

Metabolic Role of GABA in the Secretory Function of Pancreatic β-Cells: Its Hypothetical Implication in β-Cell Degradation in Type 2 Diabetes

{kind=link}
{kind=link}
{kind=link}
Abstract
:1. Metabolic Role of GABA in the Mechanism of Stimulation of Insulin Secretion
1.1. Introduction
1.2. Effect of Glucose Stimulation of Insulin Secretion on Islet GABA Content
1.3. Effects of Branched-Chain 2-Oxoacid (BCKA) Stimulation of Insulin Release on Islet GABA Content
1.4. Effects of L-Leucine Plus L-Glutamine Stimulation of Insulin Release on Islet GABA Content
1.5. Conclusions and Prospects
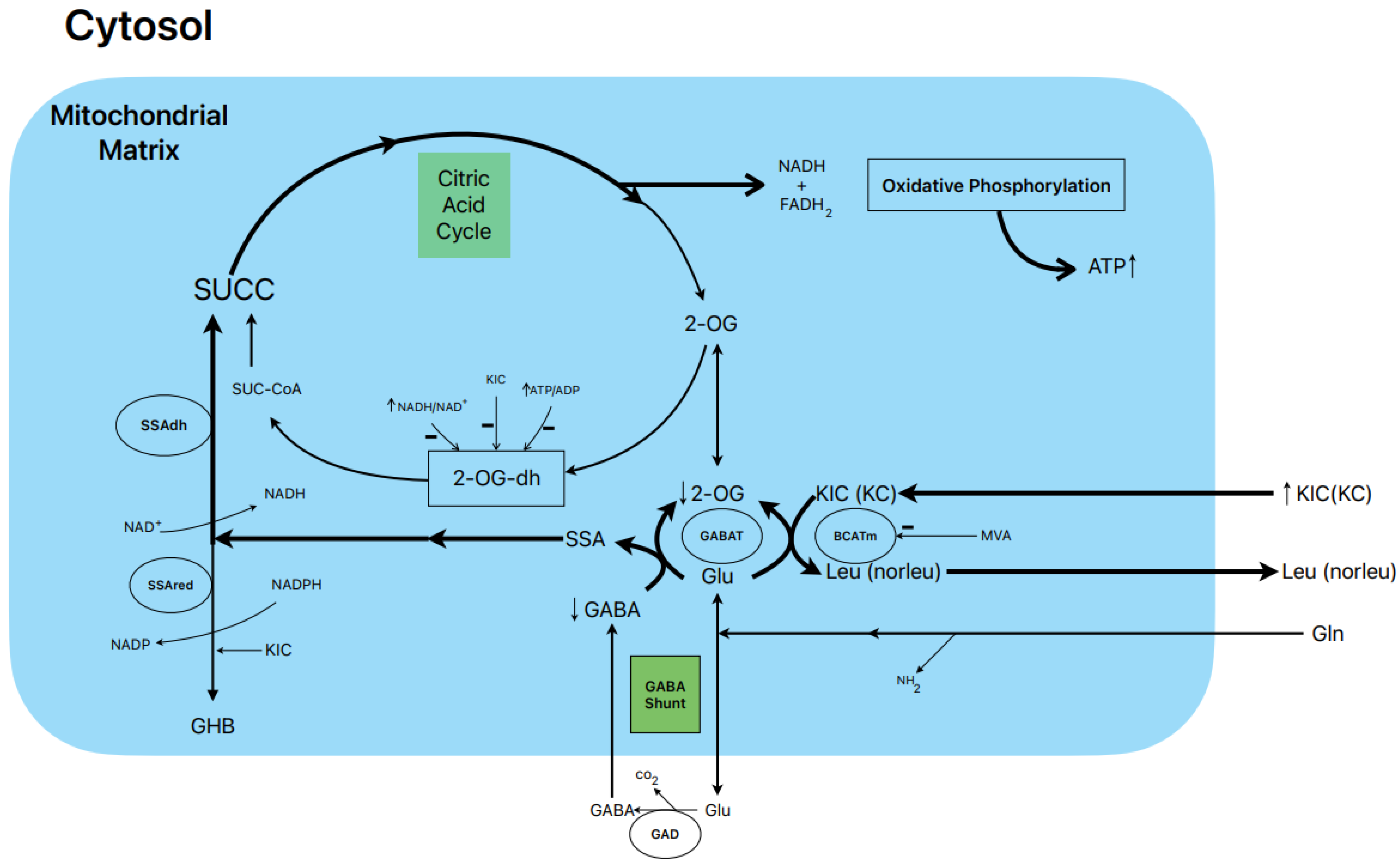
1.6. Postulated Metabolic Pathway Leading to the Stimulation of BCKA-Induced Insulin Secretion (Figure 1)
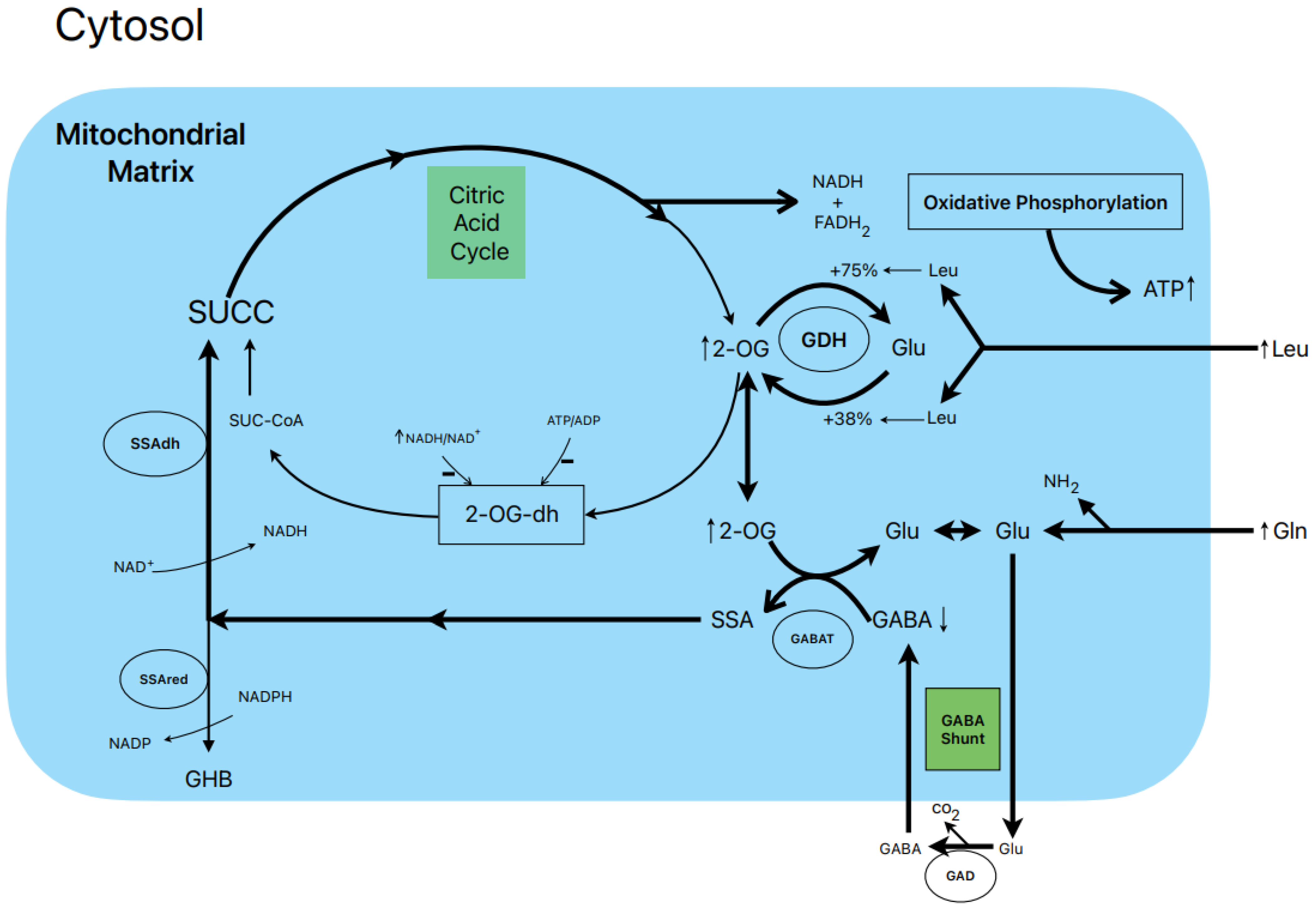
1.7. Postulated Metabolic Pathway Leading to the Stimulation of Insulin Secretion by L-Leucine plus L-Glutamine (Figure 2)
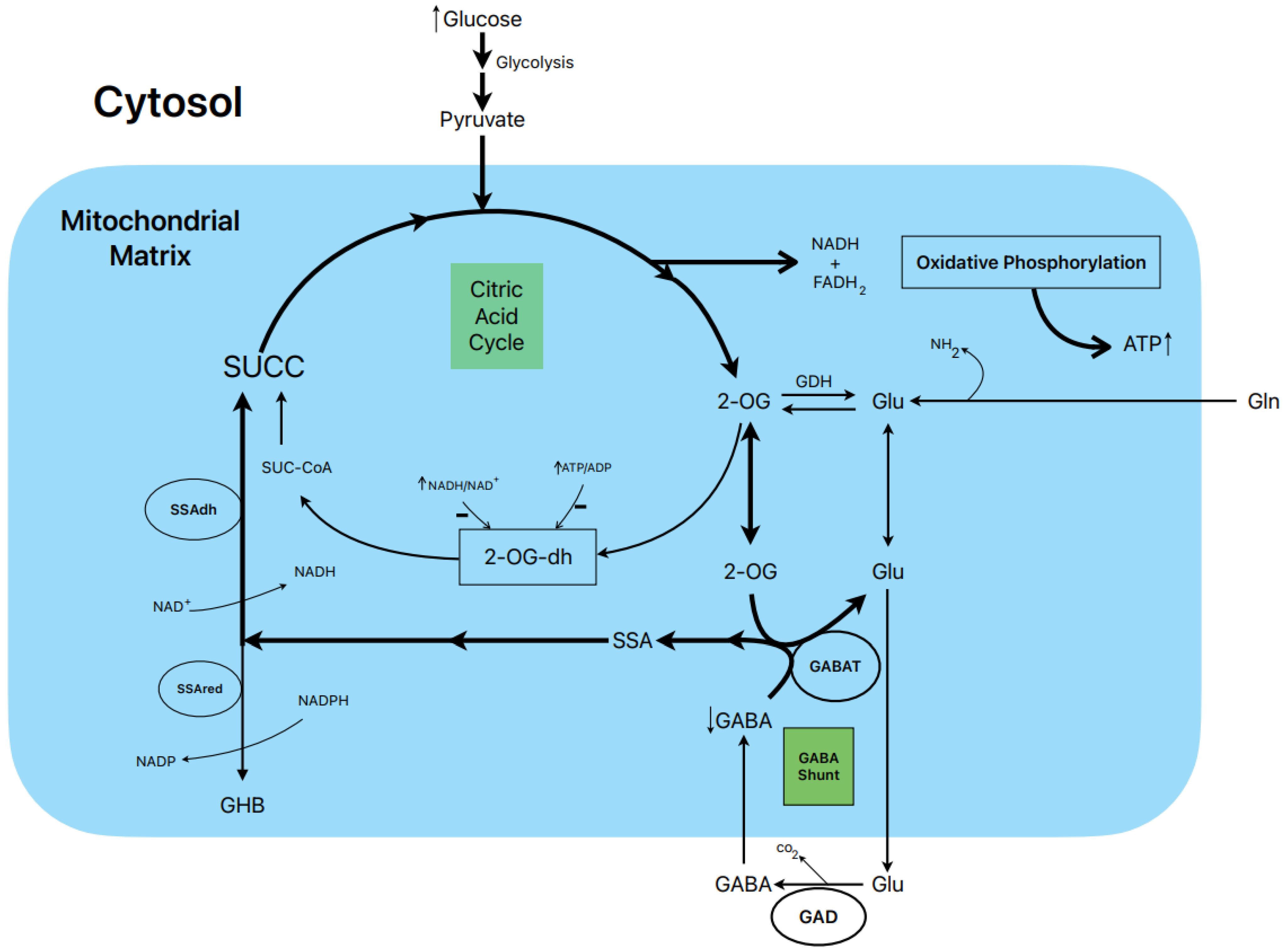
1.8. Postulated Metabolic Pathway Leading to the Stimulation of Insulin Secretion by Glucose (Figure 3)
1.9. Conclusions
1.10. Future Directions: Integral View of the Stimulus-Secretion Coupling Mechanism of Insulin Release
2. Possible Implication of the GABA Shunt in β-Cell Degradation in Type 2 Diabetes
2.1. Introduction
2.2. Intracellular vs. Extracellular Effects of GABA
2.3. Metabolic Changes Contributing to Islet Degradation in Type 2 Diabetes
2.4. Conclusions and Perspectives
Funding
Acknowledgments
Conflicts of Interest
Abbreviations
References
- Ashcroft, F.M.; Rorsman, P. Electrophysiology of the pancreatic β-cell. Prog. Biophys. Molec. Biol. 1989, 54, 87–143. [Google Scholar] [CrossRef] [PubMed]
- Gembal, M.; Gilon, P.; Henquin, J.C. Evidence that glucose can control insulin release independently from its action on ATP-sensitive K+ channels in mouse B cells. J. Clin. Investig. 1992, 89, 1288–1295. [Google Scholar] [CrossRef] [PubMed]
- Gembal, M.; Detimary, P.; Gilon, P.; Gao, Z.-Y.; Henquin, J.-C. Mechanisms by which glucose can control insulin release independently from its action on adenosine triphosphate-sensitive K+ channels in mouse B cells. J. Clin. Investig. 1993, 91, 871–880. [Google Scholar] [CrossRef]
- Kalwat, M.A.; Cobb, M.H. Mechanisms of the amplifying pathway of insulin secretion in the β cell. Pharm. Therap. 2017, 179, 17–30. [Google Scholar] [CrossRef] [PubMed]
- Pizarro-Delgado, J.; Deeney, J.T.; Martín-del-río, R.; Corkey, B.; Tamarit-Rodriguez, J. KCl-permeabilized pancreatic islets: An experimental model to explore the messenger role of ATP in the mechanism of insulin secretion. PLoS ONE 2015, 10, e0140096. [Google Scholar] [CrossRef] [PubMed]
- Sörenson, R.L.; Garry, D.G.; Brelje, T.C. Structural and functional considerations of GABA in islets of Langerhans. Diabetes 1991, 40, 1365–1374. [Google Scholar] [CrossRef]
- Hagan, D.W.; Ferreira, S.M.; Santos, G.J.; Phelps, E.A. The role of GABA on islet function. Front. Endocrinol. 2022, 13, 972115. [Google Scholar] [CrossRef]
- Menegaz, D.; Hagan, D.W.; Almaça, J.; Cianciaruso, C.; Rodriguez-Diaz, R.; Molina, J.; Dolan, R.M.; Becker, M.W.; Schwalie, P.C.; Nano, R.; et al. Mechanism and effects of pulsatile GABA secretion from cytosolic pools in the human beta cell. Nat. Metab. 2019, 1, 1110–1126. [Google Scholar] [CrossRef]
- Brice, N.L.; Varadi, A.; Ashcroft, S.J.H.; Molnar, E. Metabotropic glutamate and GABAB receptors contribute to the modulation of glucose-stimulated insulin secretion in pancreatic beta cells. Diabetologia 2002, 45, 242–252. [Google Scholar] [CrossRef]
- Wendt, A.; Birnir, B.; Buschard, K.; Gromada, J.; Salehi, A.; Sewing, S.; Rosrsman, P.; Braun, M. Glucose-inhibition of glucagon secretion from rat α-cells is mediated by GABA released from neighboring β-cells. Diabetes 2004, 53, 1038–1045. [Google Scholar] [CrossRef]
- Michalik, M.; Nelson, J.; Erecinska, M. GABA production in rat islets of Langerhans. Diabetes 1993, 42, 1506–1513. [Google Scholar] [CrossRef]
- Smismans, A.; Schuit, F.; Pipeleers, D. Nutrient regulation of gamma-aminobutyric acid release from islet beta cells. Diabetologia 1997, 40, 1411–1415. [Google Scholar] [CrossRef]
- Fernández-Pascual, S.; Mukala-Nsengu-Tshibangu, A.; Martín-del-Río, R.; Tamarit-Rodriguez, J. Conversion into GABA (γ-aminobutyric acid) may reduce the capacity of L-glutamine as an insulin secretagogue. Biochem. J. 2004, 379, 721–729. [Google Scholar] [CrossRef] [PubMed]
- Pizarro-Delgado, J.; Braun, M.; Hernández-Fisac, I.; Martín-del-Río, R.; Tamarit-Rodriguez, J. Glucose promotion of GABA metabolism contributes to the stimulation of insulin secretion in β-cells. Biochem. J. 2010, 431, 381–389. [Google Scholar] [CrossRef]
- Winnock, F.; Ling, Z.; De Proft, R.; Dejonghe, S.; Schuit, F.; Gorus, F.; Pipeleers, D. Correlation between GABA release from rat islet -cells and their metabolic state. Am. J. Physiol. Endocrinol. Metab. 2002, 282, E937–E942. [Google Scholar] [CrossRef]
- Hernández-Fisac, I.; Fernández-Pascual, S.; Ortsäter, H.; Pizarro-Delgado, J.; Martín-del-Río RBergsten, P.; Tamarit-Rodriguez, J. Oxo-4-methylpentanoic acid directs the metabolism of GABA into the Krebs cycle in rat pancreatic islets. Biochem. J. 2006, 400, 81–89. [Google Scholar] [CrossRef] [PubMed]
- Pizarro-Delgado, J.; Braun, M.; Hernández-Fisac, I.; Martín-del-Río, R.; Tamarit-Rodriguez, J. Branched-chain 2-oxoacids transamination increases GABA-shunt metabolism and insulin secretion in isolated islets. Biochem. J. 2009, 419, 359–368. [Google Scholar] [CrossRef]
- Zhou, Y.; Jetton, T.L.; Goshorn, S.; Lynch, C.J.; She, P. Transamination is required for α-ketoisocaproate but not leucine to stimulate insulin secretion. J. Biol. Chem. 2010, 285, 33718–33726. [Google Scholar] [CrossRef]
- Sener, A.; Malaisse-Lagae, F.; Malaisse, W.J. Stimulation of pancreatic islet metabolism and insulin release by a nonmetabolizable amino acid. Proc. Natl Acad. Sci. USA 1981, 78, 5460–5464. [Google Scholar] [CrossRef] [PubMed]
- Sener, A.; Somers, G.; Devis, G.; Malaisse, W.J. The stimulus-secretion coupling of amino acid-induced insulin release. Biosynthetic and secretory responses of rat pancreatic Islet to L-leucine and L-glutamine. Diabetologia 1981, 21, 135–142. [Google Scholar] [CrossRef]
- Pizarro-Delgado, J.; Deeney, J.T.; Martín-del-Río, R.; Corkey, B.; Tamarit-Rodriguez, J. Direct stimulation of islet insulin secretion by glycolytic and mitochondrial metabolites in KCl-depolarized islets. PLoS ONE 2016, 11, e0166111. [Google Scholar] [CrossRef]
- Rustenbeck, I.; Schulze, T.; Morsi, M.; Alshafei, A.; Panten, U. What is the metabolic amplification of insulin secretion and is it (still) relevant? Metabolites 2021, 11, 355. [Google Scholar] [CrossRef]
- Khan, A.; Ling, Z.C.; Landau, B.R. Quantifying the Carboxylation of Pyruvate in Pancreatic Islets. J. Biol. Chem. 1996, 271, 2539–2542. [Google Scholar] [CrossRef] [PubMed]
- Jensen, M.V.; Joseph, J.W.; Ilkayeva, O.; Burgess, S.; Lu, D.; Ronnebaum, S.M.; Odegaard, M.; Becker, T.C.; Sherry, A.D.; Newgard, C.B. Compensatory Responses to Pyruvate Carboxylase Suppression in Islet β-Cells. Preservation of glucose-stimulated insulin secretion. J. Biol. Chem. 2006, 281, 22342–22351. [Google Scholar] [CrossRef]
- Hasan, N.M.; Longacre, M.J.; Stoker, S.W.; Boonsaen, T.; Jitrapakdee, S.; Kendrick, M.A.; Wallace, J.C.; MacDonald, M.J. Impaired Anaplerosis and insulin secretion in insulinoma cells caused by small interfering RNA-mediated suppression of Pyruvate Carboxylase. J. Biol. Chem. 2008, 283, 28048–28059. [Google Scholar] [CrossRef] [PubMed]
- Zeczycki1, T.N.; Maurice, M.S.; Attwood, P.V. Inhibitors of Pyruvate Carboxylase. Open Enzym. Inhib. J. 2010, 3, 8–26. [Google Scholar] [CrossRef] [PubMed]
- MacDonald, M.J.; McKenzie, D.J.; Walker, T.M.; Kaysen, J.H. Lack of glyconeogenesis in pancreatic islets: Expression of gluconeogenic enzyme genes in islets. Horm. Metab. Res. 1992, 24, 158–160. [Google Scholar] [CrossRef]
- Tamarit-Rodriguez, J.; Vara, E.; Tamarit, J. Starvation-induced changes of palmitate metabolism and insulin secretion in isolated rat islets stimulated by glucose. Biochem. J. 1984, 221, 317–324. [Google Scholar] [CrossRef] [PubMed]
- Vara, E.; Tamarit-Rodriguez, J. Glucose stimulation of insulin secretion in islets of fed and starved rats and its dependence on lipid metabolism. Metabolism 1986, 35, 266–271. [Google Scholar] [CrossRef] [PubMed]
- Evans-Molina, C.; Sims, E.K.; DiMeglio, L.A.; Ismail, H.M.; Steck, A.K.; Palmer, J.P.; Krischer, J.P.; Geyer, S.; Xu, P.; Sosenko, J.M. Type 1 Diabetes Trialnet Study Group, β cell dysfunction exists more than 5 years before type 1 diabetes diagnosis. JCI Insight 2018, 3, e120877. [Google Scholar] [CrossRef] [PubMed]
- Misra, S. Pancreatic autoantibodies: Who to test and how to interpret the results. Pract. Diabetes 2017, 34, 221–223. [Google Scholar] [CrossRef]
- Haythorne, E.; Lloyd, M.; Walsby-Tickle, J.; Tarasov, A.I.; Sandbrink, J.; Portillo, I.; Terron Exposito, R.; Sachse, J.; Cyranka, M.; Rohm, M.; et al. Altered glycolysis impaired mitochondrial metabolism and mTORC1 activation in diabetic β-cells. Nat. Commun. 2022, 13, 6754. [Google Scholar] [CrossRef] [PubMed]
- Hellman, B.; Idahl, L.-Å.; Sehlin, J.; Täljedal, I.-B. Influence of anoxia on glucose metabolism in pancreatic islets: Lack of correlation between fructose-l,6-diphosphate, and apparent glycolytic flux. Diabetologia 1975, 11, 495–500. [Google Scholar] [CrossRef] [PubMed]
- Sekine, N.; Cirulli, V.; Regazzi, R.; Brown, L.J.; Giné, E.; Tamarit-Rodriguez, J.; Girotti, M.; Marie, S.; MacDonald, M.J.; Wollheim, C.B.; et al. Low lactate dehydrogenase and mitochondrial glycerol phosphate dehydrogenase in pancreatic β-cells. Potential role in nutrient sensing. J. Biol. Chem. 1994, 7, 4895–4902. [Google Scholar] [CrossRef]
- Medina, A.; Parween, S.; Ullsten, S.; Vishnu, N.; Siu, Y.T.; Quach, M.; Bennet, H.; Balhuizen, A.; Åkesson, L.; Wierup, N.; et al. Early deficits in insulin secretion, beta cell mass and islet blood perfusion precede onset of autoimmune type 1 diabetes in BioBreeding rats. Diabetologia 2018, 61, 896–905. [Google Scholar] [CrossRef]



Disclaimer/Publisher’s Note: The statements, opinions and data contained in all publications are solely those of the individual author(s) and contributor(s) and not of MDPI and/or the editor(s). MDPI and/or the editor(s) disclaim responsibility for any injury to people or property resulting from any ideas, methods, instructions or products referred to in the content. |
© 2023 by the author. Licensee MDPI, Basel, Switzerland. This article is an open access article distributed under the terms and conditions of the Creative Commons Attribution (CC BY) license (https://creativecommons.org/licenses/by/4.0/).
Share and Cite
Tamarit-Rodriguez, J. Metabolic Role of GABA in the Secretory Function of Pancreatic β-Cells: Its Hypothetical Implication in β-Cell Degradation in Type 2 Diabetes. Metabolites 2023, 13, 697. https://doi.org/10.3390/metabo13060697
Tamarit-Rodriguez J. Metabolic Role of GABA in the Secretory Function of Pancreatic β-Cells: Its Hypothetical Implication in β-Cell Degradation in Type 2 Diabetes. Metabolites. 2023; 13(6):697. https://doi.org/10.3390/metabo13060697
Chicago/Turabian StyleTamarit-Rodriguez, Jorge. 2023. "Metabolic Role of GABA in the Secretory Function of Pancreatic β-Cells: Its Hypothetical Implication in β-Cell Degradation in Type 2 Diabetes" Metabolites 13, no. 6: 697. https://doi.org/10.3390/metabo13060697
APA StyleTamarit-Rodriguez, J. (2023). Metabolic Role of GABA in the Secretory Function of Pancreatic β-Cells: Its Hypothetical Implication in β-Cell Degradation in Type 2 Diabetes. Metabolites, 13(6), 697. https://doi.org/10.3390/metabo13060697