
Neuroendocrine Effects on the Risk of Metabolic Syndrome in Children


{kind=link}
{kind=link}
{kind=link}
Abstract
:1. Introduction
2. Neuroendocrine Pathways of the Stress Response and the Risk of Metabolic Syndrome
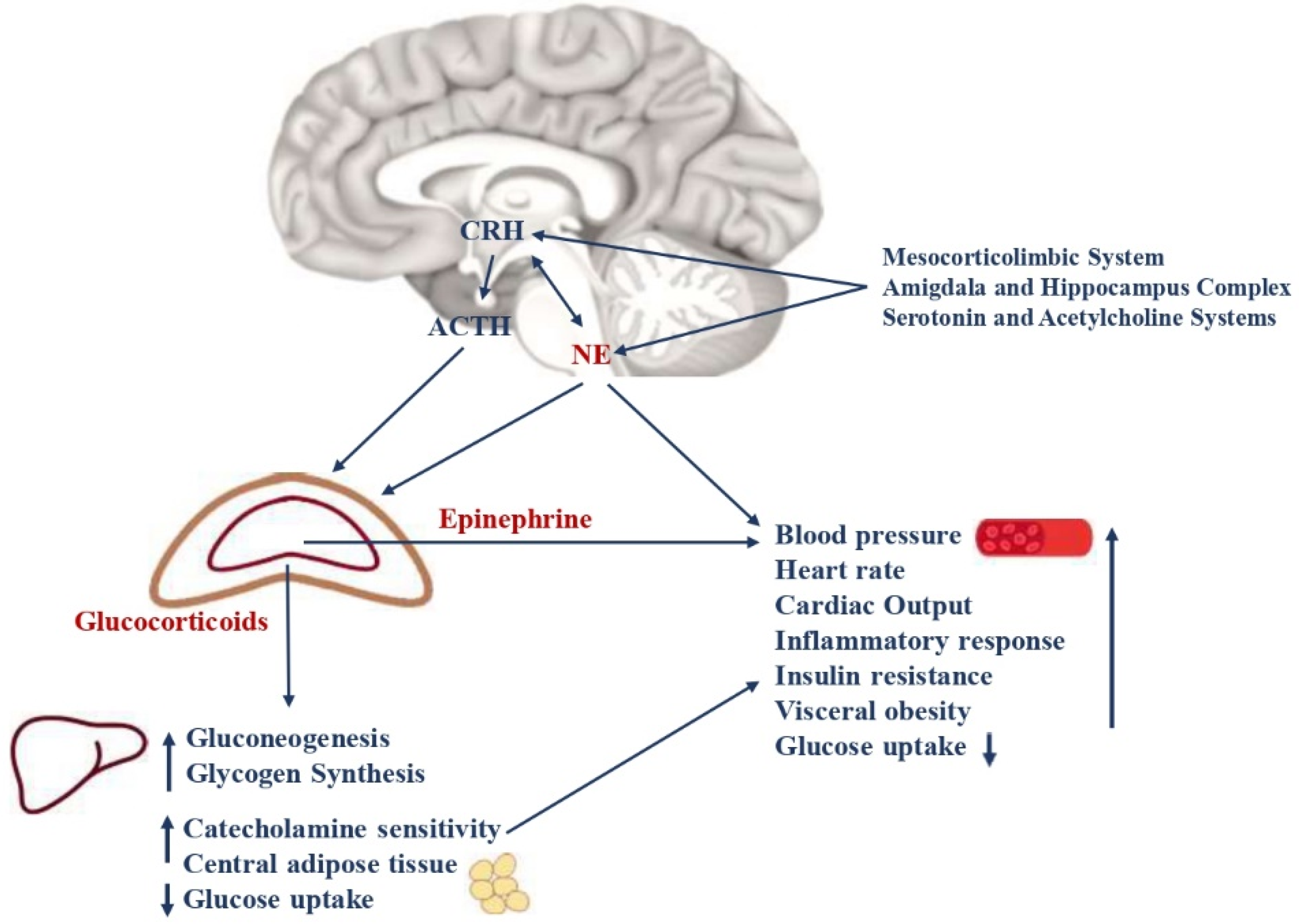
2.1. Hypothalamic-Pituitary-Adrenal Axis
2.2. Arousal and Sympathetic Nervous Systems
2.3. Genetic Variants and Epigenetic Modifications Involved in the Stress Response
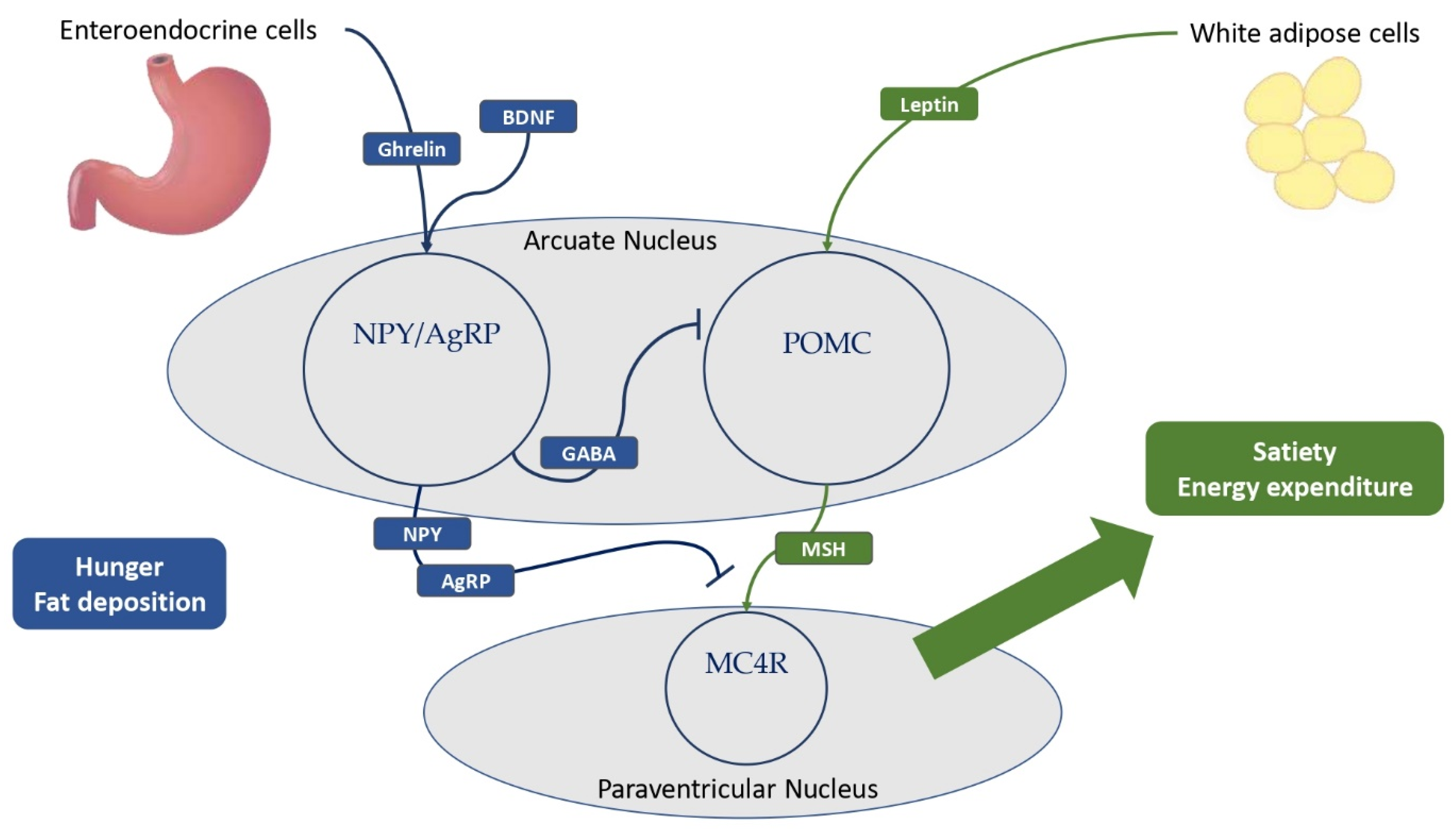
3. The Regulation of Satiety and Hunger in Central Neuroendocrine Pathways
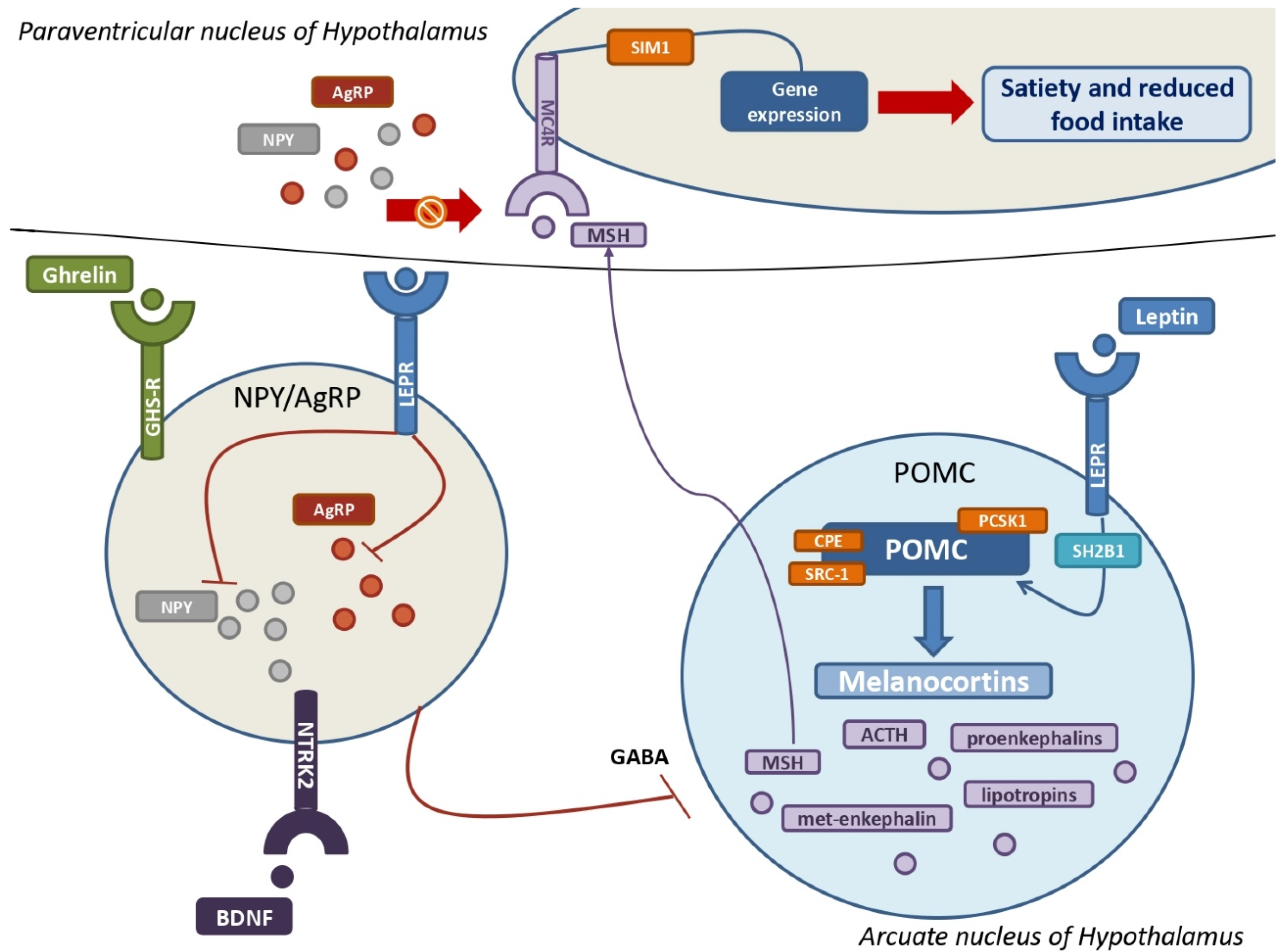
3.1. Leptin and LEPR, a Key Neuroendocrine Tandem with Metabolic Implications
3.2. The Key Role of POMC, Melanocortins, and Related Receptors in the Development of MetS
3.3. Ghrelin, the NPY–AgRP Pathway, and Metabolic Implications
4. Conclusions
Funding
Conflicts of Interest
References
- Christian Flemming, G.M.; Bussler, S.; Körner, A.; Kiess, W. Definition and Early Diagnosis of Metabolic Syndrome in Children. J. Pediatr. Endocrinol. Metab. 2020, 33, 821–833. [Google Scholar] [CrossRef]
- Alberti, K.G.M.M.; Zimmet, P.; Shaw, J. Metabolic Syndrome—A New World-Wide Definition. A Consensus Statement from the International Diabetes Federation. Diabet. Med. 2006, 23, 469–480. [Google Scholar] [CrossRef] [PubMed]
- Kumar, S.; Kelly, A.S. Review of Childhood Obesity: From Epidemiology, Etiology, and Comorbidities to Clinical Assessment and Treatment. Mayo Clin. Proc. 2017, 92, 251–265. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Mahmoud, R.; Kimonis, V.; Butler, M.G. Genetics of Obesity in Humans: A Clinical Review. Int. J. Mol. Sci. 2022, 23, 11005. [Google Scholar] [CrossRef] [PubMed]
- Hinney, A.; Körner, A.; Fischer-Posovszky, P. The Promise of New Anti-Obesity Therapies Arising from Knowledge of Genetic Obesity Traits. Nat. Rev. Endocrinol. 2022, 18, 623–637. [Google Scholar] [CrossRef]
- Kaur, Y.; de Souza, R.J.; Gibson, W.T.; Meyre, D. A Systematic Review of Genetic Syndromes with Obesity. Obes. Rev. 2017, 18, 603–634. [Google Scholar] [CrossRef]
- Sohn, Y.B. Genetic Obesity: An Update with Emerging Therapeutic Approaches. Ann. Pediatr. Endocrinol. Metab. 2022, 27, 169–175. [Google Scholar] [CrossRef]
- Kleiser, C.; Schaffrath Rosario, A.; Mensink, G.B.M.; Prinz-Langenohl, R.; Kurth, B.-M. Potential Determinants of Obesity among Children and Adolescents in Germany: Results from the Cross-Sectional KiGGS Study. BMC Public Health 2009, 9, 46. [Google Scholar] [CrossRef] [Green Version]
- Geserick, M.; Vogel, M.; Gausche, R.; Lipek, T.; Spielau, U.; Keller, E.; Pfäffle, R.; Kiess, W.; Körner, A. Acceleration of BMI in Early Childhood and Risk of Sustained Obesity. N. Engl. J. Med. 2018, 379, 1303–1312. [Google Scholar] [CrossRef]
- Qiao, Y.; Ma, J.; Wang, Y.; Li, W.; Katzmarzyk, P.T.; Chaput, J.-P.; Fogelholm, M.; Johnson, W.D.; Kuriyan, R.; Kurpad, A.; et al. Birth Weight and Childhood Obesity: A 12-Country Study. Int. J. Obes. Suppl. 2015, 5, S74–S79. [Google Scholar] [CrossRef] [Green Version]
- Lemche, E.; Chaban, O.S.; Lemche, A.V. Neuroendocrinological and Epigenetic Mechanisms Subserving Autonomic Imbalance and HPA Dysfunction in the Metabolic Syndrome. Front. Neurosci. 2016, 10, 142. [Google Scholar] [CrossRef] [Green Version]
- Charmandari, E.; Kino, T.; Souvatzoglou, E.; Chrousos, G.P. Pediatric Stress: Hormonal Mediators and Human Development. Horm. Res. Paediatr. 2003, 59, 161–179. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Womersley, J.S.; Nothling, J.; Toikumo, S.; Malan-Müller, S.; Van Den Heuvel, L.L.; McGregor, N.W.; Seedat, S.; Hemmings, S.M.J. Childhood Trauma, the Stress Response and Metabolic Syndrome: A Focus on DNA Methylation. Eur. J. Neurosci. 2022, 55, 2253–2296. [Google Scholar] [CrossRef]
- Chrousos, G.P. Stress and Disorders of the Stress System. Nat. Rev. Endocrinol. 2009, 5, 374–381. [Google Scholar] [CrossRef]
- Pervanidou, P.; Chrousos, G.P. Stress and Obesity/Metabolic Syndrome in Childhood and Adolescence. Int. J. Pediatr. Obes. 2011, 6, 21–28. [Google Scholar] [CrossRef] [PubMed]
- McEwen, B.S. Physiology and Neurobiology of Stress and Adaptation: Central Role of the Brain. Physiol. Rev. 2007, 87, 873–904. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Pervanidou, P.; Chrousos, G.P. Metabolic Consequences of Stress during Childhood and Adolescence. Metabolism 2012, 61, 611–619. [Google Scholar] [CrossRef]
- Chrousos, G.P. Organization and Integration of the Endocrine System: The Arousal and Sleep Perspective. Sleep Med. Clin. 2007, 2, 125–145. [Google Scholar] [CrossRef] [Green Version]
- Calogero, A.E.; Norton, J.A.; Sheppard, B.C.; Listwak, S.J.; Cromack, D.T.; Wall, R.; Jensen, R.T.; Chrousos, G.P. Pulsatile Activation of the Hypothalamic-Pituitary-Adrenal Axis during Major Surgery. Metabolism 1992, 41, 839–845. [Google Scholar] [CrossRef]
- Holmes, M.C.; Antoni, F.A.; Aguilera, G.; Catt, K.J. Magnocellular Axons in Passage through the Median Eminence Release Vasopressin. Nature 1986, 319, 326–329. [Google Scholar] [CrossRef]
- Pervanidou, P.; Kolaitis, G.; Charitaki, S.; Lazaropoulou, C.; Papassotiriou, I.; Hindmarsh, P.; Bakoula, C.; Tsiantis, J.; Chrousos, G.P. The Natural History of Neuroendocrine Changes in Pediatric Posttraumatic Stress Disorder (PTSD) After Motor Vehicle Accidents: Progressive Divergence of Noradrenaline and Cortisol Concentrations Over Time. Biol. Psychiatry 2007, 62, 1095–1102. [Google Scholar] [CrossRef] [PubMed]
- Ivović, M.; Marina, L.V.; Šojat, A.S.; Tančić-Gajić, M.; Arizanović, Z.; Kendereški, A.; Vujović, S. Approach to the Patient with Subclinical Cushing’s Syndrome. CPD 2020, 26, 5584–5590. [Google Scholar] [CrossRef]
- Sidibeh, C.O.; Pereira, M.J.; Abalo, X.M.; Boersma, G.J.; Skrtic, S.; Lundkvist, P.; Katsogiannos, P.; Hausch, F.; Castillejo-López, C.; Eriksson, J.W. FKBP5 Expression in Human Adipose Tissue: Potential Role in Glucose and Lipid Metabolism, Adipogenesis and Type 2 Diabetes. Endocrine 2018, 62, 116–128. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Roy, M.S.; Roy, A.; Gallucci, W.T.; Collier, B.; Young, K.; Kamilaris, T.C.; Chrousos, G.P. The Ovine Corticotropin-Releasing Hormone-Stimulation Test in Type I Diabetic Patients and Controls: Suggestion of Mild Chronic Hypercortisolism. Metabolism 1993, 42, 696–700. [Google Scholar] [CrossRef]
- Tsigos, C.; Young, R.J.; White, A. Diabetic Neuropathy Is Associated with Increased Activity of the Hypothalamic-Pituitary-Adrenal Axis. J. Clin. Endocrinol. Metab. 1993, 76, 554–558. [Google Scholar] [CrossRef] [PubMed]
- Ulrich-Lai, Y.M.; Herman, J.P. Neural Regulation of Endocrine and Autonomic Stress Responses. Nat. Rev. Neurosci. 2009, 10, 397–409. [Google Scholar] [CrossRef] [Green Version]
- Kiss, A.; Aguilera, G. Participation of α1-Adrenergic Receptors in the Secretion of Hypothalamic Corticotropin- Releasing Hormone during Stress. Neuroendocrinology 1992, 56, 153–160. [Google Scholar] [CrossRef]
- Straznicky, N.E.; Lambert, G.W.; Masuo, K.; Dawood, T.; Eikelis, N.; Nestel, P.J.; McGrane, M.T.; Mariani, J.A.; Socratous, F.; Chopra, R.; et al. Blunted Sympathetic Neural Response to Oral Glucose in Obese Subjects with the Insulin-Resistant Metabolic Syndrome. Am. J. Clin. Nutr. 2009, 89, 27–36. [Google Scholar] [CrossRef] [Green Version]
- Grassi, G.; Quarti-Trevano, F.; Seravalle, G.; Dell’Oro, R. Cardiovascular Risk and Adrenergic Overdrive in the Metabolic Syndrome. Nutr. Metab. Cardiovasc. Dis. 2007, 17, 473–481. [Google Scholar] [CrossRef]
- Pervanidou, P. Biology of Post-Traumatic Stress Disorder in Childhood and Adolescence. J. Neuroendocr. 2008, 20, 632–638. [Google Scholar] [CrossRef]
- Rubens, M.; Bruenig, D.; Adams, J.A.M.; Suresh, S.M.; Sathyanarayanan, A.; Haslam, D.; Shenk, C.E.; Mathews, B.; Mehta, D. Childhood Maltreatment and DNA Methylation: A Systematic Review. Neurosci. Biobehav. Rev. 2023, 147, 105079. [Google Scholar] [CrossRef] [PubMed]
- Bartoli, F.; Carrà, G.; Crocamo, C.; Carretta, D.; Clerici, M. Metabolic Syndrome in People Suffering from Posttraumatic Stress Disorder: A Systematic Review and Meta-Analysis. Metab. Syndr. Relat. Disord. 2013, 11, 301–308. [Google Scholar] [CrossRef] [PubMed]
- Kesebir, S. Metabolic Syndrome and Childhood Trauma: Also Comorbidity and Complication in Mood Disorder. WJCC 2014, 2, 332. [Google Scholar] [CrossRef] [PubMed]
- Napoli, E.; Tassone, F.; Wong, S.; Angkustsiri, K.; Simon, T.J.; Song, G.; Giulivi, C. Mitochondrial Citrate Transporter-Dependent Metabolic Signature in the 22q11.2 Deletion Syndrome. J. Biol. Chem. 2015, 290, 23240–23253. [Google Scholar] [CrossRef] [Green Version]
- Gao, Y.; Lin, Y.; Sun, K.; Wang, Y.; Chen, J.; Wang, H.; Zhou, X.; Fan, X.; Hui, R. Orthostatic Blood Pressure Dysregulation and Polymorphisms of β-Adrenergic Receptor Genes in Hypertensive Patients. J. Clin. Hypertens. 2014, 16, 207–213. [Google Scholar] [CrossRef]
- Guay, S.-P.; Brisson, D.; Lamarche, B.; Biron, S.; Lescelleur, O.; Biertho, L.; Marceau, S.; Vohl, M.-C.; Gaudet, D.; Bouchard, L. ADRB3 Gene Promoter DNA Methylation in Blood and Visceral Adipose Tissue Is Associated with Metabolic Disturbances in Men. Epigenomics 2014, 6, 33–43. [Google Scholar] [CrossRef]
- Wolf, E.J.; Mitchell, K.S.; Logue, M.W.; Baldwin, C.T.; Reardon, A.F.; Humphries, D.E.; Miller, M.W. Corticotropin Releasing Hormone Receptor 2 (CRHR-2) Gene Is Associated with Decreased Risk and Severity of Posttraumatic Stress Disorder in Women: CRHR-2 and PTSD. Depress. Anxiety 2013, 30, 1161–1169. [Google Scholar] [CrossRef] [Green Version]
- Häusl, A.S.; Balsevich, G.; Gassen, N.C.; Schmidt, M.V. Focus on FKBP51: A Molecular Link between Stress and Metabolic Disorders. Mol. Metab. 2019, 29, 170–181. [Google Scholar] [CrossRef]
- Pereira, M.J.; Palming, J.; Svensson, M.K.; Rizell, M.; Dalenbäck, J.; Hammar, M.; Fall, T.; Sidibeh, C.O.; Svensson, P.-A.; Eriksson, J.W. FKBP5 Expression in Human Adipose Tissue Increases Following Dexamethasone Exposure and Is Associated with Insulin Resistance. Metabolism 2014, 63, 1198–1208. [Google Scholar] [CrossRef] [PubMed]
- Kleinendorst, L.; Abawi, O.; van der Kamp, H.J.; Alders, M.; Meijers-Heijboer, H.E.J.; van Rossum, E.F.C.; van den Akker, E.L.T.; van Haelst, M.M. Leptin Receptor Deficiency: A Systematic Literature Review and Prevalence Estimation Based on Population Genetics. Eur. J. Endocrinol. 2020, 182, 47–56. [Google Scholar] [CrossRef]
- Nordang, G.B.N.; Busk, Ø.L.; Tveten, K.; Hanevik, H.I.; Fell, A.K.M.; Hjelmesæth, J.; Holla, Ø.L.; Hertel, J.K. Next-Generation Sequencing of the Monogenic Obesity Genes LEP, LEPR, MC4R, PCSK1 and POMC in a Norwegian Cohort of Patients with Morbid Obesity and Normal Weight Controls. Mol. Genet. Metab. 2017, 121, 51–56. [Google Scholar] [CrossRef]
- Obradovic, M.; Sudar-Milovanovic, E.; Soskic, S.; Essack, M.; Arya, S.; Stewart, A.J.; Gojobori, T.; Isenovic, E.R. Leptin and Obesity: Role and Clinical Implication. Front. Endocrinol. 2021, 12, 585887. [Google Scholar] [CrossRef]
- Morash, B.; Li, A.; Murphy, P.R.; Wilkinson, M.; Ur, E. Leptin Gene Expression in the Brain and Pituitary Gland. Endocrinology 1999, 140, 5995–5998. [Google Scholar] [CrossRef] [PubMed]
- Pan, W.W.; Myers, M.G. Leptin and the Maintenance of Elevated Body Weight. Nat. Rev. Neurosci. 2018, 19, 95–105. [Google Scholar] [CrossRef]
- Singh, R.K.; Kumar, P.; Mahalingam, K. Molecular Genetics of Human Obesity: A Comprehensive Review. C. R. Biol. 2017, 340, 87–108. [Google Scholar] [CrossRef] [PubMed]
- Friedman, J.M.; Halaas, J.L. Leptin and the Regulation of Body Weight in Mammals. Nature 1998, 395, 763–770. [Google Scholar] [CrossRef] [PubMed]
- Dayton, K.; Miller, J. Finding Treatable Genetic Obesity: Strategies for Success. Curr. Opin. Pediatr. 2018, 30, 526–531. [Google Scholar] [CrossRef]
- Gregoric, N.; Groselj, U.; Bratina, N.; Debeljak, M.; Zerjav Tansek, M.; Suput Omladic, J.; Kovac, J.; Battelino, T.; Kotnik, P.; Avbelj Stefanija, M. Two Cases With an Early Presented Proopiomelanocortin Deficiency—A Long-Term Follow-Up and Systematic Literature Review. Front. Endocrinol. 2021, 12, 689387. [Google Scholar] [CrossRef]
- Rui, L. SH2B1 Regulation of Energy Balance, Body Weight, and Glucose Metabolism. World J. Diabetes 2014, 5, 511–526. [Google Scholar] [CrossRef]
- Doche, M.E.; Bochukova, E.G.; Su, H.-W.; Pearce, L.R.; Keogh, J.M.; Henning, E.; Cline, J.M.; Saeed, S.; Dale, A.; Cheetham, T.; et al. Human SH2B1 Mutations Are Associated with Maladaptive Behaviors and Obesity. J. Clin. Investig. 2012, 122, 4732–4736. [Google Scholar] [CrossRef] [Green Version]
- Li, Z.; Zhou, Y.; Carter-Su, C.; Myers, M.G.; Rui, L. SH2B1 Enhances Leptin Signaling by Both Janus Kinase 2 Tyr813 Phosphorylation-Dependent and -Independent Mechanisms. Mol. Endocrinol. 2007, 21, 2270–2281. [Google Scholar] [CrossRef] [PubMed]
- Reyes, M.; Quintanilla, C.; Burrows, R.; Blanco, E.; Cifuentes, M.; Gahagan, S. Obesity Is Associated with Acute Inflammation in a Sample of Adolescents. Pediatr. Diabetes 2015, 16, 109–116. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Wasim, M.; Awan, F.R.; Najam, S.S.; Khan, A.R.; Khan, H.N. Role of Leptin Deficiency, Inefficiency, and Leptin Receptors in Obesity. Biochem. Genet. 2016, 54, 565–572. [Google Scholar] [CrossRef]
- Antunes, H.; Santos, C.; Carvalho, S. Serum Leptin Levels in Overweight Children and Adolescents. Br. J. Nutr. 2009, 101, 1262–1266. [Google Scholar] [CrossRef] [Green Version]
- Wabitsch, M.; Funcke, J.-B.; von Schnurbein, J.; Denzer, F.; Lahr, G.; Mazen, I.; El-Gammal, M.; Denzer, C.; Moss, A.; Debatin, K.-M.; et al. Severe Early-Onset Obesity Due to Bioinactive Leptin Caused by a p.N103K Mutation in the Leptin Gene. J. Clin. Endocrinol. Metab. 2015, 100, 3227–3230. [Google Scholar] [CrossRef] [Green Version]
- Barnes, M.J.; McDougal, D.H. Leptin into the Rostral Ventral Lateral Medulla (RVLM) Augments Renal Sympathetic Nerve Activity and Blood Pressure. Front. Neurosci. 2014, 8, 232. [Google Scholar] [CrossRef] [Green Version]
- Poitou, C.; Mosbah, H.; Clément, K. MECHANISMS IN ENDOCRINOLOGY: Update on Treatments for Patients with Genetic Obesity. Eur. J. Endocrinol. 2020, 183, R149–R166. [Google Scholar] [CrossRef]
- Kleinendorst, L.; Abawi, O.; van der Voorn, B.; Jongejan, M.H.T.M.; Brandsma, A.E.; Visser, J.A.; van Rossum, E.F.C.; van der Zwaag, B.; Alders, M.; Boon, E.M.J.; et al. Identifying Underlying Medical Causes of Pediatric Obesity: Results of a Systematic Diagnostic Approach in a Pediatric Obesity Center. PLoS ONE 2020, 15, e0232990. [Google Scholar] [CrossRef] [PubMed]
- Durmaz, A.; Aykut, A.; Atik, T.; Özen, S.; Ayyıldız Emecen, D.; Ata, A.; Işık, E.; Gökşen, D.; Çoğulu, Ö.; Özkınay, F. A New Cause of Obesity Syndrome Associated with a Mutation in the Carboxypeptidase Gene Detected in Three Siblings with Obesity, Intellectual Disability and Hypogonadotropic Hypogonadism. J. Clin. Res. Pediatr. Endocrinol. 2021, 13, 52–60. [Google Scholar] [CrossRef]
- Yang, Y.; van der Klaauw, A.A.; Zhu, L.; Cacciottolo, T.M.; He, Y.; Stadler, L.K.J.; Wang, C.; Xu, P.; Saito, K.; Hinton, A.; et al. Steroid Receptor Coactivator-1 Modulates the Function of Pomc Neurons and Energy Homeostasis. Nat. Commun. 2019, 10, 1718. [Google Scholar] [CrossRef] [Green Version]
- Ericson, M.D.; Lensing, C.J.; Fleming, K.A.; Schlasner, K.N.; Doering, S.R.; Haskell-Luevano, C. Bench-Top to Clinical Therapies: A Review of Melanocortin Ligands from 1954 to 2016. Biochim. Biophys. Acta Mol. Basis Dis. 2017, 1863, 2414–2435. [Google Scholar] [CrossRef] [PubMed]
- Balthasar, N. Genetic Dissection of Neuronal Pathways Controlling Energy Homeostasis. Obesity 2006, 14 (Suppl. 5), 222S–227S. [Google Scholar] [CrossRef] [PubMed]
- Farooqi, I.S.; Keogh, J.M.; Yeo, G.S.H.; Lank, E.J.; Cheetham, T.; O’Rahilly, S. Clinical Spectrum of Obesity and Mutations in the Melanocortin 4 Receptor Gene. N. Engl. J. Med. 2003, 348, 1085–1095. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Hinney, A.; Volckmar, A.-L.; Knoll, N. Melanocortin-4 Receptor in Energy Homeostasis and Obesity Pathogenesis. Prog. Mol. Biol. Transl. Sci. 2013, 114, 147–191. [Google Scholar] [CrossRef] [PubMed]
- Holder, J.L.; Butte, N.F.; Zinn, A.R. Profound Obesity Associated with a Balanced Translocation That Disrupts the SIM1 Gene. Hum. Mol. Genet. 2000, 9, 101–108. [Google Scholar] [CrossRef]
- Yeo, G.S.H.; Chao, D.H.M.; Siegert, A.-M.; Koerperich, Z.M.; Ericson, M.D.; Simonds, S.E.; Larson, C.M.; Luquet, S.; Clarke, I.; Sharma, S.; et al. The Melanocortin Pathway and Energy Homeostasis: From Discovery to Obesity Therapy. Mol. Metab. 2021, 48, 101206. [Google Scholar] [CrossRef]
- Perez-Tilve, D.; Hofmann, S.M.; Basford, J.; Nogueiras, R.; Pfluger, P.T.; Patterson, J.T.; Grant, E.; Wilson-Perez, H.E.; Granholm, N.A.; Arnold, M.; et al. Melanocortin Signaling in the CNS Directly Regulates Circulating Cholesterol. Nat. Neurosci. 2010, 13, 877–882. [Google Scholar] [CrossRef] [Green Version]
- Kühnen, P.; Krude, H.; Biebermann, H. Melanocortin-4 Receptor Signalling: Importance for Weight Regulation and Obesity Treatment. Trends Mol. Med. 2019, 25, 136–148. [Google Scholar] [CrossRef]
- Lotta, L.A.; Mokrosiński, J.; Mendes de Oliveira, E.; Li, C.; Sharp, S.J.; Luan, J.; Brouwers, B.; Ayinampudi, V.; Bowker, N.; Kerrison, N.; et al. Human Gain-of-Function MC4R Variants Show Signaling Bias and Protect against Obesity. Cell 2019, 177, 597–607.e9. [Google Scholar] [CrossRef] [Green Version]
- Krude, H.; Biebermann, H.; Luck, W.; Horn, R.; Brabant, G.; Grüters, A. Severe Early-Onset Obesity, Adrenal Insufficiency and Red Hair Pigmentation Caused by POMC Mutations in Humans. Nat. Genet. 1998, 19, 155–157. [Google Scholar] [CrossRef]
- Graves, L.E.; Khouri, J.M.; Kristidis, P.; Verge, C.F. Proopiomelanocortin Deficiency Diagnosed in Infancy in Two Boys and a Review of the Known Cases. J. Paediatr. Child Health 2021, 57, 484–490. [Google Scholar] [CrossRef] [PubMed]
- Candler, T.; Kühnen, P.; Prentice, A.M.; Silver, M. Epigenetic Regulation of POMC; Implications for Nutritional Programming, Obesity and Metabolic Disease. Front. Neuroendocr. 2019, 54, 100773. [Google Scholar] [CrossRef] [PubMed]
- Pépin, L.; Colin, E.; Tessarech, M.; Rouleau, S.; Bouhours-Nouet, N.; Bonneau, D.; Coutant, R. A New Case of PCSK1 Pathogenic Variant With Congenital Proprotein Convertase 1/3 Deficiency and Literature Review. J. Clin. Endocrinol. Metab. 2019, 104, 985–993. [Google Scholar] [CrossRef]
- Bosch, E.; Hebebrand, M.; Popp, B.; Penger, T.; Behring, B.; Cox, H.; Towner, S.; Kraus, C.; Wilson, W.G.; Khan, S.; et al. BDV Syndrome: An Emerging Syndrome With Profound Obesity and Neurodevelopmental Delay Resembling Prader-Willi Syndrome. J. Clin. Endocrinol. Metab. 2021, 106, 3413–3427. [Google Scholar] [CrossRef]
- Cacciottolo, T.M.; Henning, E.; Keogh, J.M.; Bel Lassen, P.; Lawler, K.; Bounds, R.; Ahmed, R.; Perdikari, A.; Mendes de Oliveira, E.; Smith, M.; et al. Obesity Due to Steroid Receptor Coactivator-1 Deficiency Is Associated With Endocrine and Metabolic Abnormalities. J. Clin. Endocrinol. Metab. 2022, 107, e2532–e2544. [Google Scholar] [CrossRef]
- Michaud, J.L. Sim1 Haploinsufficiency Causes Hyperphagia, Obesity and Reduction of the Paraventricular Nucleus of the Hypothalamus. Hum. Mol. Genet. 2001, 10, 1465–1473. [Google Scholar] [CrossRef]
- Xia, Q.; Grant, S.F.A. The Genetics of Human Obesity. Ann. N. Y. Acad. Sci. 2013, 1281, 178–190. [Google Scholar] [CrossRef]
- Laurila, M.; Santaniemi, M.; Kesäniemi, Y.A.; Ukkola, O. High Plasma Ghrelin Protects from Coronary Heart Disease and Leu72Leu Polymorphism of Ghrelin Gene from Cancer in Healthy Adults during the 19 Years Follow-up Study. Peptides 2014, 61, 122–129. [Google Scholar] [CrossRef]
- Thorsell, A. Brain Neuropeptide Y and Corticotropin-Releasing Hormone in Mediating Stress and Anxiety. Exp. Biol. Med. 2010, 235, 1163–1167. [Google Scholar] [CrossRef]
- Sitticharoon, C.; Chatree, S.; Churintaraphan, M. Expressions of Neuropeptide Y and Y1 Receptor in Subcutaneous and Visceral Fat Tissues in Normal Weight and Obese Humans and Their Correlations with Clinical Parameters and Peripheral Metabolic Factors. Regul. Pept. 2013, 185, 65–72. [Google Scholar] [CrossRef] [PubMed]
- Wei, Z.; Zhang, K.; Wen, G.; Balasubramanian, K.; Shih, P.B.; Rao, F.; Friese, R.S.; Miramontes-Gonzalez, J.P.; Schmid-Schoenbein, G.W.; Kim, H.-S.; et al. Heredity and Cardiometabolic Risk: Naturally Occurring Polymorphisms in the Human Neuropeptide Y(2) Receptor Promoter Disrupt Multiple Transcriptional Response Motifs. J. Hypertens. 2013, 31, 123–133. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Kuo, L.E.; Kitlinska, J.B.; Tilan, J.U.; Li, L.; Baker, S.B.; Johnson, M.D.; Lee, E.W.; Burnett, M.S.; Fricke, S.T.; Kvetnansky, R.; et al. Neuropeptide Y Acts Directly in the Periphery on Fat Tissue and Mediates Stress-Induced Obesity and Metabolic Syndrome. Nat. Med. 2007, 13, 803–811. [Google Scholar] [CrossRef] [PubMed]
- Lin, X.; Qi, Q.; Zheng, Y.; Huang, T.; Lathrop, M.; Zelenika, D.; Bray, G.A.; Sacks, F.M.; Liang, L.; Qi, L. Neuropeptide Y Genotype, Central Obesity, and Abdominal Fat Distribution: The POUNDS LOST Trial. Am. J. Clin. Nutr. 2015, 102, 514–519. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Jaakkola, U.; Kallio, J.; Heine, R.J.; Nijpels, G.; t’ Hart, L.M.; Maassen, J.A.; Bouter, L.M.; Stehouwer, C.D.A.; Dekker, J.M. Neuropeptide Y Polymorphism Significantly Magnifies Diabetes and Cardiovascular Disease Risk in Obesity: The Hoorn Study. Eur. J. Clin. Nutr. 2009, 63, 150–152. [Google Scholar] [CrossRef] [PubMed]
- Vergani, E.; Bruno, C.; Cipolla, C.; Currò, D.; Mancini, A. Plasma Levels of Neudesin and Glucose Metabolism in Obese and Overweight Children. Front. Endocrinol. 2022, 13, 881524. [Google Scholar] [CrossRef]
- Polidori, N.; Mainieri, F.; Chiarelli, F.; Mohn, A.; Giannini, C. Early Insulin Resistance, Type 2 Diabetes, and Treatment Options in Childhood. Horm. Res. Paediatr. 2022, 95, 149–166. [Google Scholar] [CrossRef]



Disclaimer/Publisher’s Note: The statements, opinions and data contained in all publications are solely those of the individual author(s) and contributor(s) and not of MDPI and/or the editor(s). MDPI and/or the editor(s) disclaim responsibility for any injury to people or property resulting from any ideas, methods, instructions or products referred to in the content. |
© 2023 by the authors. Licensee MDPI, Basel, Switzerland. This article is an open access article distributed under the terms and conditions of the Creative Commons Attribution (CC BY) license (https://creativecommons.org/licenses/by/4.0/).
Share and Cite
Scorrano, G.; La Bella, S.; Matricardi, S.; Chiarelli, F.; Giannini, C. Neuroendocrine Effects on the Risk of Metabolic Syndrome in Children. Metabolites 2023, 13, 810. https://doi.org/10.3390/metabo13070810
Scorrano G, La Bella S, Matricardi S, Chiarelli F, Giannini C. Neuroendocrine Effects on the Risk of Metabolic Syndrome in Children. Metabolites. 2023; 13(7):810. https://doi.org/10.3390/metabo13070810
Chicago/Turabian StyleScorrano, Giovanna, Saverio La Bella, Sara Matricardi, Francesco Chiarelli, and Cosimo Giannini. 2023. "Neuroendocrine Effects on the Risk of Metabolic Syndrome in Children" Metabolites 13, no. 7: 810. https://doi.org/10.3390/metabo13070810
APA StyleScorrano, G., La Bella, S., Matricardi, S., Chiarelli, F., & Giannini, C. (2023). Neuroendocrine Effects on the Risk of Metabolic Syndrome in Children. Metabolites, 13(7), 810. https://doi.org/10.3390/metabo13070810