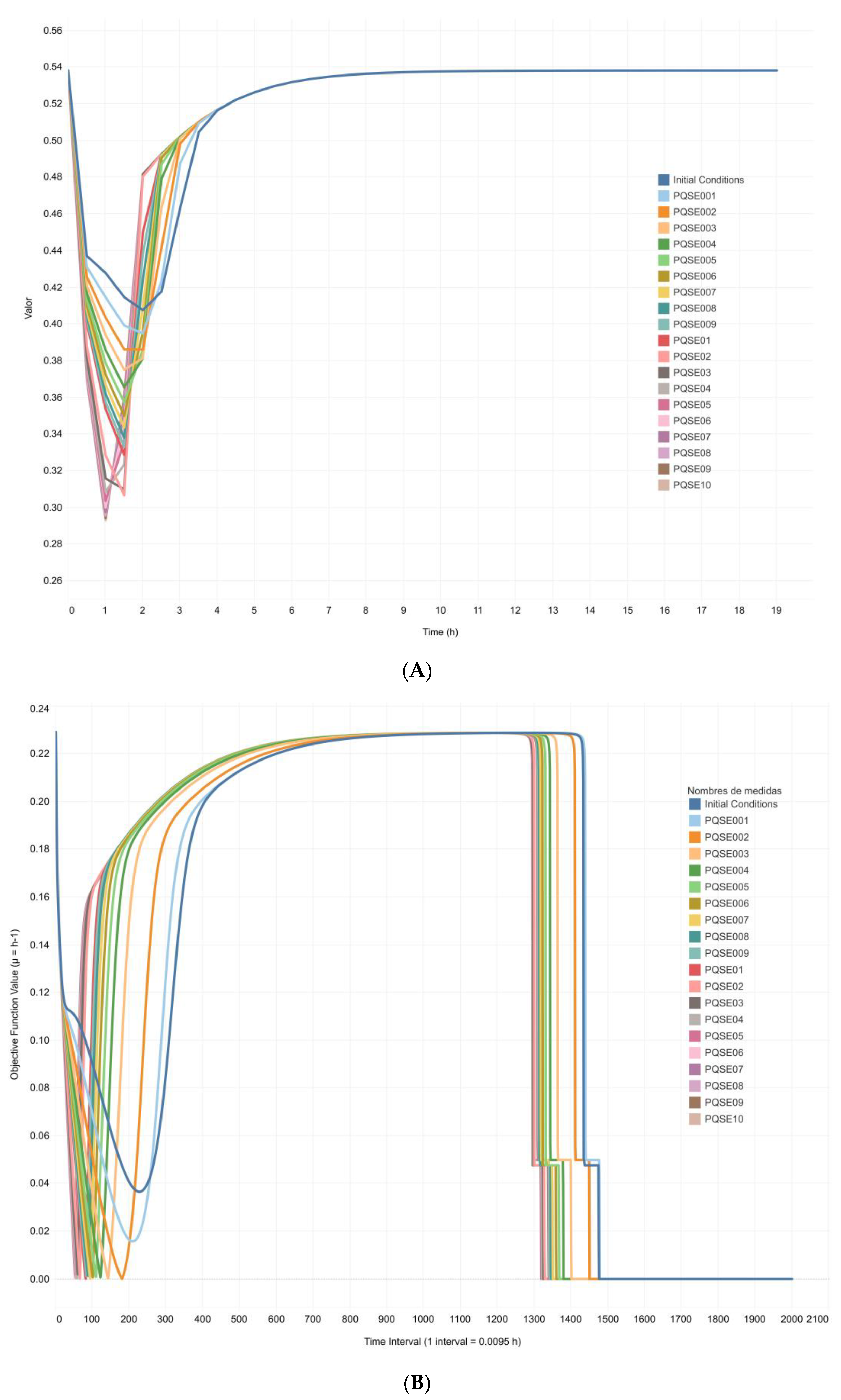
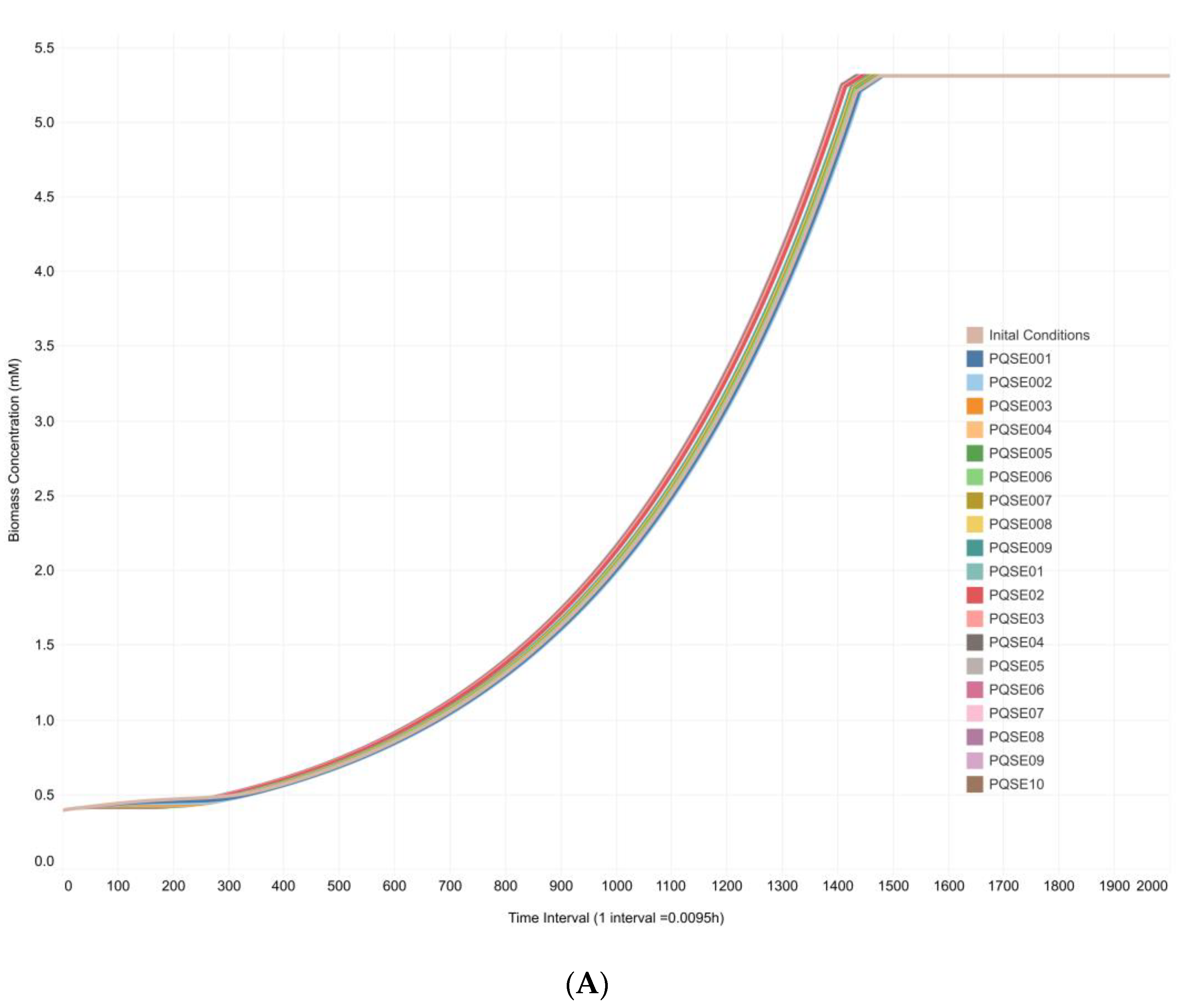
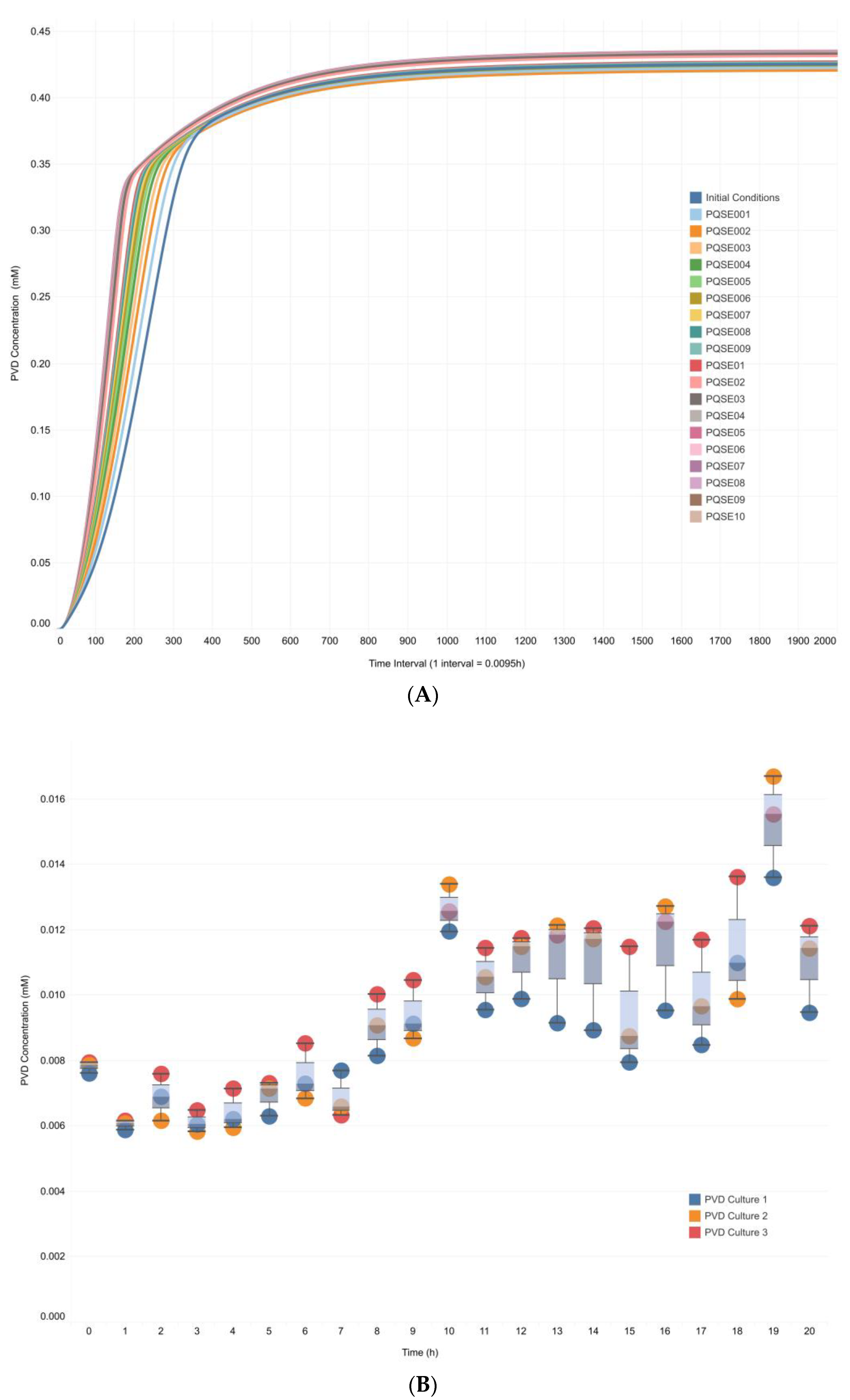
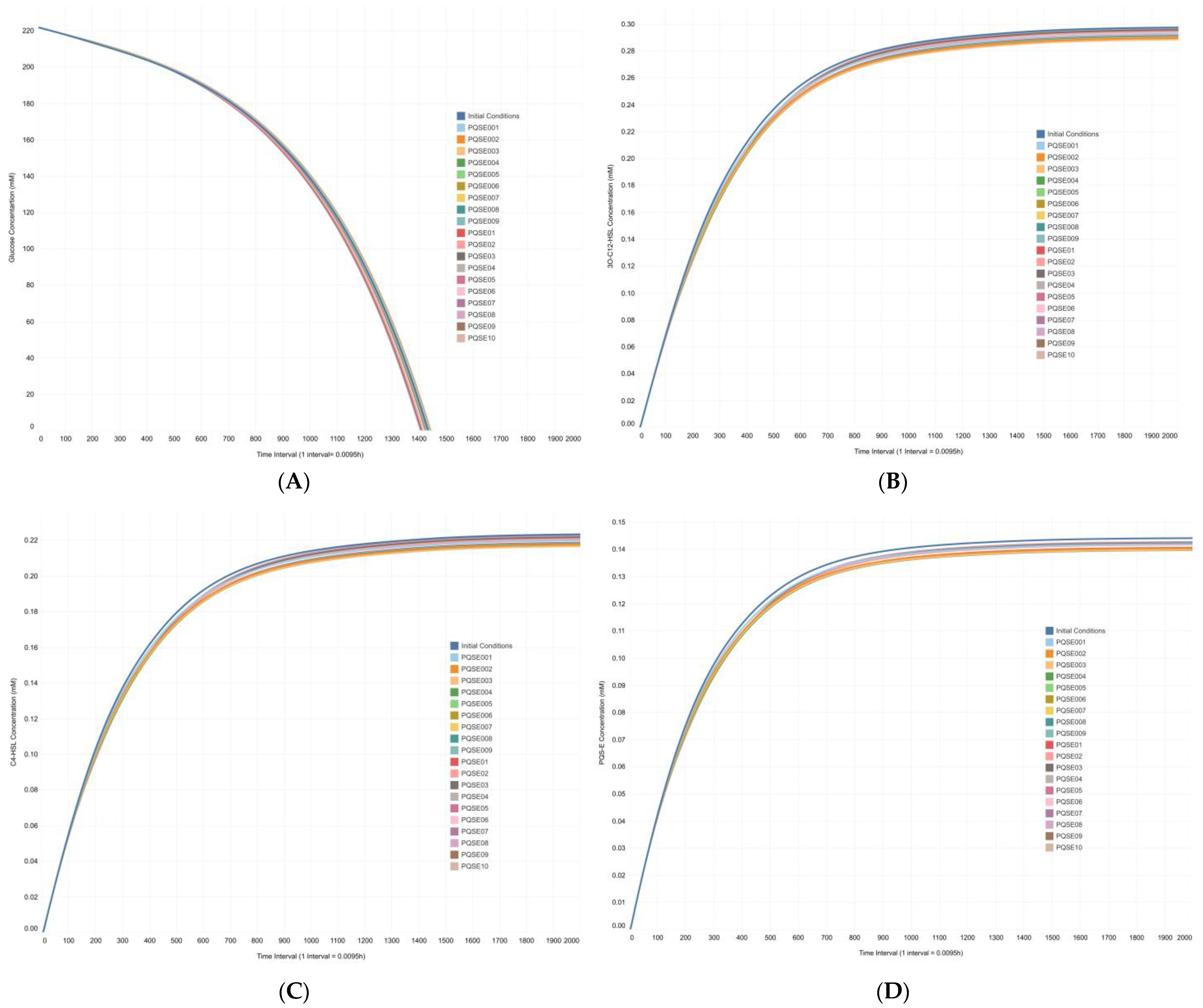
3.3. Integrative Model Simulations Evidenced the Influence of Quorum-Sensing Signaling on Pyoverdine Biosynthesis in Pseudomonas aeruginosa Cultures
4. Discussion
4.1. The QS Gene Regulatory Network Model Emulates the Natural Behavior of Pseudomonas aeruginosa
4.2. The Proposed Integrative CCBM1146 Model Helps to Infer the Influence of the QS Phenomenon on the PVD Metabolic Biosynthesis in Pseudomonas aeruginosa
5. Conclusions
Supplementary Materials
Author Contributions
Funding
Institutional Review Board Statement
Informed Consent Statement
Data Availability Statement
Conflicts of Interest
Abbreviations
Appendix A

{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
| Subset | Input Parameter Value [E-PQS] | Maximum Input Parameter Value in Subset [E-PQS] | Output Variable Value [PVD] | Average Output Variable Value in Subset [PVD] |
|---|---|---|---|---|
| Sensitivity Subset 1 = 0.68 | 1 | 1 | 0.120177986 | 0.118943557 |
| 0.9 | 0.120450180 | |||
| 0.8 | 0.120766334 | |||
| 0.7 | 0.121045165 | |||
| 0.6 | 0.121118254 | |||
| 0.5 | 0.120687054 | |||
| 0.4 | 0.119372306 | |||
| 0.3 | 0.116454998 | |||
| 0.2 | 0.110419737 | |||
| Sensitivity Subset 2 = 0.52 | 0.1 | 0.1 | 0.097173080 | 0.082671936 |
| 0.09 | 0.095075669 | |||
| 0.08 | 0.092768928 | |||
| 0.07 | 0.090219930 | |||
| 0.06 | 0.087397210 | |||
| 0.05 | 0.084268237 | |||
| 0.04 | 0.080803399 | |||
| 0.03 | 0.076979188 | |||
| 0.02 | 0.072803560 | |||
| 0.01 | 0.068338620 |
| Input Parameter Value [E-PQS] | Output Variable Value [PVD] | Rate of Change to Each Input Parameter Value [E-PQS] |
|---|---|---|
| 1 | 0.120177986 | 8.32 |
| 0.9 | 0.120450180 | 7.47 |
| 0.8 | 0.120766334 | 6.62 |
| 0.7 | 0.121045165 | 5.78 |
| 0.6 | 0.121118254 | 4.95 |
| 0.5 | 0.120687054 | 4.14 |
| 0.4 | 0.119372306 | 3.35 |
| 0.3 | 0.116454998 | 2.58 |
| 0.2 | 0.110419737 | 1.81 |
| 0.1 | 0.097173080 | 1.03 |
| 0.09 | 0.095075669 | 0.95 |
| 0.08 | 0.092768928 | 0.86 |
| 0.07 | 0.090219930 | 0.78 |
| 0.06 | 0.087397210 | 0.69 |
| 0.05 | 0.084268237 | 0.59 |
| 0.04 | 0.080803399 | 0.50 |
| 0.03 | 0.076979188 | 0.39 |
| 0.02 | 0.072803560 | 0.27 |
| 0.01 | 0.068338620 | 0.15 |
References
- Martins, M.L.; Ferreira, S.C.; Vilela, M.J. Multiscale models for biological systems. Curr. Opin. Colloid Interface Sci. 2010, 15, 18–23. [Google Scholar] [CrossRef]
- Walpole, J.; Papin, J.A.; Peirce, S.M. Multiscale computational models of complex biological systems. Annu. Rev. Biomed. Eng. 2013, 15, 137–154. [Google Scholar] [CrossRef] [PubMed]
- Warner, H.V.; Sivakumar, N.; Peirce, S.M.; Lazzara, M.J. Multiscale computational models of cancer. Curr. Opin. Biomed. Eng. 2019, 11, 137–144. [Google Scholar] [CrossRef]
- Thiele, I.; Fleming, R.M.T.; Que, R.; Bordbar, A.; Diep, D.; Palsson, B.O. Multiscale Modeling of Metabolism and Macromolecular Synthesis in E. coli and Its Application to the Evolution of Codon Usage. PLoS ONE 2012, 7, e45635. [Google Scholar] [CrossRef]
- Biggs, M.B.; Papin, J.A. Novel Multiscale Modeling Tool Applied to Pseudomonas aeruginosa Biofilm Formation. PLoS ONE 2013, 8, e78011. [Google Scholar] [CrossRef]
- Ghssein, G.; Ezzeddine, Z. A Review of Pseudomonas aeruginosa Metallophores: Pyoverdine, Pyochelin and Pseudopaline. Biology 2022, 11, 1711. [Google Scholar] [CrossRef]
- Visca, P.; Imperi, F.; Lamont, I.L. Pyoverdine siderophores: From biogenesis to biosignificance. Trends Microbiol. 2007, 15, 22–30. [Google Scholar] [CrossRef]
- Visca, P.; Imperi, F.; Lamont, I.L. Pyoverdine Synthesis and its Regulation in Fluorescent Pseudomonads. In Microbial Siderophores; Springer: Berlin/Heidelberg, Germany, 2007; Volume 12, pp. 135–163. [Google Scholar]
- Givskov, M.; Rasmussen, T.B.; Ren, D.; Balaban, N. Bacterial Cell-to-cell Communication (Quorum Sensing). In Control of Biofilm Infections by Signal Manipulation; Springer: Berlin/Heidelberg, Germany, 2008; pp. 13–38. [Google Scholar]
- Lazdunski, A.M.; Ventre, I.; Bleves, S. Cell–Cell Communication: Quorum Sensing and Regulatory Circuits in Pseudomonas aeruginosa. In Pseudomonas; Ramos, J.-L., Filloux, A., Eds.; Springer: Dordrecht, The Netherlands, 2007; pp. 279–310. [Google Scholar]
- Imperi, F.; Visca, P. Subcellular localization of the pyoverdine biogenesis machinery of Pseudomonas aeruginosa: A membrane-associated “siderosome”. FEBS Lett. 2013, 587, 3387–3391. [Google Scholar] [CrossRef]
- Ringel, M.T.; Dräger, G.; Brüser, T. PvdN Enzyme Catalyzes a Periplasmic Pyoverdine Modification. J. Biol. Chem. 2016, 291, 23929–23938. [Google Scholar] [CrossRef]
- Cobessi, D.; Celia, H.; Folschweiller, N.; Schalk, I.J.; Abdallah, M.A.; Pattus, F. The crystal structure of the pyoverdine outer membrane receptor FpvA from Pseudomonas aeruginosa at 3.6 Å resolution. J. Mol. Biol. 2005, 347, 121–134. [Google Scholar] [CrossRef]
- Greenwald, J.; Hoegy, F.; Nader, M.; Journet, L.; Mislin, G.L.; Graumann, P.L.; Schalk, I.J. Real time fluorescent resonance energy transfer visualization of ferric pyoverdine uptake in Pseudomonas aeruginosa. A role for ferrous iron. J. Biol. Chem. 2007, 282, 2987–2995. [Google Scholar] [CrossRef] [PubMed]
- Voulhoux, R.; Filloux, A.; Schalk, I.J. Pyoverdine-mediated iron uptake in Pseudomonas aeruginosa: The Tat system is required for PvdN but not for FpvA transport. J. Bacteriol. 2006, 188, 3317–3323. [Google Scholar] [CrossRef] [PubMed]
- Beare, P.A.; For, R.J.; Martin, L.W.; Lamont, I.L. Siderophore-mediated cell signalling in Pseudomonas aeruginosa: Divergent pathways regulate virulence factor production and siderophore receptor synthesis. Mol. Microbiol. 2003, 47, 195–207. [Google Scholar] [CrossRef] [PubMed]
- Orth, J.D.; Thiele, I.; Palsson, B.Ø. What is flux balance analysis? Nat. Biotechnol. 2010, 28, 245–248. [Google Scholar] [CrossRef] [PubMed]
- Raman, K.; Chandra, N. Flux balance analysis of biological systems: Applications and challenges. Brief. Bioinform. 2009, 10, 435–449. [Google Scholar] [CrossRef]
- Gianchandani, E.P.; Chavali, A.K.; Papin, J.A. The application of flux balance analysis in systems biology. Wiley Interdiscip. Rev. Syst. Biol. Med. 2010, 2, 372–382. [Google Scholar] [CrossRef]
- Maranas, C.D.; Zomorrodi, A.R. Optimization Methods in Metabolic Networks; John Wiley & Sons, Inc.: Hoboken, NJ, USA, 2016. [Google Scholar]
- Serrano-Bermúdez, L.M.; Barrios, A.F.G.; Maranas, C.D.; Montoya, D. Clostridium butyricum maximizes growth while minimizing enzyme usage and ATP production: Metabolic flux distribution of a strain cultured in glycerol. BMC Syst. Biol. 2017, 11, 58. [Google Scholar] [CrossRef]
- Mahadevan, R.; Edwards, J.S.; Doyle, F. Dynamic flux balance analysis of diauxic growth in Escherichia coli. Biophys. J. 2002, 83, 1331–1340. [Google Scholar] [CrossRef]
- Serrano-Bermúdez, L.M.; Barrios, A.F.G.; Montoya, D. Clostridium butyricum population balance model: Predicting dynamic metabolic flux distributions using an objective function related to extracellular glycerol content. PLoS ONE 2018, 13, e0209447. [Google Scholar] [CrossRef]
- Funahashi, A.; Matsuoka, Y.; Jouraku, A.; Kitano, H.; Kikuchi, N. Celldesigner: A Modeling Tool for Biochemical Networks. In Proceedings of the 2006 Winter Simulation Conference, Monterey, CA, USA, 3–6 December 2006; pp. 1707–1712. [Google Scholar]
- Kanehisa, M.; Goto, S. KEGG: Kyoto encyclopedia of genes and genomes. Nucleic Acids Res. 2000, 28, 27–30. [Google Scholar] [CrossRef]
- Kanehisa, M.; Furumichi, M.; Tanabe, M.; Sato, Y.; Morishima, K. KEGG: New perspectives on genomes, pathways, diseases and drugs. Nucleic Acids Res. 2017, 45, D353–D361. [Google Scholar] [CrossRef]
- Karp, P.D.; Billington, R.; Caspi, R.; Fulcher, C.A.; Latendresse, M.; Kothari, A.; Keseler, I.M.; Krummenacker, M.; Midford, P.E.; Ong, Q.; et al. The BioCyc collection of microbial genomes and metabolic pathways. Brief. Bioinform. 2019, 20, 1085–1093. [Google Scholar] [CrossRef]
- Caspi, R.; Altman, T.; Billington, R.; Dreher, K.; Foerster, H.; Fulcher, C.A.; Holland, T.A.; Keseler, I.M.; Kothari, A.; Kubo, A.; et al. The MetaCyc database of metabolic pathways and enzymes and the BioCyc collection of Pathway/Genome Databases. Nucleic Acids Res. 2014, 42, 473–479. [Google Scholar] [CrossRef] [PubMed]
- Bairoch, A.; Apweiler, R. The SWISS-PROT protein sequence database and its supplement TrEMBL in 2000. Nucleic Acids Res. 2000, 28, 45. [Google Scholar] [CrossRef] [PubMed]
- Kim, S.; Thiessen, P.A.; Bolton, E.E.; Chen, J.; Fu, G.; Gindulyte, A.; Han, L.; He, J.; He, S.; Shoemaker, B.A.; et al. PubChem Substance and Compound databases. Nucleic Acids Res. 2016, 44, D1202–D1213. [Google Scholar] [CrossRef] [PubMed]
- Kim, S.; Chen, J.; Cheng, T.; Gindulyte, A.; He, J.; He, S.; Li, Q.; Shoemaker, B.A.; Thiessen, P.A.; Yu, B.; et al. PubChem in 2021: New data content and improved web interfaces. Nucleic Acids Res. 2021, 49, D1388–D1395. [Google Scholar] [CrossRef] [PubMed]
- Aldridge, B.B.; Burke, J.M.; Lauffenburger, D.A.; Sorger, P.K. Physicochemical modelling of cell signalling pathways. Nat. Cell Biol. 2006, 8, 1195–1203. [Google Scholar] [CrossRef]
- Rainer, M.; Christoph, F.; Andrew, F.; Stefan, M.; James, L.; Akira, F. SoSLib; Github: Tokio, Japan, 2017. [Google Scholar]
- Machne, R.; Finney, A.; Muller, S.; Lu, J.; Widder, S.; Flamm, C. The SBML ODE Solver Library: A native API for symbolic and fast numerical analysis of reaction networks. Bioinformatics 2006, 22, 1406–1407. [Google Scholar] [CrossRef]
- Fuqua, W.C.; Winans, S.C.; Greenberg, E.P. Quorum sensing in bacteria: The LuxR-LuxI family of cell density-responsive transcriptional regulators. J. Bacteriol. 1994, 176, 269–275. [Google Scholar] [CrossRef]
- Fuqua, C.; Parsek, M.R.; Greenberg, E.P. Regulation of gene expression by cell-to-cell communication: Acyl-homoserine lactone quorum sensing. Annu. Rev. Genet. 2001, 35, 439–468. [Google Scholar] [CrossRef]
- Oberhardt, M.A.; Puchalka, J.; Fryer, K.E.; Santos, V.A.P.M.D.; Papin, J.A. Genome-Scale Metabolic Network Analysis of the Opportunistic Pathogen Pseudomonas aeruginosa PAO1. J. Bacteriol. 2008, 190, 2790–2803. [Google Scholar] [CrossRef] [PubMed]
- Seaver, S.M.D.; Liu, F.; Zhang, Q.; Jeffryes, J.; Faria, J.P.; Edirisinghe, J.N.; Mundy, M.; Chia, N.; Noor, E.; Beber, M.E.; et al. The ModelSEED Biochemistry Database for the integration of metabolic annotations and the reconstruction, comparison and analysis of metabolic models for plants, fungi and microbes. Nucleic Acids Res. 2021, 49, D575–D588. [Google Scholar] [CrossRef] [PubMed]
- Moretti, S.; Tran, V.D.T.; Mehl, F.; Ibberson, M.; Pagni, M. MetaNetX/MNXref: Unified namespace for metabolites and biochemical reactions in the context of metabolic models. Nucleic Acids Res. 2021, 49, D570–D574. [Google Scholar] [CrossRef] [PubMed]
- Elbourne, L.D.H.; Tetu, S.G.; Hassan, K.A.; Paulsen, I.T. TransportDB 2.0: A database for exploring membrane transporters in sequenced genomes from all domains of life. Nucleic Acids Res. 2017, 45, D320–D324. [Google Scholar] [CrossRef]
- Saier, M.H., Jr.; Reddy, V.S.; Moreno-Hagelsieb, G.; Hendargo, K.J.; Zhang, Y.; Iddamsetty, V.; Lam, K.J.K.; Tian, N.; Russum, S.; Wang, J.; et al. The Transporter Classification Database (TCDB): 2021 update. Nucleic Acids Res. 2021, 49, D461–D467. [Google Scholar] [CrossRef]
- Senger, R.S.; Papoutsakis, E.T. Genome-scale model for Clostridium acetobutylicum: Part I. Metabolic network resolution and analysis. Biotechnol. Bioeng. 2008, 101, 1036–1052. [Google Scholar] [CrossRef]
- Kumar, V.S.; Dasika, M.S.; Maranas, C.D. Optimization based automated curation of metabolic reconstructions. BMC Bioinform. 2007, 8, 212. [Google Scholar]
- Saadat, N.P.; van Aalst, M.; Ebenhöh, O. Network Reconstruction and Modelling Made Reproducible with moped. Metabolites 2022, 12, 275. [Google Scholar] [CrossRef]
- Schellenberger, J.; Lewis, N.E.; Palsson, B.Ø. Elimination of Thermodynamically Infeasible Loops in Steady-State Metabolic Models. Biophys. J. 2011, 100, 544–553. [Google Scholar] [CrossRef]
- Amara, A.; Takano, E.; Breitling, R. Development and validation of an updated computational model of Streptomyces coelicolor primary and secondary metabolism. BMC Genom. 2018, 19, 519. [Google Scholar] [CrossRef]
- Prigent, S.; Nielsen, J.C.; Frisvad, J.C.; Nielsen, J. Reconstruction of 24 Penicillium genome-scale metabolic models shows diversity based on their secondary metabolism. Biotechnol. Bioeng. 2018, 115, 2604–2612. [Google Scholar] [CrossRef] [PubMed]
- Kim, H.U.; Charusanti, P.; Lee, S.Y.; Weber, T. Metabolic engineering with systems biology tools to optimize production of prokaryotic secondary metabolites. Nat. Prod. Rep. 2016, 33, 933–941. [Google Scholar] [CrossRef]
- Mallmann, J.; Heckmann, D.; Bräutigam, A.; Lercher, M.J.; Weber, A.P.; Westhoff, P.; Gowik, U. The role of photorespiration during the evolution of C4 photosynthesis in the genus Flaveria. Elife 2014, 3, e02478. [Google Scholar] [CrossRef] [PubMed]
- Monod, J. The Growth of Bacterial Cultures. Annu. Rev. Microbiol. 1949, 3, 371–394. [Google Scholar] [CrossRef]
- Folsom, J.P.; Richards, L.; Pitts, B.; Roe, F.; Ehrlich, G.D.; Parker, A.; Mazurie, A.; Stewart, P.S. Physiology of Pseudomonas aeruginosa in biofilms as revealed by transcriptome analysis. BMC Microbiol. 2010, 10, 294. [Google Scholar] [CrossRef]
- Milo, R.; Phillips, R. Cell Biology by the Numbers, 1st ed.; Garland Science: New York, NY, USA, 2015. [Google Scholar]
- Fallahzadeh, V.; Ahmadzadeh, M.; Sharifi, R. Growth and pyoverdine production kinetics of Pseudomonas aeruginosa 7NSK2 in an experimental fermentor. J. Agric. Technol. 2010, 6, 107–115. [Google Scholar]
- Imperi, F.; Tiburzi, F.; Visca, P. Molecular basis of pyoverdine siderophore recycling in Pseudomonas aeruginosa. Proc. Natl. Acad. Sci. USA 2009, 106, 20440–20445. [Google Scholar] [CrossRef]
- Meyer, J.M.; Abdallah, M.A. The Fluorescent Pigment of Pseudomonas fluorescens: Biosynthesis, Purification and Physicochemical Properties. J. Gen. Microbiol. 1978, 107, 319–328. [Google Scholar] [CrossRef]
- Pearson, J.P.; Gray, K.M.; Passador, L.; Tucker, K.D.; Eberhard, A.; Iglewski, B.H.; Greenberg, E.P. Structure of the autoinducer required for expression of Pseudomonas aeruginosa virulence genes [density-dependent transcription/gene activation/las genes/N-acyl homoserine lactone/3-oxo-N-(tetrahydro-2-oxo-3-furanyl)dodecanamide. J. Biochem. 1994, 91, 197–201. [Google Scholar]
- Venturi, V. Regulation of quorum sensing in Pseudomonas. FEMS Microbiol. Rev. 2006, 30, 274–291. [Google Scholar] [CrossRef]
- Venturi, V.; Weisbeek, P.; Koster, M. Gene regulation of siderophore-mediated iron acquisition in Pseudomonas: Not only the Fur repressor. Mol. Microbiol. 1995, 17, 603–610. [Google Scholar] [CrossRef] [PubMed]
- Gilbert, D.; Heiner, M.; Ghanbar, L.; Chodak, J. Spatial quorum sensing modelling using coloured hybrid Petri nets and simulative model checking. BMC Bioinform. 2019, 20, 173. [Google Scholar] [CrossRef] [PubMed]
- Nadal-Jimenez, P.; Koch, G.; Reis, C.R.; Muntendam, R.; Raj, H.; Jeronimus-Stratingh, C.M.; Cool, R.H.; Quax, W.J. PvdP is a tyrosinase that drives maturation of the pyoverdine chromophore in Pseudomonas aeruginosa. J. Bacteriol. 2014, 196, 2681–2690. [Google Scholar] [CrossRef]
- Twycross, J.; Band, L.R.; Bennett, M.J.; King, J.R.; Krasnogor, N. Stochastic and deterministic multiscale models for systems biology: An auxin-transport case study. BMC Syst. Biol. 2010, 4, 34. [Google Scholar] [CrossRef] [PubMed]
- Barrios, A.F.G.; Achenie, L.E.K. Escherichia coli autoinducer-2 uptake network does not display hysteretic behavior but AI-2 synthesis rate controls transient bifurcation. Biosystems 2010, 99, 17–26. [Google Scholar] [CrossRef]
- Leoni, L.; Orsi, N.; De Lorenzo, V.; Visca, P. Functional Analysis of PvdS, an Iron Starvation Sigma Factor of Pseudomonas aeruginosa Functional Analysis of PvdS, an Iron Starvation Sigma Factor of Pseudomonas aeruginosa. California Institu. J. Bacteriol. 2000, 182, 1481–1491. Available online: http://jb.asm.org/ (accessed on 17 September 2013). [CrossRef] [PubMed]
- Moon, C.D.; Zhang, X.-X.; Matthijs, S.; Schäfer, M.; Budzikiewicz, H.; Rainey, P.B. Genomic, genetic and structural analysis of pyoverdine-mediated iron acquisition in the plant growth-promoting bacterium Pseudomonas fluorescens SBW25. BMC Microbiol. 2008, 8, 7. [Google Scholar] [CrossRef]
- Vasil, M.L.; Ochsner, U.A.; Johnson, Z.; Colmer, J.A.; Hamood, A.N. The Fur-regulated gene encoding the alternative sigma factor PvdS is required for iron-dependent expression of the LysR-type regulator PtxR in pseudomonas aeruginosa. J. Bacteriol. 1998, 180, 6784–6788. [Google Scholar] [CrossRef]
- Diggle, S.P.; Matthijs, S.; Wright, V.J.; Fletcher, M.P.; Chhabra, S.R.; Lamont, I.L.; Kong, X.; Hider, R.C.; Cornelis, P.; Cámara, M.; et al. The Pseudomonas aeruginosa 4-Quinolone Signal Molecules HHQ and PQS Play Multifunctional Roles in Quorum Sensing and Iron Entrapment. Chem. Biol. 2007, 14, 87–96. [Google Scholar] [CrossRef]
- Jimenez, P.N.; Koch, G.; Thompson, J.A.; Xavier, K.B.; Cool, R.H.; Quax, W.J. The multiple signaling systems regulating virulence in Pseudomonas aeruginosa. Microbiol. Mol. Biol. Rev. 2012, 76, 46–65. [Google Scholar] [CrossRef]
- Chincholkar, S.B.; Chaudhari, B.L.; Rane, M.R. Microbial Siderophore: A State of Art. In Microbial Siderophores; Varma, A., Chincholkar, S.B., Eds.; Springer: Berlin/Heidelberg, Germany, 2007; pp. 233–242. [Google Scholar]
- Das, A.; Prasad, R.; Srivastava, A.; Giang, P.H.; Bhatnagar, K.; Varma, A. Fungal Siderophores: Structure, Functions and Regulation. In Microbial Siderophores; Springer: Berlin/Heidelberg, Germany, 2007; pp. 1–42. [Google Scholar]
- Arevalo-Ferro, C.; Hentzer, M.; Reil, G.; Görg, A.; Kjelleberg, S.; Givskov, M.; Riedel, K.; Eberl, L. Identification of quorum-sensing regulated proteins in the opportunistic pathogen Pseudomonas aeruginosa by proteomics. Environ. Microbiol. 2003, 5, 1350–1369. [Google Scholar] [CrossRef] [PubMed]
- Williams, P.; Cámara, M. Quorum sensing and environmental adaptation in Pseudomonas aeruginosa: A tale of regulatory networks and multifunctional signal molecules. Curr. Opin. Microbiol. 2009, 12, 182–191. [Google Scholar] [CrossRef] [PubMed]
- Wintermute, E.H.; Lieberman, T.D.; Silver, P.A. An objective function exploiting suboptimal solutions in metabolic networks. BMC Syst. Biol. 2013, 7, 98. [Google Scholar] [CrossRef] [PubMed]
- Schultz, A.; Qutub, A.A. Predicting internal cell fluxes at sub-optimal growth. BMC Syst. Biol. 2015, 9, 18. [Google Scholar] [CrossRef]
- Kim, D.; Chung, S.; Lee, S.; Choi, J. Relation of microbial biomass to counting units for Pseudomonas aeruginosa. Afr. J. Microbiol. Res. 2012, 6, 4620–4622. [Google Scholar]
- Ghssein, G.; Matar, S. Chelating Mechanisms of Transition Metals by Bacterial Metallophores ‘Pseudopaline and Staphylopine’: A Quantum Chemical Assessment. Computation 2018, 6, 56. [Google Scholar] [CrossRef]
- Lhospice, S.; Gomez, N.O.; Ouerdane, L.; Brutesco, C.; Ghssein, G.; Hajjar, C.; Liratni, A.; Wang, S.; Richaud, P.; Bleves, S.; et al. Pseudomonas aeruginosa zinc uptake in chelating environment is primarily mediated by the metallophore pseudopaline. Sci. Rep. 2017, 7, 17132. [Google Scholar] [CrossRef]











| Simulation Scenario | E-PQS [μM] | ID Simulation Scenario | Simulation Scenario | E-PQS [μM] | ID Simulation Scenario |
|---|---|---|---|---|---|
| Sc1 | 0.00 | Initial conditions | Sc11 | 0.1 | PQSE01 |
| Sc2 | 0.01 | PQSE001 | Sc12 | 0.2 | PQSE02 |
| Sc3 | 0.02 | PQSE002 | Sc13 | 0.3 | PQSE03 |
| Sc4 | 0.03 | PQSE003 | Sc14 | 0.4 | PQSE04 |
| Sc5 | 0.04 | PQSE004 | Sc15 | 0.5 | PQSE05 |
| Sc6 | 0.05 | PQSE005 | Sc16 | 0.6 | PQSE06 |
| Sc7 | 0.06 | PQSE006 | Sc17 | 0.7 | PQSE07 |
| Sc8 | 0.07 | PQSE007 | Sc18 | 0.8 | PQSE08 |
| Sc9 | 0.08 | PQSE008 | Sc19 | 0.9 | PQSE09 |
| Sc10 | 0.09 | PQSE009 | Sc20 | 1.0 | PQSE10 |
| Quorum-Sensing Network | Metabolic Network | EC/TC Number | Reaction Equation in the Metabolic Network Model |
|---|---|---|---|
| 3O-C12-HSL production | 3O-C12-HSL synthesis | EC 2.3.1.184 | [c]: 3oxddACP + amet <==> 5mta + ACP + h + n3oxdd-hsl |
| C4-HSL production | C4-HSL synthesis | EC 2.3.1.184 | [c]: amet + butACP <==> 5mta + ACP + h + nb-hsl |
| PQS production | PQS synthesis | EC 1.14.13.182 | [c]: fadh2 + h + hhq + o2 --> nad + h2o + pqs |
| C4-HSL diffusion | C4-HSL transport | - | nb-hsl[c] <==> nb-hsl[e] |
| 3O-C12-HSL diffusion | 3O-C12-HSL transport | - | n3oxdd-hsl[c] <==> n3oxdd-hsl[e] |
| PQS diffusion | PQS transport | - | pqs[c] <==> pqs[e] |
| Ferribactin production | Ferribactin synthesis | EC 6.3.2. | [c]: glu-L + tyr-L + (2) ser-L + arg-L + 24dab + (2) fohorn + lys-L + (2) thr-L --> fbn + (12) h2o + (2) h |
| PVD production | PVD synthesis | EC 1.14.18. | [c]: fbn + o2 --> pvd1 + h2o |
| PVD export | PVD transport | TC-1.B.14.1.6 | pvd1[c] --> pvd1[e] |
| Metabolic Reaction | Sc1 | Sc2 | Sc3 | Sc4 | Sc5 | Sc6 |
|---|---|---|---|---|---|---|
| 3O-C12-HSL synthesis | X | X | X | |||
| C4-HSL synthesis | X | X | X | |||
| PQS synthesis | X | X | X | |||
| C4-HSL transport | X | X | X | |||
| 3O-C12-HSL transport | X | X | X | |||
| PQS transport | X | X | X | |||
| Ferribactin synthesis | X | X | ||||
| PVD synthesis | X | X | ||||
| PVD transport | X | X |
| Reactions and Components | Model | |
|---|---|---|
| iMO1056 | CCBM1146 | |
| Metabolic reactions | 728 | 774 |
| Transport reactions | 150 | 146 |
| Biomass reaction | 1 | 1 |
| Maintenance reaction | 1 | 1 |
| Exchange reactions | 118 | 120 |
| Reactions for metabolite input from the culture medium | 84 | 81 |
| Total reactions | 1082 | 1123 |
| Total metabolites | 760 | 880 |
| Total genes | 1056 | 1146 |
Disclaimer/Publisher’s Note: The statements, opinions and data contained in all publications are solely those of the individual author(s) and contributor(s) and not of MDPI and/or the editor(s). MDPI and/or the editor(s) disclaim responsibility for any injury to people or property resulting from any ideas, methods, instructions or products referred to in the content. |
© 2023 by the authors. Licensee MDPI, Basel, Switzerland. This article is an open access article distributed under the terms and conditions of the Creative Commons Attribution (CC BY) license (https://creativecommons.org/licenses/by/4.0/).
Share and Cite
Clavijo-Buriticá, D.C.; Arévalo-Ferro, C.; González Barrios, A.F. A Holistic Approach from Systems Biology Reveals the Direct Influence of the Quorum-Sensing Phenomenon on Pseudomonas aeruginosa Metabolism to Pyoverdine Biosynthesis. Metabolites 2023, 13, 659. https://doi.org/10.3390/metabo13050659
Clavijo-Buriticá DC, Arévalo-Ferro C, González Barrios AF. A Holistic Approach from Systems Biology Reveals the Direct Influence of the Quorum-Sensing Phenomenon on Pseudomonas aeruginosa Metabolism to Pyoverdine Biosynthesis. Metabolites. 2023; 13(5):659. https://doi.org/10.3390/metabo13050659
Chicago/Turabian StyleClavijo-Buriticá, Diana Carolina, Catalina Arévalo-Ferro, and Andrés Fernando González Barrios. 2023. "A Holistic Approach from Systems Biology Reveals the Direct Influence of the Quorum-Sensing Phenomenon on Pseudomonas aeruginosa Metabolism to Pyoverdine Biosynthesis" Metabolites 13, no. 5: 659. https://doi.org/10.3390/metabo13050659
APA StyleClavijo-Buriticá, D. C., Arévalo-Ferro, C., & González Barrios, A. F. (2023). A Holistic Approach from Systems Biology Reveals the Direct Influence of the Quorum-Sensing Phenomenon on Pseudomonas aeruginosa Metabolism to Pyoverdine Biosynthesis. Metabolites, 13(5), 659. https://doi.org/10.3390/metabo13050659