
The Selective LAT1 Inhibitor JPH203 Enhances Mitochondrial Metabolism and Content in Insulin-Sensitive and Insulin-Resistant C2C12 Myotubes
 and
and
Abstract
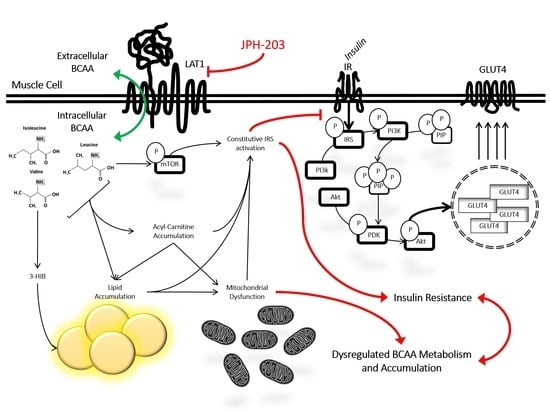
:
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
1. Introduction
2. Experimental Methods
2.1. Cell Culture
2.2. Cell Viability
2.3. Quantitative Real-Time Polymerase Chain Reaction (qRT-PCR)
2.4. Immunoblotting
2.5. Seahorse Metabolic Assays
2.6. Fluorescent Staining and Microscopy
2.7. Liquid Chromatography–Mass Spectrometry (LC–MS)
2.8. Statistical Analyses
3. Results
3.1. JPH at High Levels Reduces Insulin Sensitivity in Previously Insulin-Resistant Cells
3.2. JPH Improves Mitochondrial Function without Altering Mitochondrial Biogenic Signaling
3.3. Effect of JPH on Extracellular BCAA Content with and without Insulin Resistance
4. Discussion
5. Conclusions
Supplementary Materials
Author Contributions
Funding
Institutional Review Board Statement
Informed Consent Statement
Data Availability Statement
Acknowledgments
Conflicts of Interest
References
- Arany, Z.; Neinast, M. Branched Chain Amino Acids in Metabolic Disease. Curr. Diab. Rep. 2018, 18, 76. [Google Scholar] [CrossRef] [PubMed]
- Gannon, N.P.; Schnuck, J.K.; Vaughan, R.A. BCAA Metabolism and Insulin Sensitivity—Dysregulated by Metabolic Status? Mol. Nutr. Food Res. 2018, 62, e1700756. [Google Scholar] [CrossRef] [PubMed]
- Holeček, M. Why Are Branched-Chain Amino Acids Increased in Starvation and Diabetes? Nutrients 2020, 12, 3087. [Google Scholar] [CrossRef] [PubMed]
- Lynch, C.J.; Adams, S.H. Branched-chain amino acids in metabolic signalling and insulin resistance. Nat. Rev. Endocrinol. 2014, 10, 723–736. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Newgard, C.B. Interplay between lipids and branched-chain amino acids in development of insulin resistance. Cell Metab. 2012, 15, 606–614. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Newgard, C.B. Metabolomics and Metabolic Diseases: Where Do We Stand? Cell Metab. 2017, 25, 43–56. [Google Scholar] [CrossRef] [Green Version]
- Bishop, C.A.; Machate, T.; Henning, T.; Henkel, J.; Püschel, G.; Weber, D.; Grune, T.; Klaus, S.; Weitkunat, K. Detrimental effects of branched-chain amino acids in glucose tolerance can be attributed to valine induced glucotoxicity in skeletal muscle. Nutr. Diabetes 2022, 12, 20. [Google Scholar] [CrossRef]
- Haufe, S.; Engeli, S.; Kaminski, J.; Witt, H.; Rein, D.; Kamlage, B.; Utz, W.; Fuhrmann, J.C.; Haas, V.; Mahler, A.; et al. Branched-chain amino acid catabolism rather than amino acids plasma concentrations is associated with diet-induced changes in insulin resistance in overweight to obese individuals. Nutr. Metab. Cardiovasc. Dis. 2017, 27, 858–864. [Google Scholar] [CrossRef]
- Jang, C.; Oh, S.F.; Wada, S.; Rowe, G.C.; Liu, L.; Chan, M.C.; Rhee, J.; Hoshino, A.; Kim, B.; Ibrahim, A.; et al. A branched-chain amino acid metabolite drives vascular fatty acid transport and causes insulin resistance. Nat. Med. 2016, 22, 421–426. [Google Scholar] [CrossRef] [Green Version]
- Mardinoglu, A.; Gogg, S.; Lotta, L.A.; Stancakova, A.; Nerstedt, A.; Boren, J.; Bluher, M.; Ferrannini, E.; Langenberg, C.; Wareham, N.J.; et al. Elevated Plasma Levels of 3-Hydroxyisobutyric Acid Are Associated With Incident Type 2 Diabetes. EBioMedicine 2018, 27, 151–155. [Google Scholar] [CrossRef] [Green Version]
- He, B.; Moreau, R. Lipid-regulating properties of butyric acid and 4-phenylbutyric acid: Molecular mechanisms and therapeutic applications. Pharmacol. Res. 2019, 144, 116–131. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Crossland, H.; Smith, K.; Idris, I.; Phillips, B.E.; Atherton, P.J.; Wilkinson, D.J. Exploring mechanistic links between extracellular BCAA & muscle insulin resistance: An in vitro approach. Am. J. Physiol. Cell Physiol. 2020, 319, C1151–C1157. [Google Scholar] [CrossRef] [PubMed]
- Crossland, H.; Smith, K.; Idris, I.; Phillips, B.E.; Atherton, P.J.; Wilkinson, D.J. Phenylbutyrate, a branched-chain amino acid keto dehydrogenase activator, promotes branched-chain amino acid metabolism and induces muscle catabolism in C2C12 cells. Exp. Physiol. 2021, 106, 585–592. [Google Scholar] [CrossRef] [PubMed]
- Beriault, D.R.; Werstuck, G.H. Detection and quantification of endoplasmic reticulum stress in living cells using the fluorescent compound, Thioflavin T. Biochim. Biophys. Acta 2013, 1833, 2293–2301. [Google Scholar] [CrossRef] [Green Version]
- Dai, F.; Jiang, T.; Bao, Y.Y.; Chen, G.J.; Chen, L.; Zhang, Q.; Lu, Y.X. Fenofibrate improves high-fat diet-induced and palmitate-induced endoplasmic reticulum stress and inflammation in skeletal muscle. Life Sci. 2016, 157, 158–167. [Google Scholar] [CrossRef] [PubMed]
- Vanweert, F.; Neinast, M.; Tapia, E.E.; van de Weijer, T.; Hoeks, J.; Schrauwen-Hinderling, V.B.; Blair, M.C.; Bornstein, M.R.; Hesselink, M.K.C.; Schrauwen, P.; et al. A randomized placebo-controlled clinical trial for pharmacological activation of BCAA catabolism in patients with type 2 diabetes. Nat. Commun. 2022, 13, 3508. [Google Scholar] [CrossRef]
- Okunushi, K.; Furihata, T.; Morio, H.; Muto, Y.; Higuchi, K.; Kaneko, M.; Otsuka, Y.; Ohno, Y.; Watanabe, Y.; Reien, Y.; et al. JPH203, a newly developed anti-cancer drug, shows a preincubation inhibitory effect on L-type amino acid transporter 1 function. J. Pharmacol. Sci. 2020, 144, 16–22. [Google Scholar] [CrossRef]
- Okano, N.; Naruge, D.; Kawai, K.; Kobayashi, T.; Nagashima, F.; Endou, H.; Furuse, J. First-in-human phase I study of JPH203, an L-type amino acid transporter 1 inhibitor, in patients with advanced solid tumors. Investig. New Drugs 2020, 38, 1495–1506. [Google Scholar] [CrossRef]
- Akasu, R.; Miyazaki, T.; Elhussiny, M.Z.; Sugiura, Y.; Tomitsuka, Y.; Haraguchi, S.; Otsu, K.; Chowdhury, V.S.; Miyazaki, A. Calpain-mediated proteolytic production of free amino acids in vascular endothelial cells augments obesity-induced hepatic steatosis. J. Biol. Chem. 2022, 298, 101953. [Google Scholar] [CrossRef]
- Owada, T.; Kurasawa, K.; Endou, H.; Fujita, T.; Anzai, N.; Hayashi, K. LAT1-specific inhibitor ameliorates severe autoimmune arthritis in SKG mouse. Int. Immunopharmacol. 2022, 109, 108817. [Google Scholar] [CrossRef]
- Bröer, A.; Gauthier-Coles, G.; Rahimi, F.; van Geldermalsen, M.; Dorsch, D.; Wegener, A.; Holst, J.; Bröer, S. Ablation of the ASCT2 (SLC1A5) gene encoding a neutral amino acid transporter reveals transporter plasticity and redundancy in cancer cells. J. Biol. Chem. 2019, 294, 4012–4026. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Cormerais, Y.; Massard, P.A.; Vucetic, M.; Giuliano, S.; Tambutté, E.; Durivault, J.; Vial, V.; Endou, H.; Wempe, M.F.; Parks, S.K.; et al. The glutamine transporter ASCT2 (SLC1A5) promotes tumor growth independently of the amino acid transporter LAT1 (SLC7A5). J. Biol. Chem. 2018, 293, 2877–2887. [Google Scholar] [CrossRef] [Green Version]
- Poncet, N.; Mitchell, F.E.; Ibrahim, A.F.; McGuire, V.A.; English, G.; Arthur, J.S.; Shi, Y.B.; Taylor, P.M. The catalytic subunit of the system L1 amino acid transporter (slc7a5) facilitates nutrient signalling in mouse skeletal muscle. PLoS ONE 2014, 9, e89547. [Google Scholar] [CrossRef] [Green Version]
- Collao, N.; Akohene-Mensah, P.; Nallabelli, J.; Binet, E.R.; Askarian, A.; Lloyd, J.; Niemiro, G.M.; Beals, J.W.; van Vliet, S.; Rajgara, R.; et al. The role of L-type amino acid transporter 1. Am. J. Physiol. Cell Physiol. 2022, 323, C595–C605. [Google Scholar] [CrossRef] [PubMed]
- Mazzulla, M.; Hodson, N.; Lees, M.; Scaife, P.J.; Smith, K.; Atherton, P.J.; Kumbhare, D.; Moore, D.R. LAT1 and SNAT2 Protein Expression and Membrane Localization of LAT1 Are Not Acutely Altered by Dietary Amino Acids or Resistance Exercise Nor Positively Associated with Leucine or Phenylalanine Incorporation in Human Skeletal Muscle. Nutrients 2021, 13, 3906. [Google Scholar] [CrossRef]
- Lyon, E.S.; Rivera, M.E.; Johnson, M.A.; Sunderland, K.L.; Vaughan, R.A. Actions of chronic physiological 3-hydroxyisobuterate treatment on mitochondrial metabolism and insulin signaling in myotubes. Nutr. Res. 2019, 66, 22–31. [Google Scholar] [CrossRef] [PubMed]
- Rivera, M.E.; Lyon, E.S.; Johnson, M.A.; Sunderland, K.L.; Vaughan, R.A. Effect of valine on myotube insulin sensitivity and metabolism with and without insulin resistance. Mol. Cell Biochem. 2020, 468, 169–183. [Google Scholar] [CrossRef]
- Rivera, M.E.; Lyon, E.S.; Johnson, M.A.; Vaughan, R.A. Leucine increases mitochondrial metabolism and lipid content without altering insulin signaling in myotubes. Biochimie 2020, 168, 124–133. [Google Scholar] [CrossRef]
- Kumar, N.; Dey, C.S. Metformin enhances insulin signalling in insulin-dependent and-independent pathways in insulin resistant muscle cells. Br. J. Pharmacol. 2002, 137, 329–336. [Google Scholar] [CrossRef]
- Kumar, N.; Dey, C.S. Development of insulin resistance and reversal by thiazolidinediones in C2C12 skeletal muscle cells. Biochem. Pharmacol. 2003, 65, 249–257. [Google Scholar] [CrossRef]
- Rivera, C.N.; Kamer, M.M.; Rivera, M.E.; Watne, R.M.; Macgowan, T.C.; Wommack, A.J.; Vaughan, R.A. Insulin resistance promotes extracellular BCAA accumulation without altering LAT1 content, independent of prior BCAA treatment in a myotube model of skeletal muscle. Mol. Cell Endocrinol. 2023, 559, 111800. [Google Scholar] [CrossRef] [PubMed]
- Ye, L.; Varamini, B.; Lamming, D.W.; Sabatini, D.M.; Baur, J.A. Rapamycin has a biphasic effect on insulin sensitivity in C2C12 myotubes due to sequential disruption of mTORC1 and mTORC2. Front. Genet. 2012, 3, 177. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Yoon, M.S. The Role of Mammalian Target of Rapamycin (mTOR) in Insulin Signaling. Nutrients 2017, 9, 1176. [Google Scholar] [CrossRef]
- Maimaiti, M.; Sakamoto, S.; Yamada, Y.; Sugiura, M.; Rii, J.; Takeuchi, N.; Imamura, Y.; Furihata, T.; Ando, K.; Higuchi, K.; et al. Expression of L-type amino acid transporter 1 as a molecular target for prognostic and therapeutic indicators in bladder carcinoma. Sci. Rep. 2020, 10, 1292. [Google Scholar] [CrossRef] [Green Version]
- Wang, Q.; Holst, J. L-type amino acid transport and cancer: Targeting the mTORC1 pathway to inhibit neoplasia. Am. J. Cancer Res. 2015, 5, 1281–1294. [Google Scholar]
- Menon, S.; Dibble, C.C.; Talbott, G.; Hoxhaj, G.; Valvezan, A.J.; Takahashi, H.; Cantley, L.C.; Manning, B.D. Spatial control of the TSC complex integrates insulin and nutrient regulation of mTORC1 at the lysosome. Cell 2014, 156, 771–785. [Google Scholar] [CrossRef] [Green Version]
- Mesquita, P.H.C.; Vann, C.G.; Phillips, S.M.; McKendry, J.; Young, K.C.; Kavazis, A.N.; Roberts, M.D. Skeletal Muscle Ribosome and Mitochondrial Biogenesis in Response to Different Exercise Training Modalities. Front. Physiol. 2021, 12, 725866. [Google Scholar] [CrossRef]




Disclaimer/Publisher’s Note: The statements, opinions and data contained in all publications are solely those of the individual author(s) and contributor(s) and not of MDPI and/or the editor(s). MDPI and/or the editor(s) disclaim responsibility for any injury to people or property resulting from any ideas, methods, instructions or products referred to in the content. |
© 2023 by the authors. Licensee MDPI, Basel, Switzerland. This article is an open access article distributed under the terms and conditions of the Creative Commons Attribution (CC BY) license (https://creativecommons.org/licenses/by/4.0/).
Share and Cite
Rivera, C.N.; Smith, C.E.; Draper, L.V.; Ochoa, G.E.; Watne, R.M.; Wommack, A.J.; Vaughan, R.A. The Selective LAT1 Inhibitor JPH203 Enhances Mitochondrial Metabolism and Content in Insulin-Sensitive and Insulin-Resistant C2C12 Myotubes. Metabolites 2023, 13, 766. https://doi.org/10.3390/metabo13060766
Rivera CN, Smith CE, Draper LV, Ochoa GE, Watne RM, Wommack AJ, Vaughan RA. The Selective LAT1 Inhibitor JPH203 Enhances Mitochondrial Metabolism and Content in Insulin-Sensitive and Insulin-Resistant C2C12 Myotubes. Metabolites. 2023; 13(6):766. https://doi.org/10.3390/metabo13060766
Chicago/Turabian StyleRivera, Caroline N., Carly E. Smith, Lillian V. Draper, Gabriela E. Ochoa, Rachel M. Watne, Andrew J. Wommack, and Roger A. Vaughan. 2023. "The Selective LAT1 Inhibitor JPH203 Enhances Mitochondrial Metabolism and Content in Insulin-Sensitive and Insulin-Resistant C2C12 Myotubes" Metabolites 13, no. 6: 766. https://doi.org/10.3390/metabo13060766
APA StyleRivera, C. N., Smith, C. E., Draper, L. V., Ochoa, G. E., Watne, R. M., Wommack, A. J., & Vaughan, R. A. (2023). The Selective LAT1 Inhibitor JPH203 Enhances Mitochondrial Metabolism and Content in Insulin-Sensitive and Insulin-Resistant C2C12 Myotubes. Metabolites, 13(6), 766. https://doi.org/10.3390/metabo13060766